

Abstract
Purpose
DNA Ligase 4 (LIG4) is a key factor in the non-homologous end-joining (NHEJ) DNA double-strand break repair pathway needed for V(D)J recombination and the generation of the T receptor and immunoglobulin molecules. Defects in LIG4 result in a variable syndrome of growth retardation, pancytopenia, combined immunodeficiency, cellular radiosensitivity, and developmental delay.
Methods
We diagnosed a patient with LIG4 syndrome by radiosensitivity testing on peripheral blood cells, and established that two of her four healthy siblings carried the same compound heterozygous LIG4 mutations. An extensive analysis of the immune phenotype, cellular radiosensitivity, telomere length, and T and B cell antigen receptor repertoire was performed in all siblings.
Results
In the three genotypically affected individuals, variable severities of radiosensitivity, alterations of T and B cell counts with an increased percentage of memory cells, and hypogammaglobulinemia, were noticed. Analysis of T and B cell antigen receptor repertoires demonstrated increased usage of alternative microhomology-mediated end-joining (MHMEJ) repair, leading to diminished N nucleotide addition and shorter CDR3 length. However, overall repertoire diversity was preserved.
Conclusions
We demonstrate that LIG4 syndrome presents with high clinical variability even within the same family, and that distinctive immunologic abnormalities may be observed also in yet asymptomatic individuals.
Keywords: Ligase 4 deficiency, DNA repair, Telomere length, Immune repertoire, Microhomology-mediated end-joining
Introduction
DNA Ligase 4 (LIG4) is a component of the ubiquitous non-homologous end-joining (NHEJ) DNA double-strand break (DSB) repair pathway that is also involved in the process of V(D)J recombination [1]. After the Variable (V), Diversity (D), and Joining (J) elements of the T cell receptor (TCR) and immunoglobulin genes have been targeted at recombination signal sequences by the recombination-activating gene 1 (RAG1) and RAG2 endonuclease complex [2], DNA ends are stabilized by the Ku70 and Ku80 proteins. The holoenzyme DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is then recruited to stabilize the complex and to activate the endonuclease ARTEMIS, which opens the hairpins formed at the DNA coding ends [3]. Before these are joined, nucleotides may be added or deleted at the junctions between coding elements, thus contributing further to diversity. In particular, nucleotides resulting from asymmetric resolution of the hairpin lead to introduction of palindromic sequences (P-nucleotides), whereas additional nucleotides may be added by the enzyme terminal deoxynucleotidyl transferase (TdT) [4]. The nucleotides inserted between V and D, or V and J segments, are referred to as N1, and those introduced between D and J segments, are referred to as N2 nucleotides. Eventually, the coding ends are ligated by DNA Ligase 4 (LIG4) and its co-factors XRCC4 and XLF [5, 6].
The V(D)J recombination process is specific for lymphocytes and mandatory for T and B cell development. Consequently, severe defects of the NHEJ pathway lead to absence of T and B cells, and manifest as severe combined immunodeficiency (SCID). However, defects allowing partial function may result in milder presentations of combined immunodeficiency (CID) or “leaky SCID” [7]. In addition, defects of the NHEJ pathway cause increased and generalized cellular sensitivity to ionizing radiation (IR).
The LIG4 syndrome (OMIM#606593) is an autosomal recessive genetic disorder caused by hypomorphic mutations in the LIG4 gene. Complete absence of LIG4 function causes late embryonic lethality due to impaired neuronal development [8, 9]. Patients with LIG4 deficiency manifest short stature, microcephaly, variable degrees of CID, developmental delay, pancytopenia, and severe cellular radiosensitivity [10–15]. LIG4 deficiency is also associated with chromosomal instability [16], and an increased risk of malignancies [11, 13, 17–19]. The fact that some patients have a more severe immunodeficiency, consistent with SCID or Omenn syndrome (OS) [17, 18, 20, 21], and present severe neurological abnormalities [22], while others have less pronounced immune defects and develop normally[19, 23], is illustrative of the variable clinical expressivity of LIG4 syndrome. In some patients, the diagnosis of LIG4 deficiency is made even during adulthood [24].
The severity of the clinical presentation of LIG4 deficiency has been correlated with the nature and the position of the mutations. In particular, mutations affecting the XRCC4-binding domain are associated with “microcephalic primordial dwarfism” (MPD), resembling what recently reported also in XRCC4-deficient patients [25], whereas earlier truncations on one or both alleles of the LIG4 gene result in more severe phenotypes [26]. Besides growth failure, patients mutated in XRCC4 showed neurological presentations, severe radiosensitivity, but no immunodeficiency, and no abnormalities in V(D)J recombination in particular [27].
In this manuscript we demonstrate intrafamilial phenotypic variability of LIG4 deficiency. Among three siblings who shared the same bi-allelic LIG4 mutations, the index patient manifested moderately severe features of LIG4 syndrome, whereas the remaining two mutated siblings were largely asymptomatic. This clinical heterogeneity was associated with variable abnormalities of the immunological phenotype, of cellular sensitivity to IR, and of composition of the T cell receptor β (TRB) and immunoglobulin heavy chain (IGH) repertoires.
Methods
Patients and samples
Blood samples from all patients and controls were obtained upon informed consent according to Boston Children’s Hospital IRB protocol 04-09-113R. Generation of a fibroblast cell line established from P2 has been previously reported [16].
DNA sequencing
DNA was isolated from PBMCs using the Puregene blood kit from Qiagen (Qiagen Inc., Valencia CA, USA). The two LIG4 mutations in P2 have been reported previously [16]. For sequencing of the other siblings, the regions around the mutations were amplified using Choice Taq™ DNA polymerase (Denville Scientific Inc., South Plainfield, NJ, USA); primer sequences are reported in the Supplementary Material. Samples were gel-purified and Sanger sequencing was performed by Eton Bioscience Inc. (Charlestown, MA, USA).
Analysis of lymphocyte subsets
Analysis of B and T cell subsets was performed by flow cytometry on whole blood after red blood cell lysis (BD Pharm lyse, BD Biosciences, San Jose, CA, USA), using mouse anti-human CD3 (FITC, Biolegend, San Diego, CA), CD4 (PE, Biolegend), CD8 (APC, Biolegend), CD19 (PerCP-Cy5.5, Biolegend), CD21 (PE, BD Bioscience), CD24 (APC, Biolegend), CD27 (APC, eBioscience, San Diego, CA), CD38 (FITC, eBioscience), CD45RA (FITC, Biolegend), CD45RO (PerCP-Cy5.5, Biolegend), CCR7 (Pacific Blue, Biolegend), IgM (PE, Southern-Biotec, Birmingham, AL), IgD (FITC, BD Bioscience). Data were acquired on an LSR-Fortessa (BD Bioscience) and analyzed with FlowJo software (Version 9.6.4).
Radiosensitivity Testing
Radiosensitivity testing was performed on fibroblasts from P2 upon irradiation with 5Gy as described previously16. Confocal imaging was performed on a Fluoview FV 1000 microscope (UplanApo 10x and 60x/1.2na water lenses, Olympus). The numbers of nuclear foci per cell were counted manually or with Volocity software (PerkinElmer) after careful setup.
PBMCs were irradiated with 10Gy, fixed after 1h, 6h, 24h and 40h using the Fix&Perm permeabilization kit (Invitrogen Inc/Life Technologies, Grand Island, NY, USA) and chilled methanol, and stained for CD45 (mouse anti-human CD45, BD Pharmigen™ #557833), CD3 (BV421 mouse anti-human CD3, BD Horizon™ #562426) and phospho Histon H2A.X (clone JBW301, mouse monoclonal FITC, Millipore, #16-202A) or Isotype (Mouse IgG1 k, clone MOPC-21, FITC-conjugate, Milli-Mark™, # FCMAB299F), respectively. Mean fluorescence intensities (MFI) of γH2AX were evaluated on gated CD45+CD3+ lymphocytes. The fold inductions were calculated from the MFIs of unirradiated cells and irradiated cells after given time points.
Telomere length
Telomere length was measured on peripheral blood mononuclear cells using flow cytometry and fluorescence in situ hybridization (FISH) [28]. The telomere length in lymphocytes and granulocytes was individually plotted relative to a nomogram of healthy controls.
Next generation sequencing of TRB and IGHC rearranged products
CD3+ lymphocytes were isolated from PBMCs by MACS purification (Miltenyi Biotec, San Diego, CA, USA). Genomic DNA was isolated from CD3+ lymphocytes using the Purgene blood kit (Qiagen Inc.). Next generation sequencing [29] at the TRB locus was performed according to a protocol by Adaptive® Biotechnologies (Seattle, WA, USA) [29].
NGS of IGH products was performed on cDNA obtained from isolated CD19+ B cells, using primers for the human immunoglobulin heavy chain (HIGH) from iRepertoire® (iRepertoire Inc, Huntsville, AL, USA) as reported previously29. Libraries were sequenced using GS Junior 454 (Roche, Mannheim, Germany).
Raw data for TRB and IGH CDR3 sequences were provided by iAdaptive® and iRepertoire®, respectively. Cumulative sequences were analyzed in Microsoft Excel, and diversity indices were calculated with the software PAST as described [30]. Heat maps were generated with GENE-E (www.broadinstitute.org) using raw data formatted in Excel. For all other analysis, raw data were formatted from .csv files in Excel into FASTA files using Genious® software. Subsequently, FASTA files were uploaded into the ImMunoGeneTics (IMGT) database and analyzed with the IGMT/high V quest tool [31], followed by the IgAT analysis tool [32]. For IGH sequencing, the two healthy brothers and one unrelated healthy individual served as controls; for TRB sequencing, data from age matched controls (18–35y) provided from Adaptive® were used. Since the controls from Adaptive® had too large data sets to be analyzed by IgAT (more than 300,000 unique sequences), the 25,000 most abundant unique sequences were selected for analysis.
Statistical analysis
Distributions of TRB and IGH sequences were compared based on a two-tailed unpaired students t test, and statistical significance was determined at p ≤ 0.05.
Results
Clinical features
The index patient (P2) (Fig. 1) is the fourth child of a Caucasian Northern American family. She presented at the age of 7 years with recurrent pneumonias and sinusitis. Laboratory investigations at the age of 13 years showed severe panhypogammaglobulinemia associated with lymphopenia, and low B cell count in particular. The patient was started on IVIG treatment and antibiotic prophylaxis with trimethoprim-sulfamethoxazole. At the age of 13 years, her short stature (Fig. S1) and small head circumference (occipital frontal circumference – OFC - < 2nd percentile) prompted investigation for a DNA repair defect. Upon irradiation of peripheral blood mononuclear cells (PBMC) with 10Gy, increased DNA damage and delayed kinetics of DNA repair, as shown by elevated levels of phosphorylated H2AX (γH2X) (Fig. S2), were consistent with what we had previously demonstrated for various LIG4-deficient cell lines [16], and were therefore suggestive of LIG4 syndrome.
Fig. 1. Two compound heterozygous LIG4 mutations are found in the index patient and two of her siblings.

Diagram of the pedigree and annotation of LIG4 genotypes. The mutation c.1345A>C (K449Q) was found on one allele in the father, and c.2440C>T (R814X) in the mother. Both mutations were inherited by siblings P1, P2, and P3. III.3 is heterozygous for the c.2440C>T mutation. The symbol “+” denotes the wild type allele.
By contrast, P1 and P3 did not show increased susceptibility to infections (other than one episode of walking pneumonia in P1). Whereas P1 was small for his age (OFC is not known), P3 has normal height and weight and an OFC on the 25th percentile (Fig. S1 and data not shown).
Genetic diagnosis
Sequencing of the LIG4 gene in P2 revealed two compound heterozygous mutations, c.1345A>C (p.K449Q), and c.2440C>T (p.R814*) (Fig. 1, Fig. S3). Screening of family members revealed that P2’s father and mother were heterozygous for the p.K449Q and the p.R814* mutation, respectively. Unexpectedly, the older brother (P1) and the younger sister (P3) were compound heterozygous for both mutations. Finally, two other healthy brothers (III.2 and III.3) were either heterozygous for one LIG4 mutation or homozygous for wild-type alleles.
The p.R814* mutation, leading to a late truncation of the protein in the XRCC4-binding domain, has been frequently reported in patients with LIG4 syndrome [26]. Homozygosity for this mutation is associated with mild disease, whereas compound heterozygosity with another mutation affecting the catalytic domain has been reported in patients with moderately severe disease. The p.K449Q mutation affects the enzymatic domain and has not been reported previously. It is predicted to have a deleterious effect according to PolyPhen2 [33] and SIFT [34] and has a scaled CADD score of 24.9 [35]. According to the genome browser of the Exome Aggregation Consortium (ExAC), Cambridge, MA (URL: http://exac.broadinstitute.org) [02/2016], p.K449Q is a single nucleotide variant (SNV) (rs138156481) with an allele frequency of 8.238E-6, compared to 8.237E-5 for the p.R814* mutant.
Laboratory investigations
Immunological studies revealed lymphopenia with low CD3+ T cell count in both P2 and P3, whereas P1 had a normal lymphocyte count, and his T cell count was modestly decreased (Table 1). However, the proportion of naïve CD4+ and CD8+ T cells was markedly decreased in all three siblings. T cell proliferation to mitogen stimulation was modestly impaired in P2 and P3, and was not analyzed in P1.
Table 1.
Cell counts and antibody titers of LIG4-deficient siblings.
| P1 | P2 | P3 | Reference Values | |
|---|---|---|---|---|
| Age at evaluation (y) | 23 | 17 (12a) | 12 | |
| WBC (x103/μl) |
4.41

|
8.72 |
3.08
|
P2/P3: 5.5–9.3 P1: 5.0–10.0 |
| Lymphocytes (x103/μl) | 1.15 |
0.82
|
0.52
|
P2/P3: 1.17–2.3 P1: 0.95–2.04 |
| Neutrophils (x103/μl) |
2.47
|
7.3
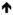
|
2.17
|
P2/P3: 3.04–6.06 P1: 3.54–7.52 |
| Hemoglobin (g/dl) | 16.7 | 12.5 |
10.6
|
P2/P3: 11.3–13.4 P1: 11.4–15.1 |
| MCV (fl) |
93
|
92.4
|
95.5
|
P2/P3: 82.1–87.7 P1: 82–92 |
| Platelet (x103/μl) | 228 | 238 | 197 | P2/P3: 189–342 P1: 150–450 |
| Immunoglobulins | ||||
| IgG (mg/dL) | 942 | 2380b (149a
) |
628
|
639–1344 (584–1509) P1: 751–1560 |
| IgA (mg/dL) | n.a.c |
<7
(10a
)
|
321
|
70–312 (45–237) |
| IgM (mg/dL) | n.a. |
<5
(18a
)
|
44 | 40–240 (49–230) |
| IgE (units/ml) | n.a. | <1 | 211 | P2: <200 P3: <500 |
| Lymphocyte subsets | ||||
| T cells | ||||
| CD3+ (x103/μl) (%) |
891
(77%) |
771
(82%) |
444
(69%) |
1000–2600 (56–84%) |
| CD3+CD4+ (x103/μl) (%) |
396
(34%) |
308
(33%) |
213
(33%) |
530–1500 (31–52%) |
| CD3+CD8+ (x103/μl) (%) | 469 (41%
) |
410 (44%
) |
195 (30%) | 330–1100 (18–35%) |
| Naive CD4+CD45RA+CCR7+ (%) |
8.65
|
2.1
|
10.7
|
P2/P3: 31.3–69.6 P1: 21–61.4 |
| Effector memory CD4+CD45RA−CCR7− (%) |
26.6
|
23.9 |
30.9
|
P2/P3: 7.8–25.9 P1: 7.6–25.1 |
| Central memory CD4+CD45RA−CCR7+ (%) |
63
|
73.7
|
56.8
|
P2/P3: 21–41.3 P1: 26.8–62.1 |
| Naïve CD8+CD45RA+CCR7+ (%) |
4.32
|
2.1
|
7.7
|
P2/P3: 31.1–73.2 P1: 11.4–66.5 |
| Effector memory CD8+CD45RA−CCR7− (%) | 42.3 |
47.8
|
63.2
|
P2/P3: 8.8–44.4 P1: 16.8–54.6 |
| Central memory CD8+CD45RA−CCR7+ (%) |
41.8
|
48.5
|
24
|
P2/P3: 2.6–8.7 P1: 3.7–23.2 |
| B cells | ||||
| CD19+/μl (%) |
53
(5%
)
|
12
(1%
)
|
10
(2%
) |
110–570 (6–23%) |
| Naïve IgD+CD27− B cells (%) |
46.7
|
76.3 | 63.8 | P2/P3: 51.3–82.5 P1: 48.4–79.7 |
| Switched memory IgD−CD27+ B cells (%) | 22.5 |
7
|
14.7 | P2/P3: 8.7–25.6 P1: 8.3–27.8 |
| Unswitched memory IgD+CD27+ B cells (%) |
25.3
|
1.1
|
16.2 | P2/P3: 4.6–18.2 P1: 7–23.8 |
| Transitional CD24hiCD38hi B cells (%) | 7.05 | 5.8 | 13.3 | P2/P3: 5.3–18.9 |
| Plasmablasts CD24lowCD38hi (%) | 2.4 | 1.6 |
7.9
|
P2/P3: 0.3–2.5 |
| CD19+CD21lowCD38low B cells (%) |
16.6
|
39.7
|
25.7
|
P2/P3:1.1–11.9 |
| Marginal zone-like CD24hiCD38low B cells (%) | 35.3 | 32.4 | 22.7 | P2/P3: 10.7–39.1 |
| NK cells | ||||
| CD3+CD56+/μl (%) | 202 (17%) | 140 (15%) | 171 (27%
) |
70–480 (3–22%) |
| T cell response to mitogen stimulation | ||||
| Con A (CPM) | n.a. | 69450 | 43145 | 74586-1943 |
| PHA (CPM) | n.a. | 55617 | 89873 | 104415-319 |
| Anti-CD3 (CPM) | n.a. | 97836 | 57283 | 78423-2054 |
| T cell mitogen background (CPM) | n.a. | 2496 | 1742 | 321–2510 |
| Response to immunization | ||||
| Tetanus (IU/ml) | n.a. | 0.471 | 2.19 | 0.15–7 |
| Pneumococcal titers (# serotypes with protective titers / 23 serotypes) after vaccination with PSV23 | n.a. | 2/231 | 5/23 |
Before start IVIG treatment
Under IVIG treatment
Not available
While P2 had been diagnosed with panhypogammaglobulinemia, P3 had slightly reduced levels of serum IgG, but increased levels of IgA, and normal serum IgM. P1 had normal levels of IgG, and was not investigated for IgA and IgM at his own request. P2 and P3 had protective titers of antibodies to tetanus toxoid; however upon immunization with a 23-valent pneumococcal polysaccharide (PPSV23) vaccine, protective antibody responses were detected against only 2 and 5 of 23 serotypes in P2 and P3, respectively. All three siblings manifested severe B cell lymphopenia, but some differences were noticed in the distribution of B cell subsets. In particular, the percentage of switched and unswitched memory B cells was markedly reduced in P2, but was normal in P3. All three siblings had an increased proportion of CD21lowCD38low CD19+ B cells, which are enriched for self-reactive specificities [36]. However, autoantibodies were not detected in any of the three siblings (data not shown). Moderate anemia was present in P3, and the red cell mean corpuscular volume was elevated in all three siblings. The absolute neutrophil count (ANC) was slightly decreased in P1 and P3.
Cellular radiosensitivity testing and telomere length
Compared to a fibroblast cell line obtained from a patient with severe LIG4 syndrome (411BR) and a healthy control, P2’s fibroblasts showed intermediate radiosensitivity, suggesting a mild cellular phenotype caused by the p.K449Q and the p.R814* mutations (Fig. 2a). Following the unexpected discovery that also P1 and P3 carried the same bi-allelic LIG4 mutations, comparative testing of IR sensitivity was performed on T cells from all three siblings. Upon exposure of PBMCs to 10Gy, induction and resolution of DNA damage were measured at various time points. As shown in Fig. 2b and Fig. S2, increased DNA damage and delayed kinetics of DNA repair were observed in P2 and P3, as compared to what observed in T cells from the healthy siblings III.2 (C1) and III.3 (C2). However, a milder phenotype was detected in P1.
Fig. 2. Increased sensitivity to ionizing radiation (IR) and short telomere length in genotypically affected siblings.

Fibroblasts of P2 were irradiated with 5Gy and γH2AX foci were counted in at least 50 cells using a Fluoview FV 1000 confocal microscope (Olympus) with an UplanApo 60x/1.2na water lens (Olympus) at given time points after IR. Numbers of γH2AX foci/cell are compared to the established LIG4-deficient cell line 411BR [11] and a healthy control (a). PBMCs from patients P1-P3 and two controls C1-C2 (healthy siblings III.2 and III.3) were irradiated with 10Gy, and γH2AX was assessed by flow-cytometry. The γH2AX fold induction was calculated by dividing the mean fluorescence intensities (MFI) of unirradiated cells by the MFI of irradiated cells of each time point (* p≤0.05) (b).
Telomere length in lymphocytes (c) and granulocytes (d) is expressed relative to healthy age-matched controls. The colored lines represent percentile thresholds. In two controls, granulocyte event numbers were too low to measure the telomere length with confidence.
Telomere length was markedly reduced (<1st percentile) in both lymphocytes and granulocytes of all three genotypically affected siblings, but not in their healthy brothers C1 and C2 (Fig. 2c–d).
Analysis of TRB and IGHC repertoire diversity and composition
In order to investigate whether the increased susceptibility to infections in P2, but not in P1 or P3, could also reflect differences of T and B cell repertoire diversity and composition, we performed NGS of TRB and IGH rearrangements. The two genotypically unaffected siblings (C1 and C2) and an unrelated healthy subject (C3) served as internal controls. Ecology parameters were used to analyze diversity of amino acid sequences used in the TRB and IGH repertoire (Shannon’s H index) and unevenness of their distribution among the T and B cell populations (frequency of top 100 clones). As shown in Fig. 3a, TRB repertoire diversity was significantly different between controls and patients, however, this was not the case for the IGH repertoire (Fig. 3b). To further document the presence of clonotypic expansions, we selected the top 100 most abundant unique clones, and analyzed what fraction of the total sequences they represented. The most abundantly expressed TRB clonotypes accounted for a much higher fraction of all clonotypes in each of three LIG4-mutated siblings as compared to controls (Fig. 3c), whereas expansion of IGH clonotypes was observed in P2 and P3 only (Fig. 3d). The presence of TRB clonotypic expansions in all three LIG4-mutated siblings was confirmed when measuring the D50 diversity index, which estimates the minimal percentage of unique sequences that account for 50% of the total sequences expressed (Fig. 3e). By contrast, abnormalities of evenness of the IGH repertoire were more subtle (Fig. 3f).
Fig. 3. Evaluation of TRB and IGH repertoire diversity shows modest clonal expansions.

The Shannon Entropy index (H) was used to measure repertoire diversity in unique TRB (a) and IGH (b) sequences in each patient and controls. Groups of 3 controls and 3 patients (P1-P3) are shown, black bars indicate mean values, colored bars the SD. The top 100 most abundant unique TRB (c) and IGH (d) clones were considered, and their frequency was reported as referred to the cumulative number of sequences in the whole data set. Horizontal bars indicate mean value (***, p≤0.001). The cumulative frequencies of total TRB (e) and IGH (f) sequences are plotted against the cumulative frequencies of unique sequences in controls (C1-C3) and patients (P1-P3).
Next, we analyzed pairing of individual V and J genes in unique TRB and IGH sequences. As compared to controls, reduced TRBV/TRBJ pairing diversity was observed in P1, P2, and P3 (Fig. 4). Along with lack or marked reduction in the frequency of rearrangements containing certain TRBV genes (6.8, 7.7, 6.9, 16.1), increased frequency of other TRBV/TRBJ gene pairing (V5.1-J1.1/J1.2; V6.5-J1.1/J1.2; V5.1-J2.7; V7.2-J2.7) was appreciated, especially in P2 and P3. When analyzed at the gene family level, increased frequency of unique sequences containing TRBV5 and TRBJ1, and reduced frequency of sequences with no TRBD gene and of those containing TRBJ2, were present. Interestingly, these abnormalities were either detected in P2 and P3 only, or were more pronounced in them than in P1 (Fig. S4). By contrast, no distinctive restriction of individual IGHV/IGHJ gene rearrangements was observed in the LIG4-mutated siblings (Fig. S5), although there was increased frequency of unique sequences using no IGHD gene and of sequences containing IGHJ3 gene family, and decreased usage of those with IGHJ4 (Fig. S6).
Fig. 4. The TRB repertoire of LIG4-deficient patients P1-P2 shows minimal skewing.

The global usage of unique TRB sequences is demonstrated for 3 healthy controls and P1-P3 on a heat map showing TCRBV genes joint with TCRBJ genes. The frequency of gene usage is color-coded, using yellow and red to indicate rare and most common pairing of V-J genes, respectively. Blank boxes indicate absence of that specific rearrangement in the total population of sequences obtained.
The complementarity determining region 3 (CDR3) of T and B cell receptor molecules plays a critical role in antigen binding [29], and abnormalities of IGH CDR3 length and hydrophobicity profile have a major impact on immune competence and self/non-self discrimination [37, 38]. A shorter CDR3 length was observed for both total and unique TRB (Fig. 5a–b) and IGH (Fig. 5c–d) sequences in all three affected siblings, but was more pronounced in P2 and P3. Possible mechanisms accounting for shorter CDR3 length include: reduced D gene usage, preferential usage of shorter D gene segments, increased nucleotide deletions, or reduced nucleotide additions. No evidence for increased deletion of nucleotides at coding joins was observed in TRB and IGH sequences from the affected siblings (data not shown). By contrast, N nucleotide addition was consistently reduced. In particular, an increased proportion of unique TRB-CDR3 sequences without N nucleotide addition were detected in P2 (N1: 2.5 fold more than in controls, ± SD 9.38; N2: 1.2 fold, ± SD 5.48), and a similar trend was observed also for P3 (Fig. 6a–b). On the other hand, there were fewer sequences with larger amounts of N nucleotides added, especially for P2 and P3. A similar phenotype was observed for IGH-CDR3 sequences, especially in P2 and P3 (Fig. 6c–d).
Fig. 5. Shorter length of the CDR3 regions in LIG4-deficient individuals.

The relative frequency with which CDR3 sequences of indicated length (expressed as number of nucleotides, nt) occur is expressed as percentage of total (a) and unique (b) TRB CDR3 sequences, and total (c) and unique (d) IGH CDR3 sequences for P1-P2, as compared to mean control values. Error bars on control columns indicate SD.
Fig. 6. Sequences obtained from LIG4-deficient individuals show significantly reduced N nucleotide addition between joined V(D)J gene elements.

The percentage of unique sequences with indicated numbers of N nucleotides added are shown for TRB (a, b) and IGH (c, d). N1 nucleotides are added between V and D or V and J gene elements, and N2 nucleotides between D and J elements. No N nucleotide addition is indicated as 0, each 3 additional N nucleotides added are shown in one bar up to more than 16 (16+) N nucleotides; error bars indicate SD (* p≤0.05; ** p≤0.01).
On average, the CDR3 region of unique TRB clonotypes contained 1.2 less N1 and 1.2 less N2 nucleotide insertions in the patients vs. controls (Fig. S7a). In TRB clones characterized by direct V-J joining (without D gene contribution), N1 was on average 2.9 nucleotides shorter in patients than in controls, and this difference was even higher for P2 and P3 (3.4 nucleotides) (Fig. S7b). Similarly, the CDR3 region from unique IGH sequences contained on average 0.8 less N1 and 1.0 less N2 nucleotides in the three affected siblings than in controls (Fig. S8a), and 0.4 less N1 nucleotides in clones without D gene usage (Fig. S8b).
Small alterations of amino acid composition of the CDR3 region of IGH transcripts were present in the patients as compared to controls, with reduced abundance of Tyrosine (Y) residues in P2 and P3, and greater abundance of the hydrophobic Alanine (A) in all patients (data not shown). However, these differences did not significantly alter the IGH-CDR3 hydrophobicity profile (data not shown).
Increased usage of microhomology-mediated end-joining in TRB and IGH sequences from LIG4-mutated siblings
In the absence of LIG4, DNA DSBs can be repaired by an alternative end-joining pathway involving microhomology-mediated end joining (MHMEJ) and the enzymes LIG3 and PARP1, or another back-up pathway involving LIG1 [39]. In this process, short stretches of microhomology (as little as 2bp) are paired with the complementary DNA strand and overhangs are deleted. To analyze the frequency of MHMEJ during V(D)J recombination, we focused our attention on the most abundantly used V-(D)-J rearrangements in both TRB and IGH unique clonotypes from the LIG4-mutated siblings (as an example, frequencies of all possible TRBV-D joins are shown in Fig. S9). Sequences joined by MHMEJ do not involve N nucleotide addition, so only sequences without N nucleotides were analyzed. MHMEJ was defined as the use of at least 2bp microhomology, followed by at least 5bp of the clearly identifiable adjacent segment. The frequency of unique clonotypes with evidence of MHMEJ was assessed for each of these V-(D)-J joins in the patients, and in corresponding joins from healthy controls. In the TRB repertoire, an increased frequency of MHMEJ was observed in all three LIG4-mutated siblings (P1<P2/P3), as compared to controls (Fig. 7a). In IGH, evidence of MHMEJ was mainly found in P2 and in fewer clonotypes (Fig. 7b). Interestingly, we observed increased direct joining between IGHV and IGHJ elements in P2 and P3. In two cases, direct V-J joining in P2 was the result of MHMEJ. However, MHMEJ represented a very rare event, occurring in about 0.3–0.4% of overall unique sequences analyzed in P2.
Fig. 7. MHMEJ of V(D)J elements is more frequently used in LIG4-deficient patients than in controls.

MHMEJ was found in joins of V-D and D-J elements of TRB sequences (a). The diagram shows the frequency of specific joins mediated by MH, expressed as percentage of all unique sequences. Corresponding sequences found in the joint region are shown below. Some joins of TRBV5.1, TRBV5.4, TRBV5.6 with TRBD1 had 1nt deletion.
In contrast, MHMEJ was found only in three joins of the IGH locus, and almost exclusively in P2 (b). Some sequences had a 1nt insertion (indicated in blue) in the corresponding J segment.
Discussion
We report on three siblings who shared bi-allelic mutations in LIG4, but had a variable clinical presentation. In particular, two of them (P1, P3) had no significant history of infections, and P3 had normal growth. This variability of clinical severity was paralleled by differences in the degree of immunological abnormalities. In particular, while all three affected siblings manifested B cell lymphopenia, this was more pronounced in P2 and P3 than in P1. Moreover, P2 suffered from panhypogammaglobulinemia, whereas IgG serum levels were modestly reduced in P3 and normal in P1. Yet, severe B cell lymphopenia was observed in all three siblings, and is a typical finding also in patients with other hypomorphic NHEJ defects [40]. DNA rearrangements in the B cell lineage are not restricted to V(D)J recombination, but also include class switch recombination (CSR). Insufficient repair of DNA DSBs in LIG4-mutated B cells may lead to apoptosis and contribute to severe B cell lymphopenia. In the T cell compartment, depletion of naïve cells and increased proportion of memory T cells may reflect compensatory mechanisms of homeostatic proliferation.
Pancytopenia is a common feature of LIG4-deficiency, and can mark progression to bone marrow failure, which occurs in about 70% of the patients [26]. Some degree of pancytopenia was documented in P3. It is likely that environmental or infectious triggers may lead to accumulation of DNA DSBs in myeloid progenitors affecting cell survival. Early exhaustion of the hematopoietic stem cell niche has been documented in a mouse model [41]. The extent of exposure to such triggers could contribute to phenotypic variability, even in patients sharing the same genetic defects.
We documented abnormally short telomere length in both lymphocytes and granulocytes from the LIG4-mutated siblings. It is likely that the reduced telomere length in LIG4 syndrome reflects replicative stress in the setting of poor cell survival. In addition to short telomere length, the overlap between primary telomeropathies and LIG4 syndrome also includes bone marrow failure and radiosensitivity [42, 43], and therefore these features do not allow differential diagnosis.
Analysis of cellular radiosensitivity demonstrated that all three LIG4-mutated siblings showed increased sensitivity towards ionizing radiation with delayed repair kinetics characteristic for LIG4 deficiency, but with slightly different levels of severity. Correlation between the magnitude of DNA DSB repair capacities and the severity of clinical features has also been observed by others [44].
Hypomorphic mutations in genes involved in V(D)J recombination may affect composition, richness, and diversity of T and B cell receptor repertoires. In particular, restriction and skewing of T and B cell repertoires have been documented in patients with RAG mutations [45, 46]. Although diversified TRB and IGH repertoires were documented in the three LIG4-deficient patients studied here, clonotypic expansions and preferential usage of some gene families were noticed, especially in the TRB repertoire. In particular, the Vβ5 family was used significantly more often by P2 and P3, and Jβ1 was preferred over Jβ2.
Previous studies had shown that CSR junctions of Lig4/LIG4-deficient mouse and human B cells are biased towards MH usage [47, 48]. We have observed an increased rate of MHMEJ in the patients – particularly in P2, and less prominently in P1 - than in controls. When MHMEJ is used during V(D)J recombination, the addition of N nucleotides to facilitate joining cannot take place. Indeed, reduced N nucleotide addition, including a significantly increased percentage of joins without any N nucleotides, was observed in the TRB and IGH repertoires of LIG4-mutated patients, thus contributing to the shorter length of the CDR3 sequences. Although shorter CDR3 length and reduced N nucleotide addition have been previously described in patients with LIG4 syndrome18, to our knowledge this is the first time that evidence for increased MHMEJ in freshly isolated T and B lymphocytes from these patients has been documented. We also found that gene families preferentially used in the patients show an advantage for MH when joined with certain D or J genes. This was particularly true for the TRB repertoire, where increased usage of the TRBV5.1, TRBV6.5 and TRBV7.2 genes was documented in the LIG4-mutated patients.
Aberrant direct IGHV-IGHJ joins were observed at higher frequency in the LIG4-mutated siblings, especially P2. In two cases, we obtained evidence that the direct V-J join was the product of MHMEJ. This pathway may allow joining of DNA ends flanking targeted IGHV-IGHJ segments that cannot be joined by NHEJ, because such joining would contradict the 12–23 rule. The increased usage of MHMEJ in LIG4-mutated patients has important implications also for genomic instability. In fact, it is well established that MHMEJ may allow joining of unrepaired RAG-induced DNA DSB with a reciprocal genomic partner, thereby increasing the risk for chromosomal translocations [49] (in particular, interchromosomal fusions with MH at the fusion points mediated by LIG3 [50]) and cancer development. Finally, although MHMEJ accounted only for a minority of joins detected in the LIG4-mutated patients reported here, it is important to recognize that this may represent an underestimate of the real frequency with which MHMEJ operates, because this mechanism of DNA repair is often associated with the introduction of deletions, which may impede recognition of MH. Furthermore, MH may happen after N nucleotides have been added.
The identification of immunological abnormalities in clinically asymptomatic subjects represents an important challenge in medicine. Together with recent reports of adult patients with mild clinical presentation, our description of LIG4 mutations in asymptomatic individuals indicates that the clinical spectrum of LIG4-deficiency is broader than previously anticipated. Shorter telomere length, reduced proportion of naïve T cells, B cell lymphopenia, and increased cellular radiosensitivity represent important laboratory biomarkers that should prompt considering LIG4 deficiency even in yet asymptomatic subjects. Presymptomatic identification of LIG4 deficiency may have important prognostic implications, because progressive accumulation of DNA DSBs in stem and progenitor cells may eventually lead to cell exhaustion and development of clinical symptoms, including infections, bone marrow failure, and malignancies. It may further impact treatment decisions in regard to DNA-damaging alkylating agents, since increased toxicity can be anticipated [19]. As clinical manifestations develop later in life, it could be assumed that only severe cases will be identified at newborn screening.
Supplementary Material
Acknowledgments
We thank all our patients and controls for participating in this study, and Dr. Frederick Alt for helpful comments and advice. This work was supported by a grant (5R01AI100887) from the National Institute of Allergy and Infectious Diseases – NIH to L.D.N.
Footnotes
Author contributions
K Felgentreff performed flow-cytometry analysis on P1, genetic sequencing of the siblings, radiosensitivity testing and NGS, and wrote the manuscript. S Baxi is the immunologist caring for the patients; he identified the index patient, and provided samples and all clinical data. YN Lee helped with the analysis of the TRB and IGH repertoire. K Dobbs performed high throughput sequencing and helped with DNA sequencing and flow-cytometry. L Henderson helped with analysis of the TRB NGS data. K Csomos was involved in sample preparation for NGS sequencing. E Tsitsikov analyzed the lymphocyte subsets of P2 and P3. M Armanios analyzed the telomere length in granulocytes and lymphocytes. J Walter and L.D. Notarangelo planned the study and wrote the manuscript. In addition, L.D. Notarangelo supervised K Felgentreff in this project.
Disclosure of conflict of interest
None of the authors have any conflicts of interest to declare.
References
- 1.Lieber MR, Ma Y, Pannicke U, Schwarz K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA repair. 2004;3(8–9):817–26. doi: 10.1016/j.dnarep.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 2.Rucci F, Notarangelo LD, Fazeli A, Patrizi L, Hickernell T, Paganini T, et al. Homozygous DNA ligase IV R278H mutation in mice leads to leaky SCID and represents a model for human LIG4 syndrome. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3024–9. doi: 10.1073/pnas.0914865107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108(6):781–94. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 4.Janeway CA, Jr, TP, Walport M, et al. The generation of diverstity in immunoglobulins. New York: Garland Science; 2001. [Google Scholar]
- 5.Grawunder U, Wilm M, Wu X, Kulesza P, Wilson TE, Mann M, et al. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388(6641):492–5. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 6.Buck D, Malivert L, de Chasseval R, Barraud A, Fondaneche MC, Sanal O, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124(2):287–99. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 7.Shearer WT, Dunn E, Notarangelo LD, Dvorak CC, Puck JM, Logan BR, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. The Journal of allergy and clinical immunology. 2014;133(4):1092–8. doi: 10.1016/j.jaci.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frank KM, Sekiguchi JM, Seidl KJ, Swat W, Rathbun GA, Cheng HL, et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396(6707):173–7. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 9.Gatz SA, Ju L, Gruber R, Hoffmann E, Carr AM, Wang ZQ, et al. Requirement for DNA ligase IV during embryonic neuronal development. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(27):10088–100. doi: 10.1523/JNEUROSCI.1324-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Molecular cell. 2001;8(6):1175–85. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 11.Ben-Omran TI, Cerosaletti K, Concannon P, Weitzman S, Nezarati MM. A patient with mutations in DNA Ligase IV: clinical features and overlap with Nijmegen breakage syndrome. American journal of medical genetics Part A. 2005;137A(3):283–7. doi: 10.1002/ajmg.a.30869. [DOI] [PubMed] [Google Scholar]
- 12.Gruhn B, Seidel J, Zintl F, Varon R, Tonnies H, Neitzel H, et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet journal of rare diseases. 2007;2:5. doi: 10.1186/1750-1172-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toita N, Hatano N, Ono S, Yamada M, Kobayashi R, Kobayashi I, et al. Epstein-Barr virus-associated B-cell lymphoma in a patient with DNA ligase IV (LIG4) syndrome. American journal of medical genetics Part A. 2007;143A(7):742–5. doi: 10.1002/ajmg.a.31644. [DOI] [PubMed] [Google Scholar]
- 14.Unal S, Cerosaletti K, Uckan-Cetinkaya D, Cetin M, Gumruk F. A novel mutation in a family with DNA ligase IV deficiency syndrome. Pediatric blood & cancer. 2009;53(3):482–4. doi: 10.1002/pbc.22031. [DOI] [PubMed] [Google Scholar]
- 15.Tamura S, Higuchi K, Tamaki M, Inoue C, Awazawa R, Mitsuki N, et al. Novel compound heterozygous DNA ligase IV mutations in an adolescent with a slowly-progressing radiosensitive-severe combined immunodeficiency. Clinical immunology. 2015 doi: 10.1016/j.clim.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Felgentreff K, Du L, Weinacht KG, Dobbs K, Bartish M, Giliani S, et al. Differential role of nonhomologous end joining factors in the generation, DNA damage response, and myeloid differentiation of human induced pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(24):8889–94. doi: 10.1073/pnas.1323649111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. European journal of immunology. 2006;36(1):224–35. doi: 10.1002/eji.200535401. [DOI] [PubMed] [Google Scholar]
- 18.Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. Journal of immunology. 2006;176(8):5060–8. doi: 10.4049/jimmunol.176.8.5060. [DOI] [PubMed] [Google Scholar]
- 19.Riballo E, Critchlow SE, Teo SH, Doherty AJ, Priestley A, Broughton B, et al. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Current biology : CB. 1999;9(13):699–702. doi: 10.1016/s0960-9822(99)80311-x. [DOI] [PubMed] [Google Scholar]
- 20.van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. The Journal of clinical investigation. 2006;116(1):137–45. doi: 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grunebaum E, Bates A, Roifman CM. Omenn syndrome is associated with mutations in DNA ligase IV. The Journal of allergy and clinical immunology. 2008;122(6):1219–20. doi: 10.1016/j.jaci.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 22.HIJ, Warris A, van der Flier M, Reisli I, Keles S, Chishimba S, et al. Clinical spectrum of LIG4 deficiency is broadened with severe dysmaturity, primordial dwarfism, and neurological abnormalities. Human mutation. 2013;34(12):1611–4. doi: 10.1002/humu.22436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riballo E, Doherty AJ, Dai Y, Stiff T, Oettinger MA, Jeggo PA, et al. Cellular and biochemical impact of a mutation in DNA ligase IV conferring clinical radiosensitivity. The Journal of biological chemistry. 2001;276(33):31124–32. doi: 10.1074/jbc.M103866200. [DOI] [PubMed] [Google Scholar]
- 24.Yue J, Lu H, Lan S, Liu J, Stein MN, Haffty BG, et al. Identification of the DNA repair defects in a case of Dubowitz syndrome. PloS one. 2013;8(1):e54389. doi: 10.1371/journal.pone.0054389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murray JE, van der Burg M, HIJ, Carroll P, Wu Q, Ochi T, et al. Mutations in the NHEJ component XRCC4 cause primordial dwarfism. American journal of human genetics. 2015;96(3):412–24. doi: 10.1016/j.ajhg.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murray JE, Bicknell LS, Yigit G, Duker AL, van Kogelenberg M, Haghayegh S, et al. Extreme growth failure is a common presentation of ligase IV deficiency. Human mutation. 2014;35(1):76–85. doi: 10.1002/humu.22461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo C, Nakazawa Y, Woodbine L, Bjorkman A, Shimada M, Fawcett H, et al. XRCC4 deficiency in human subjects causes a marked neurological phenotype but no overt immunodeficiency. The Journal of allergy and clinical immunology. 2015;136(4):1007–17. doi: 10.1016/j.jaci.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH) Nat Protoc. 2006;1(5):2365–76. doi: 10.1038/nprot.2006.263. [DOI] [PubMed] [Google Scholar]
- 29.Danska JS, Livingstone AM, Paragas V, Ishihara T, Fathman CG. The presumptive CDR3 regions of both T cell receptor alpha and beta chains determine T cell specificity for myoglobin peptides. The Journal of experimental medicine. 1990;172(1):27–33. doi: 10.1084/jem.172.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Connell AE, Volpi S, Dobbs K, Fiorini C, Tsitsikov E, de Boer H, et al. Next generation sequencing reveals skewing of the T and B cell receptor repertoires in patients with wiskott-Aldrich syndrome. Frontiers in immunology. 2014;5:340. doi: 10.3389/fimmu.2014.00340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604. doi: 10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
- 32.Rogosch T, Kerzel S, Hoi KH, Zhang Z, Maier RF, Ippolito GC, et al. Immunoglobulin analysis tool: a novel tool for the analysis of human and mouse heavy and light chain transcripts. Frontiers in immunology. 2012;3:176. doi: 10.3389/fimmu.2012.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 35.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, et al. Expansion of autoreactive unresponsive CD21-/low B cells in Sjogren’s syndrome-associated lymphoproliferation. Arthritis Rheum. 2013;65(4):1085–96. doi: 10.1002/art.37828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frankl JA, Thearle MS, Desmarais C, Bogardus C, Krakoff J. T-cell receptor repertoire variation may be associated with type 2 diabetes mellitus in humans. Diabetes Metab Res Rev. 2015 doi: 10.1002/dmrr.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.James LC, Tawfik DS. The specificity of cross-reactivity: promiscuous antibody binding involves specific hydrogen bonds rather than nonspecific hydrophobic stickiness. Protein science : a publication of the Protein Society. 2003;12(10):2183–93. doi: 10.1110/ps.03172703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simsek D, Brunet E, Wong SY, Katyal S, Gao Y, McKinnon PJ, et al. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLoS genetics. 2011;7(6):e1002080. doi: 10.1371/journal.pgen.1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Volk T, Pannicke U, Reisli I, Bulashevska A, Ritter J, Bjorkman A, et al. DCLRE1C (ARTEMIS) mutations causing phenotypes ranging from atypical severe combined immunodeficiency to mere antibody deficiency. Hum Mol Genet. 2015 doi: 10.1093/hmg/ddv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nijnik A, Woodbine L, Marchetti C, Dawson S, Lambe T, Liu C, et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature. 2007;447(7145):686–90. doi: 10.1038/nature05875. [DOI] [PubMed] [Google Scholar]
- 42.Stanley SE, Armanios M. The short and long telomere syndromes: paired paradigms for molecular medicine. Curr Opin Genet Dev. 2015;33:1–9. doi: 10.1016/j.gde.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stanley SE, Rao AD, Gable DL, McGrath-Morrow S, Armanios M. Radiation Sensitivity and Radiation Necrosis in the Short Telomere Syndromes. Int J Radiat Oncol Biol Phys. 2015;93(5):1115–7. doi: 10.1016/j.ijrobp.2015.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA repair. 2014;16:84–96. doi: 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 45.Yu X, Almeida JR, Darko S, van der Burg M, DeRavin SS, Malech H, et al. Human syndromes of immunodeficiency and dysregulation are characterized by distinct defects in T-cell receptor repertoire development. The Journal of allergy and clinical immunology. 2014;133(4):1109–15. doi: 10.1016/j.jaci.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.HIJ, Driessen GJ, Moorhouse MJ, Hartwig NG, Wolska-Kusnierz B, Kalwak K, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. The Journal of allergy and clinical immunology. 2014;133(4):1124–33. doi: 10.1016/j.jaci.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449(7161):478–82. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 48.Pan-Hammarstrom Q, Jones AM, Lahdesmaki A, Zhou W, Gatti RA, Hammarstrom L, et al. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. The Journal of experimental medicine. 2005;201(2):189–94. doi: 10.1084/jem.20040772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferguson DO, Sekiguchi JM, Chang S, Frank KM, Gao Y, DePinho RA, et al. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(12):6630–3. doi: 10.1073/pnas.110152897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jones RE, Oh S, Grimstead JW, Zimbric J, Roger L, Heppel NH, et al. Escape from telomere-driven crisis is DNA ligase III dependent. Cell reports. 2014;8(4):1063–76. doi: 10.1016/j.celrep.2014.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.