

Abstract
Cell lines derived from the fall armyworm, Spodoptera frugiperda (Sf), are widely used as hosts for recombinant protein production in the baculovirus-insect cell system (BICS). However, it was recently discovered that these cell lines are contaminated with a virus, now known as Sf-rhabdovirus [1]. The detection of this adventitious agent raised a potential safety issue that could adversely impact the BICS as a commercial recombinant protein production platform. Thus, we examined the properties of Sf-RVN, an Sf-rhabdovirus-negative Sf cell line, as a potential alternative host. Nested RT-PCR assays showed Sf-RVN cells had no detectable Sf-rhabdovirus over the course of 60 passages in continuous culture. The general properties of Sf-RVN cells, including their average growth rates, diameters, morphologies, and viabilities after baculovirus infection, were virtually identical to those of Sf9 cells. Baculovirus-infected Sf-RVN and Sf9 cells produced equivalent levels of three recombinant proteins, including an intracellular prokaryotic protein and two secreted eukaryotic glycoproteins, and provided similar N-glycosylation patterns. In fact, except for the absence of Sf-rhabdovirus, the only difference between Sf-RVN and Sf9 cells was SF-RVN produced higher levels of infectious baculovirus progeny. These results show Sf-RVN cells can be used as improved, alternative hosts to circumvent the potential safety hazard associated with the use of Sf-rhabdovirus-contaminated Sf cells for recombinant protein manufacturing with the BICS.
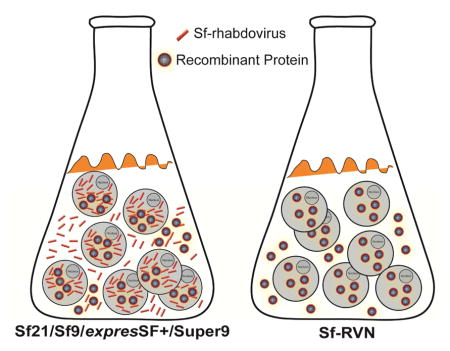
Graphical Abstract
1. Introduction
Since it was first described in the peer-reviewed literature in the early 1980’s [2, 3], the baculovirus-insect cell system (BICS) has become a widely recognized and heavily utilized recombinant protein production platform [4, 5]. The advantages of the BICS include its flexibility, speed, simplicity, eukaryotic protein processing capacity, and ability to produce multi-subunit protein complexes. For nearly 30 years, the BICS was used mainly to produce recombinant proteins for basic research in academic and industrial labs. More recently, however, the BICS has emerged as a bona fide commercial manufacturing platform, which is now being used to produce several biologics licensed for use in human (Cervarix®, Provenge®, Glybera® and Flublok®) or veterinary (Porcilis® Pesti, BAYOVAC CSF E2®, Circumvent® PCV, Ingelvac CircoFLEX® and Porcilis® PCV) medicine [6]. In addition, the BICS is being used to produce several other biologic candidates, including potential vaccines against norovirus, parvovirus, Ebola virus, respiratory syncytial virus, and hepatitis E virus, which are in various stages of human clinical trials [6].
The insect cell lines most commonly used as hosts in the BICS are derived from the cabbage looper, T. ni, or fall armyworm, Spodoptera frugiperda (Sf), and most biologics manufactured with the BICS are produced using the latter. The original Sf cell line, designated IPLB-SF-21, also known as Sf-21, was derived from pupal ovaries in 1977 [7]. Other commonly used Sf cell lines include Sf9, a subclone of IPLB-SF-21 [8], and its daughter subclones, Super 9 [9] and Sf900+, also known as expresSF+ [10].
Recently, Ma and coworkers [1] discovered that every Sf cell line tested, including Sf21 and Sf9 cells obtained from two reputable commercial sources, was contaminated with a novel rhabdovirus. This adventitious agent, which is now known as Sf-rhabdovirus, was discovered in a general effort to evaluate the biosafety of the BICS platform. A research group at Takeda Vaccines, Inc. independently confirmed the presence of Sf-rhabdovirus in the Sf9 cells used to produce their norovirus vaccine candidate [11]. In addition, we found that all our lab Sf-21, Sf9, and expresSF+ cell stocks, obtained from a variety of sources, are contaminated with this virus (data not shown). Thus, Sf-rhabdovirus appears to be a very common, if not universal, contaminant of the Sf cell lines commonly used as hosts for recombinant protein production in the BICS.
It is important to note that Sf-rhabdovirus is unlikely to be harmful for humans, as it cannot replicate in human or monkey cell lines [1]. Moreover, the only detectable phylogenetic relationship between Sf-rhabdovirus and previously recognized viruses was found in a short sequence, which was related to the L genes of insect and plant rhabdoviruses [1]. Therefore, the potential impact of Sf-rhabdovirus on biosafety of the BICS platform, if any, remains speculative at this time. Nevertheless, whenever adventitious agents are discovered in human biologics, the usual response is to take steps to remove them from the product. Ideally, this involves eradicating the adventitious agent from all stages of the manufacturing process. One notable precedent was the elimination of porcine circovirus from an attenuated live rotavirus vaccine and the Vero cells used to produce that vaccine [12, 13].
In this study, we characterized the relevant properties of an Sf-rhabdovirus-negative Sf cell line, Sf-RVN, as a potential alternative host for the BICS. We detected no Sf-rhabdovirus in these cells for 60 continuous passages in culture. Compared to Sf9 cells, Sf-RVN cells had virtually identical growth rates, sizes, morphologies, viabilities after baculovirus infection, and provided equivalent recombinant protein production levels and N-glycosylation profiles. Interestingly, Sf-RVN cells produced ~5–10 fold higher levels of infectious baculovirus progeny. These results validate Sf-RVN cells as an alternative host that can be used to circumvent the potential safety hazard associated with the use of Sf-rhabdovirus-contaminated Sf cells for recombinant protein manufacturing in the BICS.
2. Materials and methods
2.1. Insect cell culture
The isolation of Sf9 cells has been described [8] and the isolation of Sf-RVN cells will be described (Geisler, C., Maghodia, A.B., and Jarvis, D.L., submitted) elsewhere. Both cell lines were routinely maintained as shake-flask cultures at 28°C in ESF 921 medium (Expression Systems, Woodland, CA).
2.2. Sf-rhabdovirus and mycoplasma detection
Samples of Sf9 and Sf-RVN cultures containing 1 x 106 cells were harvested and the cells were pelleted by low speed centrifugation. The cell-free supernatants were filtered through a 0.22μm filter (CELLTREAT Scientific, Shirley, MA) and then ultracentrifuged at 131,000 x g for 22 h at 4°C. Total RNA was extracted from both the low speed cell and high speed cell-free pellets using the RNASolv reagent (Omega Bio-Tek, Inc., Norcross, GA), according to the manufacturer’s protocol. The RNAs were then quantified and used as templates for cDNA synthesis with the ProtoScript II First Strand cDNA synthesis kit (New England Biolabs, Ipswich, MA) and either an Sf-rhabdovirus-specific (320-SP1) primer or the oligo(dT)23-VN primer included in the kit, according to the manufacturer’s protocol. Equivalent amounts of each cDNA preparation were then used for PCR’s with Taq DNA polymerase, ThermoPol reaction buffer (New England Biolabs), and either Sf-rhabdovirus- (Mono-1 and Mono-2; [1]) or Sf ribosomal protein L3- (SfRPL3-SP and SfRPL3-ASP) specific primers, respectively. The reaction mixtures were incubated at 94°C for 3 min, cycled 35 times at 94°C for 30s, 55 °C for 1 min, and 72 °C for 1 min, and finally incubated at 72 °C for 10 min. For Sf-rhabdovirus-specific RNA detection, one μL of each primary PCR was then used as the template for secondary PCR’s under the same conditions, except the primers were nested Sf-rhabdovirus-specific primers (Mono-1i and Mono-2i; [1]). The sequence of each primer used for these assays is given in Table 1, which is a modified version of Table 1 in the publication by Ma and coworkers [1]. Primary and secondary PCR products were analyzed by agarose gel electrophoresis with ethidium bromide staining. In addition, the gel-purified primary PCR product obtained with primers Mono-1 and -2 was further purified on a HiBind DNA Mini Column (Omega-Biotek, Norcross, GA) according to the manufacturer’s protocol, and then directly sequenced using the same primers (Genewiz, South Plainfield, NJ).
Table 1.
Sequences of primers usfed in this study1.
| Primer | Sequence (5′ to 3′) | Position2 | Product size (bp)3 |
|---|---|---|---|
| Mono-1 | GGCAAGGCTGTTTGGATTACTGACC | 8365–8389 | |
| Mono-2 | ACAGGTTTGCAGCTAAGGAGGACA | 9158–9135 | 794 |
| Mono-3 | TGGCGAGGGACTGCTTACAGAAGG | 10630–10653 | |
| Mono-4 | CACAGCCGGGGGTGCAATCA | 11359–11340 | 730 |
| Mono-5 | ACAGGAGATGCGGAAGACCCCTC | 12137–12159 | |
| Mono-6 | ATCTCGCAGGTGGGACAACCCC | 12962–12941 | 826 |
| Mono-1i | ATATGAGAGCCCCAGACACACAGCC | 8571–8595 | |
| Mono-2i | ACGATGTGGTGAGAGAAACACCTCCT | 9071–9046 | 501 |
| 320-SP1 | CACATCTAGAGCTTGAAGACC | 6210–6230 | |
| 320-ASP1 | ACCATCACAGCCAGTGCTG | 6991–7010 | 481 |
| SfRPL3-SP | ACATCGAAACTCCTCATGGTCT | ||
| SfRPL3-ASP | TCTTGATAACCTTGCCATCCTT | 627 | |
| SfCOX1-SP | TGGAGCAGGAACTGGATG | ||
| SfCOX1-ASP | TCGACGAGGTATACCTGC | 958 | |
| SfGAPDH-SP | TCGGTATCAACGGTTTCG | ||
| SfGAPDH-ASP | GATCGATAACGCGGTTGG | 960 |
Mono- primers were adapted from Ma et al. (2014).
Numbers refer to starting and ending nucleotides in the Sf-rhabdovirus genome sequence (GenBank accession number KF94707).
Size of the amplification products resulting from PCR with odd/even primer pairs.
We also tested samples containing about 105 Sf-RVN and Sf9 cells for mycoplasma using the Universal Mycoplasma Detection kit from American Type Culture Collection (Manassas, VA), according to the manufacturer’s protocol.
2.3. Cell growth properties, morphologies, and diameters
Sf-RVN and Sf9 cells were seeded at a starting density of 1.0 x 106 cells/mL in 50-mL shake flask cultures, triplicate samples were removed every 24 h for 4 days, and viable cell densities and sizes were measured using a Countess® automated cell counter (ThermoFisher Scientific, Inc.). Doubling times were calculated using the formula: Td = T x Log2/Log(Q2/Q1) where Td = doubling time, T = time (h) elapsed since the last passage, Q1 = cell seeding density, and Q2 = viable cell count. Cell morphologies were documented by collecting phase contrast images at a magnification of 10X using an Olympus FSX-100 microscope and FSX-BSW imaging software.
2.4. Baculovirus expression vectors
A baculovirus expression vector designated BacPAK6-ΔChi/Cath encoding full-length, untagged E. coli β-galactosidase (β-gal) was produced in two sequential steps. In the first step, BacPAK6 viral DNA was recombined with a plasmid encoding E. coli β-glucuronidase under the control of the baculovirus p6.9 promoter. In this plasmid, the p6.9-β-glucuronidase gene was inserted in place of the AcMNPV chiA and v-cath genes and embedded within wild type AcMNPV flanking sequences. The desired recombinant was tentatively identified by its blue plaque phenotype in the presence of X-GlcA (RPI Corp., Mount Prospect, IL). The recombination site was confirmed by PCR with primers specific for the β-glucuronidase gene and 5′ UTR of the AcMNPV gp64 gene, which were internal and external to the transfer plasmid, respectively. This virus was amplified and viral DNA was isolated and digested with I-SceI to delete the entire β-glucuronidase expression cassette. In the second step, Sf9 cells were transfected with the I-SceI-digested viral DNA. The resulting progeny were resolved by plaque assay in the presence of X-GlcA and the final recombinant baculovirus, BacPAK6-ΔChi/Cath, was identified by its white plaque phenotype.
The recombinant baculovirus expression vectors designated AcP(−)p6.9hSEAP and AcP(−)p6.9hEPO encoded 8X HIS-tagged forms of human secreted alkaline phosphatase (hSEAP) and human erythropoietin (hEPO), respectively, under the control of AcMNPV p6.9 promoters and honeybee prepromellitin signal peptides. Synthetic genes encoding mature SEAP and EPO (Genbank NP_001623.3 amino acids 23-511 and Genbank NP_000790.2 amino acids 28-193, respectively) with N-terminal TEV protease cleavage sites (ENLYFQG) were designed using OPTIMIZER [14] to match AcMNPV codon usage (http://www.kazusa.or.jp). These sequences were synthesized, cloned, and sequenced by Genscript (Piscataway, N.J.) and error-free clones were used to produce recombinant baculovirus expression vectors by in vitro recombination with Ac6.9GT, as described previously [15].
Standard methods were used to plaque-purify, amplify, and titer recombinant baculovirus expression vectors in Sf9 cells, as described previously [16]. In addition, Sf-rhabdovirus-negative stocks were produced for this study. First, Sf9 cells were infected with working stocks of each baculovirus vector, and then baculoviral DNA was isolated using a standard method [17]. This method includes proteinase K, SDS, and RNaseA treatments, followed by phenol/chloroform/isoamyl alcohol extraction and DNA precipitation with isopropanol, which was expected to eliminate Sf-rhabdovirus and Sf-rhabdoviral RNA. The resulting baculoviral DNA preparations were then used to transfect Sf-RVN cells and the progeny were plaque-purified, amplified, and titered as described previously [16], except Sf-RVN were used as the hosts for plaque-purification and amplification, instead of Sf9 cells. During this process, we tested the baculoviral DNA-transfected and baculovirus-infected Sf-RVN cell extracts, as well as the pellets obtained by ultracentrifuging samples of the final working virus stocks, for the presence of Sf-rhabdoviral RNA using the RT-PCR/nested PCR assay, as described above, and detected no Sf-rhabdovirus contamination.
2.5. Recombinant protein expression
Sf-RVN and Sf9 cells in ESF 921 were seeded into six-well plates at densities of 1 x 106 cells/well. The cells were then mock-infected with ESF 921 or infected with Sf-rhabdovirus-negative stocks of BacPAK6-ΔChi/Cath, AcP(−)p6.9hSEAP, or AcP(−)p6.9hEPO at multiplicities of infection (MOIs) of either 0.1 or 5 plaque-forming units (pfu)/cell. At various times post infection, the infected cells were harvested, cell densities were measured, and the cells were pelleted by low speed centrifugation. The cells and cell-free media were then processed in various ways, depending upon the nature of the model protein being expressed and purpose of the experiment, as described below. In each case, however, the levels of recombinant protein in cell extracts and/or cell-free media were measured by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting [18, 19] with protein- or tag-specific primary antibodies and alkaline phosphatase-conjugated secondary antibodies, as specified below. Immunoreactive proteins were visualized using a standard alkaline phosphatase-based color reaction [20] and relative intensities were estimated by scanning and quantitating the bands using Image J software.
For β-gal, infected cell pellets were used to prepare cytoplasmic extracts for enzyme activity assays, as described previously [21]. Immunoblotting was performed using rabbit anti-β-gal (EMD Millipore Corporation, Germany) and alkaline phosphatase conjugated goat anti-rabbit IgG (Sigma-Aldrich, St. Louis, MO) as the primary and secondary probes, respectively.
For hSEAP, infected cell-free media were prepared for enzyme activity assays, as described previously [22] and immunoblotting was performed using mouse anti-penta-His (ThermoFisher) and alkaline phosphatase conjugated rabbit anti-mouse IgG (Sigma-Aldrich) as the primary and secondary probes, respectively.
For hEPO, infected cell-free media were prepared for immunoblotting with rabbit anti-hEPO (U-CyTech, Utrecht, The Netherlands) and alkaline phosphatase conjugated goat anti-rabbit IgG (Sigma-Aldrich) as the primary and secondary probes, respectively.
2.6. N-glycan analysis
Fifty mL shake flask cultures of Sf-RVN and Sf9 cells were infected with Sf-rhabdovirus-negative stocks of AcP(−)p6.9hEPO and hEPO was affinity purified from the cell- and virus-free supernatants using Ni-NTA resin (ThermoFisher), as described previously [23]. N-glycans were enzymatically released from the purified hEPO preparations by digestion with PNGaseF (New England Biolabs), and the released N-glycans were purified, derivatized, and analyzed by MALDI-TOF-MS as described previously [23]. Structures were assigned to peaks based on predicted masses and knowledge of the N-glycans produced in Sf cells. Relative quantification of different structures was accomplished by dividing the combined peak intensities from isotopic clusters of individual permethylated N-glycan structures by the total intensity of all annotated N-glycan peaks.
2.7. Species confirmation
Portions of the RPL-3, COX-1, and GAPDH genes were PCR amplified from Sf9 or Sf-RVN cell cDNAs, as described in section 2.2, with SfRPL3-SP and SfRPL3-ASP, SfCOX1-SP and SfCOX1-ASP, and SfGAPDH-SP and SfGAPDH-ASP as the primers, respectively (Table 1). The gel-purified PCR products were further purified using HiBind DNA Mini Columns (Omega-Biotek) according to the manufacturer’s protocol, directly sequenced (Genewiz), and the sequences were assembled and aligned using Vector NTI 10.3.1 (Invitrogen). The reference sequences used in the multiple sequence alignments and Genbank Accession numbers were Sf21 COX-1 (GCTM01000731.1), B. mori COX-1 (AK383601.1), T. ni COX-1 (KP200882.1 and AB533499.1), Sf21 GAPDH (GCTM01012875), B. mori GAPDH (XM_012690444.1), T. ni GAPDH (GBKU01013056.1 and GBKU01009778.1), and Sf21 RPL-3 (AY072287.1), B. mori RPL-3 (NM_001043661.1), and T. ni RPL-3 (GBKU01065447.13).
Results
3.1. Sf-RVN cells have no detectable Sf-rhabdovirus
Total RNA was isolated from Sf-RVN cell extracts at various passage levels and tested for the presence of Sf-rhabdovirus using RT-PCR/nested PCR, as described in Materials and methods. A strong amplification product of the expected size was observed when total RNA from Sf9 cells was used as a positive control for this assay (Fig. 1A), as expected [1]. In contrast, no products were observed when we used total RNAs isolated from Sf-RVN cells every five passages during the course of 60 sequential passages in our lab (Fig. 1A). We also observed no products in negative controls with total RNA isolated from D. melanogaster S2 cells, which reportedly do not support Sf-rhabdovirus replication [1]. Approximately equal amounts of the expected amplification product were observed when we used the same RNA preparations for RT-PCRs with an oligodT primer and Sf ribosomal protein L3-specific primers (Fig. 1B). In an extension of these assays, we observed strong amplification products of the expected sizes using two other Sf-rhabdovirus-specific primer pairs, Mono-3/Mono-4 and Mono-5/Mono-6 (Table 1), which were derived from other regions of the L-protein coding sequence [1], for RT-PCRs with total RNA isolated from Sf9, but not Sf-RVN cells (data not shown). We observed a strong amplification product of the expected size in RT-PCR/nested PCR assays with total RNA isolated from pellets obtained by ultracentrifuging Sf9, but not Sf-RVN cell-free media, as described in Materials and methods (Fig. 1C). Finally, we confirmed the diagnostic specificity of our PCR assay for Sf-rhabdovirus RNA by sequencing the primary PCR product obtained using Mono-1/Mono-2 primers with Sf9 cell cDNA as the template and performing a multiple sequence alignment (Fig. S1), which demonstrated the amplimer sequence matched the previously reported Sf-rhabdovirus sequence [1]. Together, these results demonstrated there was no detectable Sf-rhabdovirus RNA in Sf-RVN cells or cell-free media over the course of 60 passages, strongly suggesting these cells are Sf-rhabdovirus-free.
Fig. 1.

Sf-rhabdovirus assays. Total RNA isolated from Sf-RVN cells at various passage levels (from P0 to P60) was assayed for (A) Sf-rhabdovirus- or (B) Sf ribosomal protein L3-specific RNAs by RT-PCR/nested PCR or RT-PCR, respectively, as described in Materials and methods. In parallel, total RNA isolated from the pellet obtained by ultracentrifuging Sf-RVN cell-free media at P60 was assayed for (C) Sf-rhabdovirus-specific RNA by RT-PCR/nested PCR, as described in Materials and methods. Total RNAs from Sf9 cells (Sf9) or the pellet obtained by ultracentrifuging Sf9 cell-free media (CFM) were used as positive controls and total RNA from S2R+ cells (S2) was used as a negative control. An additional negative control reaction was performed with no template (H2O) and the lanes marked M show the 100-bp markers, with selected sizes indicated on the left.
3.2. Sf-RVN cells have no detectable mycoplasma
We also tested Sf-RVN and Sf9 cells for mycoplasma using the Universal Mycoplasma Detection kit from the American Type Culture Collection, as described in Materials and methods. This PCR-based assay uses primers complementary to sequences conserved in the 16S rRNA genes of over 60 different mycoplasma, acholeplasma, spiroplasma and ureaplasma species, including eight species frequently found as cell culture contaminants. The results demonstrated that neither the Sf9 nor the Sf-RVN cells used for this assay were detectably contaminated with mycoplasma. They also showed the absence of a PCR product was not due to inhibition of the PCR reaction by the insect cell lysates, as amplimers of the expected sizes were observed in PCRs performed using lysates spiked with the control template (lanes marked + in Fig. 2).
Fig. 2.

Mycoplasma assays. Sf-RVN and Sf9 cell extracts (−) were assayed for mycoplasma contamination using the PCR-based Universal Mycoplasma Detection Kit (American Type Culture Collection), as described in Materials and methods. A plasmid encoding an M. arginini rRNA target sequence was used as the positive control. Additional controls were performed using Sf-RVN and Sf9 cell lysates alone or spiked with the positive control plasmid (+) to determine if the lysate interfered with the assay. A negative control reaction was performed with no template (H2O). The lane marked M shows the 100-bp markers, with selected sizes indicated on the left.
3.3. Sf-RVN and Sf9 cells have the same general properties
The next set of experiments was performed to compare several general properties of Sf-RVN and Sf9 cells, including their culture densities, diameters, doubling times, morphologies, and viabilities in response to baculovirus infection. The results showed that Sf-RVN and Sf9 cells achieved virtually identical average densities over the course of four days after being seeded into parallel shake flask cultures in ESF-921 medium (Fig. 3A). This time frame encompassed the 2–3 days of growth typically allowed between serial passages during routine insect cell line maintenance. The results also revealed no significant differences in the average diameters (Fig. 3B), doubling times (Fig. 3C), or morphologies (Fig. 3D) of Sf-RVN and Sf9 cells during the course of these experiments. Finally, we found no significant differences in the viabilities of Sf-RVN and Sf9 cells in response to baculovirus infection, which were indistinguishable over 4 days after infection at multiplicities of either 0.1 (Fig. 4A) or 5 (Fig. 4B) pfu/cell. The time frame and two different MOIs used in this experiment encompassed the conditions typically used to produce either working baculovirus stocks or recombinant proteins, respectively. Overall, the results of these experiments demonstrated the general properties of Sf-RVN and Sf9 cells examined in this study are indistinguishable.
Fig. 3.

Cell growth and morphology. Sf-RVN and Sf9 cells were seeded into shake flasks at a density of 1.0 x 106 cells/mL in ESF 921 medium. Triplicate samples were harvested at various times after seeding and viable cell counts and diameters were measured with an automated cell counter, as described in Materials and methods. The Figure shows the (A) average viable cell densities, (B) diameters, and (C) doubling times measured in three independent experiments, as well as (D) phase contrast micrographs of Sf-RVN and Sf9 cells at a magnification of 10X. The error bars represent the confidence intervals (P<0.05).
Fig. 4.

Cell viability after baculovirus infection. Sf-RVN and Sf9 cells were infected with an Sf-rhabdovirus-negative stock of AcP(−)p6.9hSEAP at an MOI of (A) 0.1 or (B) 5 pfu/cell. Triplicate samples were harvested at various times post-infection and viability was measured using an automated cell counter, as described in Materials and methods. The plots show the average percent viability measured in two independent experiments. The error bars represent the confidence intervals (P<0.05).
3.4. Sf-RVN and Sf9 cells produce recombinant proteins at nearly identical levels
We subsequently compared the levels of baculovirus-mediated recombinant protein production supported by Sf-RVN and Sf9 cells using E. coli β-gal, a model bacterial, intracellular protein; hSEAP, a model human, secreted glycoprotein; and hEPO, a model human, secreted glycoprotein of biotechnological significance. It is important to emphasize that we prepared and used Sf-rhabdovirus-negative working stocks of each recombinant baculovirus for these studies, as described in Materials and methods.
The E. coli β-gal expression experiments showed there were no significant differences in the intracellular enzyme activity levels produced by Sf-RVN and Sf9 cells during 4 days of infection with the recombinant baculovirus (Fig. 5A). The representative immunoblotting results in Fig. 5B–5C indicated Sf-RVN produced slightly more total intracellular β-gal protein. An independent biological replicate in which we stained the gel with Coomassie Brilliant blue yielded the same result, but as in the experiment shown in Fig. 5, the increase in intracellular β-gal levels produced by Sf-RVN cells was not particularly remarkable (data not shown). Finally, we noted the levels of enzyme activity and immunoreactive intracellular β-gal produced by Sf-RVN and Sf9 cells were both lower at 4 days post-infection, as compared to earlier time points, which might reflect baculovirus-induced cytopathic effects, perhaps resulting in product degradation by cellular proteases at this very late time of infection.
Fig. 5.

Recombinant β-gal production. Sf-RVN and Sf9 cells were infected with an Sf-rhabdovirus-negative stock of BacPAK6-ΔChi/Cath at an MOI of 5 pfu/cell. Triplicate samples were harvested at various times post-infection, and clarified intracellular extracts were assayed for (A) β-gal activity, as described in Materials and methods. This plot shows the average results with error bars representing the confidence intervals (P<0.05). One set of extracts was also used to measure (B) total intracellular β-gal production levels by immunoblotting analysis, with (C) scanning laser densitometry to estimate relative immunoreactive band intensities, as described in Materials and methods.
Our analysis of hSEAP production and secretion during 4 days of infection yielded essentially the same results. In this case, we expanded the experiment to include both low (0.1 pfu/cell; Fig. 6A, 6B, 6C) and high (5 pfu/cell; Fig. 6D, 6E, 6F) MOI infections because some investigators have reported higher productivity with low, rather than conventional high MOI infections in the BICS [24]. The results showed there were no statistically significant differences in the levels of hSEAP activity produced by Sf-RVN and Sf9 cells infected at either low (Fig. 6A) or high (Fig. 6D) MOIs. The representative immunoblotting results shown in Fig. 6 indicated Sf9 produced slightly more hSEAP when infected at low (Fig. 6B–6C) and Sf-RVN produced slightly more hSEAP when infected at high (Fig. 6E–6F) MOI. However, these differences were neither remarkable nor completely reproducible in an independent biological replicate of this experiment (data not shown). Both Sf-RVN and Sf9 cells produced more hSEAP activity and immunoreactive extracellular protein at 1 day and about 3-fold more hSEAP activity by 4 days post-infection when infected at high MOI. We also noted there were no differences in the viabilities of Sf-RVN and Sf9 cells during 4 days of infection at either MOI, which were measured as part of the hSEAP expression and secretion experiments described here (Fig. 4).
Fig. 6.

Recombinant hSEAP production. Sf-RVN and Sf9 cells were infected with an Sf-rhabdovirus-negative stock of AcP(−)p6.9hSEAP at an MOI of (A, B, C) 0.1 or (D, E, F) 5 pfu/cell. Triplicate samples were harvested at various times post-infection, cell-free media were prepared and assayed for (A, D) hSEAP activity, as described in Materials and methods, and the average results were plotted with error bars representing the confidence intervals (P<0.05). One set of cell-free media was also used to measure (B, E) total extracellular hSEAP production levels by immunoblotting analysis, with (C, F) scanning laser densitometry to estimate relative immunoreactive band intensities, as described in Materials and methods.
Finally, we also obtained the same general results when we compared the levels of hEPO production and secretion by Sf-RVN and Sf9 cells. As there is no simple functional assay for this product, our analysis was limited to simply comparing the levels of immunoreactive hEPO secreted into the extracellular media by the two different cell types during a 4-day infection. The results of two independent biological replicates of this experiment revealed no major reproducible differences in the levels of secreted hEPO produced by Sf-RVN and Sf9 cells (Fig. 7A–7B).
Fig. 7.

Recombinant hEPO production. Sf-RVN and Sf9 cells were infected with an Sf-rhabdovirus-negative stock of AcP(−)p6.9hEPO at an MOI of 5 pfu/cell. Samples were harvested at various times post-infection and cell-free media were prepared and assayed for (A) total extracellular hEPO production levels by immunoblotting analysis, with (B) scanning laser densitometry used to estimate relative immunoreactive band intensities, as described in Materials and methods.
Together, these results demonstrated that Sf-RVN and Sf9 cells produce and secrete three different recombinant proteins at identical or nearly identical levels.
3.5. Sf-RVN and Sf9 cells provide similar protein N-glycosylation profiles
Another important factor to assess in comparing Sf-RVN and Sf9 cells is their protein N-glycosylation patterns, as those provided by different cell lines can be quite different. Thus, we infected Sf9 and Sf-RVN cells with AcP(−)p6.9hEPO, purified the secreted hEPO from the cell-free media, enzymatically released total N-glycans, and analyzed the permethylated products by MALDI-TOF MS, as described in Materials and methods. The results showed the vast majority of the N-glycans linked to hEPO produced by both cell lines had trimannosyl core structures (labeled 2 and 3 in Fig. 8A), as expected. We also observed small proportions of hybrid-type structures with a terminal N-acetylglucosamine residue (labeled 4 and 5 in Fig. 8A), as expected. By quantifying these different structures, we determined the hEPO N-glycosylation profiles provided by Sf-RVN and Sf9 cells were highly similar, though the Sf-RVN cell product had slightly more fucosylated N-glycans (Fig. 8B).
Fig. 8.

N-glycosylation profiles. Sf-RVN and Sf9 cells were infected with an Sf-rhabdovirus-negative stock of AcP(−)p6.9hEPO at MOI of 3 pfu/cell and hEPO-His was affinity-purified from the cell free media, as described in Materials and methods. N-glycans were enzymatically released, recovered, permethylated, and (A) analyzed by MALDI-TOF MS, as described in Materials and methods, with molecular ions detected as [M + Na]+ assigned structures, annotated using the standard cartoon symbolic representations, numbered for simplicity, and (B) presented as percentages of total.
3.6. Sf-RVN cells produce more infectious baculovirus progeny
In addition to their utility as hosts for recombinant protein production, Sf9 cells are widely considered to be among the best hosts for production of working baculovirus stocks. Thus, it was of interest to compare the amounts of infectious recombinant baculoviral vector progeny produced by Sf9 and Sf-RVN cells. This experiment involved infecting both cell types with two different Sf-rhabdovirus-negative baculovirus stocks, AcP(−)p6.9hEPO and AcP(−)p6.9hSEAP, harvesting the budded viral progeny from all four infections, and comparing the infectious viral titers in plaque assays, as described in Materials and methods. The results of three independent biological replicates showed working stocks of both baculoviruses had ~5–10 fold higher titers when produced by Sf-RVN, as compared to those produced by Sf9 cells (Fig. 9).
Fig. 9.

Recombinant baculovirus production. Sf-RVN and Sf9 cells were infected with Sf-rhabdovirus-negative stocks of AcP(−)p6.9hSEAP or AcP(−)p6.9hEPO. The resulting progeny were harvested and titered by plaque assays, as described in Materials and methods. The resulting titers were plotted as the average viral titers obtained in three independent experiments, with error bars representing confidence intervals (P<0.05 or 0.001). and methods.
3.7. Sf-RVN is an S. frugiperda cell line
In a final set of experiments, we confirmed Sf-RVN is an S. frugiperda cell line by directly sequencing PCR amplimers corresponding to portions of the ribosomal protein L3 (RPL-3), cytochrome c oxidase 1 (COX-1) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) genes. Each amplimer was obtained using gene-specific primers designed using the Sf21 genome sequence (Table 1) and Sf-RVN or Sf9 cell cDNA as the template. Multiple sequence alignments including the experimentally determined Sf-RVN and Sf9 amplimer sequences and the Sf21, Trichoplusia ni, and Bombyx mori RPL-3, COX-1, and GAPDH sequences deposited in GenBank are shown in Fig. S2, S3, and S4, respectively. The results demonstrated the Sf-RVN RPL-3 and GAPDH sequences perfectly matched those from Sf9 and Sf21 and the Sf-RVN COX-1 sequence perfectly matched the COX-1 sequence from Sf9 and Sf21, except for a 3 bp deletion in the latter. In contrast, there were multiple differences between the Sf-RVN, T. ni, and B. mori RPL-3, COX-1, and GAPDH sequences. Thus, these results demonstrate Sf-RVN is, indeed, an S. frugiperda cell line.
4. Discussion
The recent surge of regulatory approvals for the use of BICS-derived biologics in human and veterinary patients is a critically important milestone in the emergence of the BICS as a bona fide commercial biologics manufacturing platform [6]. However, the discovery of adventitious agents in the Sf cell lines used most frequently as hosts for baculovirus vectors [1] raises concerns about the safety of BICS-produced biologics. In this context, it is important to emphasize that there is no evidence that Sf-rhabdovirus poses a threat to human or veterinary patients; in fact, there is evidence to the contrary [1]. Nevertheless, the clear response to the identification of any adventitious agent in any biologic manufacturing platform is to eliminate the agent to create an inherently safer system. Thus, in this report, we characterized Sf-RVN, which does not appear to be contaminated with Sf-rhabdovirus.
This conclusion is based on the results of a highly sensitive RT-PCR/nested PCR assay, which we used to demonstrate that Sf-RVN cells and cell-free media had no detectable Sf-rhabdovirus RNA over the course of 60 passages in our lab. We recognize these results do not prove there is no Sf-rhabdovirus in Sf-RVN cells. However, this claim is strongly supported by the duration of our RT-PCR/nested PCR testing regimen, which spanned 60 serial passages of Sf-RVN cells over the course of nearly half a year, as described herein. In fact, this experiment is still ongoing and, at this time, we still have not detected any trace of Sf-rhabdovirus in Sf-RVN cells after 80 continuous serial passages over the past eight months (data not shown). If these cells had a low level of Sf-rhabdoviral contamination, we would expect this virus to replicate fairly quickly to detectable levels, considering the reportedly high level of contamination (2 X 109 particles/mL of extracellular growth medium) in other Sf cell cultures [1].
The second major conclusion from this study is that the essential properties of Sf-RVN cells, in context of their potential as an alternative host for the BICS, are highly similar to those of Sf9 cells, which we used as a “gold standard” host for the BICS due to their widespread use in the field. We found that neither our Sf-RVN nor our Sf9 cells are detectably contaminated with mycoplasma. We also found that the basic growth properties of Sf-RVN and Sf9 cells, examined within the parameters of standard cell culture maintenance protocols, were indistinguishable. Their average diameters, doubling times, and morphologies, which were measured during the course of the comparative growth experiments, were indistinguishable, as well. Finally, we found both cell lines had the same general response to baculovirus infection, at least in terms of the impact of infection on cell viability during 4 days of infection.
The third major conclusion from this study is that Sf-RVN cells can function at least as well as Sf9 cells as the host component of the BICS. This conclusion was supported by the finding that Sf-RVN and Sf9 cells supported approximately equal levels of recombinant protein and glycoprotein production, secretion, and enzyme activity. Formally, this conclusion may be applied only to the three different products used as models in our study. While we used an intracellular bacterial protein and two secreted human N-glycoproteins in an effort to broaden our analysis, it is possible that Sf-RVN cells will be found to produce higher or lower levels of other recombinant proteins in the future. We also found that Sf-RVN and Sf9 cells provided highly similar N-glycosylation patterns. While Sf-RVN cells produced hEPO with a slightly higher proportion of core-fucosylated N-glycans, this was a relatively minor quantitative, not a major qualitative difference, and we have observed this level of variation in N-glycosylation of multiple preparations of hEPO produced in individual insect cell lines. In fact, we previously reported an hEPO preparation from Sf9 cells had >95% core fucosylated N-glycans [25]. Thus, we consider the minor differences observed in core fucosylation of hEPO by Sf-RVN and Sf9 cells to be unremarkable. Formally, our conclusions about the N-glycosylation patterns provided by Sf-RVN and Sf9 cells formally apply only to hEPO, as this was the model used in our study. However, compared to the potential for variation in recombinant protein production levels, Sf-RVN and Sf9 are far less likely to differentially N-glycosylate another glycoprotein because the analytical results obtained with any given product reflect endogenous N-glycan processing capabilities. If they existed, differences in the extent of N-glycan processing would most likely have been detected in our analysis of hEPO glycosylation by the two different cell lines.
The last important functional capability of Sf-RVN and Sf9 cells examined in this study was their ability to produce infectious recombinant baculovirus progeny. Interestingly, we found Sf-RVN cells produced ~5–10 fold more infectious progeny when used to propagate two different recombinant baculoviruses, as compared to Sf9 cells. This difference was statistically significant and demonstrates a clear advantage of Sf-RVN over Sf9 cells.
We found the absence of any major differences in the growth properties or recombinant protein production levels observed in Sf-RVN and Sf9 cells surprising. We expected even a relatively innocuous persistent infection, such as the Sf-rhabdovirus infection, would impose a metabolic load and/or other deleterious effects that would reduce the growth and or productivity of Sf9 cells. Apparently, however, infection with Sf-rhabdovirus has little impact, except for a reduction in the ability to support production of infectious baculovirus progeny. Perhaps this reflects competition between some aspect of the Sf-rhabdovirus and baculovirus replication cycles. While we have no direct information on the nature of the Sf-rhabdovirus replication cycle in Sf9 cells, it is reasonable to speculate that it replicates in the cytoplasm, like other rhabdoviruses [26]. In addition, we know that Sf-rhabdovirus is a negative strand RNA virus with no requirement for viral DNA replication [26], while the baculovirus used in this study is a double-stranded DNA virus that replicates in the nucleus and requires viral DNA replication [27]. Given these likely differences in subcellular sites of viral replication, viral genomes and genome replication mechanisms, and the absence of any clear impact of Sf-rhabdovirus contamination on recombinant protein production, it seems unlikely these two viruses compete in the early stages of replication required to produce their respective viral proteins, genomes, and intracellular progeny. However, both viruses ultimately must bud from the plasma membrane of the infected cell. So, in the final analysis, we might speculate that the most likely step at which Sf-rhabdovirus impacts the baculovirus replication cycle is budding, which is the final step in the production of infectious baculovirus progeny.
Finally, we believe it is important to report that recent bioinformatic analyses of the published Sf-21 genome [28] and transcriptome [29] in our lab indicated these cells harbor additional sequences distinct from, but related to Sf-rhabdovirus. We have experimentally confirmed the presence of these newly identified sequences in genomic DNA and total RNA isolated from Sf9 and Sf-RVN cells. However, we found sequences from only four of the five canonical Rhabdoviral genes and only two included intact open reading frames. Therefore, unlike the Sf-rhabdovirus sequences discovered by Ma and coworkers [1], these newly identified sequences cannot produce a virus or virus-like particle. We also learned from a review of the literature that integration of partial copies of Rhabdovirus transcripts into the host cell germline, a phenomenon termed “fossilization”, is common and widespread in insect species [30]. We conclude that the new Sf-rhabdovirus-related sequences are not relevant to the present study. They are most likely fossil sequences, not sequences derived from a second replicating, particle-producing rhabdovirus in Sf cells and, as such, has no impact on Sf-RVN cells as improved alternative hosts for the BICS. The new rhabdovirus-related sequences identified in our follow-up study and their evolutionary relationship to Sf-rhabdovirus will be described elsewhere (Geisler, C. and Jarvis, D.L., manuscript in preparation).
In summary, this report described the relevant properties of Sf-RVN, an Sf-rhabdovirus-negative Sf cell line, as a potential replacement for Sf9 cells as the host component of the BICS. The striking similarities between Sf-RVN and Sf9 cells suggest this would be a seamless replacement, as any bioprocess previously established using Sf9 and/or other Sf cells would likely be directly applicable to Sf-RVN. Thus, Sf-RVN is a compelling alternative that can be used to enhance the safety profile of the BICS and restore confidence in this emerging biologics manufacturing platform to the level enjoyed before the discovery of Sf-rhabdovirus as an adventitious agent.
Supplementary Material
Sf-rhabdovirus PCR product sequence. The diagnostic Sf-rhabdovirus PCR product obtained using primers Mono-1 and -2 and Sf9 cell cDNA as the template was directly sequenced, as described in Materials and methods, and the resulting DNA sequence was aligned with the Sf-rhabdovirus genome sequence originally published by Ma and coworkers [1].
Sf-RVN RPL-3 gene sequence. The Sf RPL-3 gene fragments amplified using Sf-RVN or Sf9 cell cDNA as the templates were sequenced and aligned with the Sf21, T. ni, and B. mori RPL-3 sequences from GenBank, as described in Materials and methods.
Sf-RVN COX-1 gene sequence. The Sf COX-1 gene fragments amplified using Sf-RVN or Sf9 cell cDNA as the templates were sequenced and aligned with the Sf21, T. ni, and B. mori COX-1 sequences from GenBank, as described in Materials and methods.
Sf-RVN GAPDH gene sequence. The Sf GAPDH gene fragments amplified using Sf-RVN or Sf9 cell cDNA as the templates were sequenced and aligned with the Sf21, T. ni, and B. mori GAPDH sequences from GenBank, as described in Materials and methods.
Highlights.
Spodoptera frugiperda (Sf) cells are widely used as hosts in the baculovirus expression system.
All Sf cell lines tested have been found to be contaminated with a novel Rhabdovirus.
This study introduces Sf-RVN, an Sf-rhabdovirus-negative cell line.
The relevant properties of these cells are nearly identical to contaminated Sf9 cells.
Thus, the use of Sf-RVN cells will enhance the biosafety of the baculovirus expression system.
Acknowledgments
We sincerely thank Dr. Hideaki Mabashi-Asazuma (University of Wyoming) for providing BacPAK6-ΔChi/Cath for use in this study. We also thank Dr. Jesse Gatlin (University of Wyoming) for constructive comments during the course of this study. This work was supported by Awards R43 GM102982 and R43 AI112118 from the National Institutes of Health, Institutes of General Medical Sciences and Allergy and Infectious Diseases, respectively.
Abbreviations
- MALDI-TOF MS
matrix assisted laser desorption ionization time of flight mass spectrometry
- PCR
polymerase chain reaction
- PNGase
peptide-N4-(N-acetyl-β-glucosaminyl) asparagine amidase
- Sf
Spodoptera frugiperda
Footnotes
Conflict of Interest
A.B.M. and C.G. are employees and D.L.J. is the President of GlycoBac, LLC. Accordingly, all authors declare potential conflicts of interest as we expect GlycoBac will provide the new cell line reported herein as a commercial product, which will be of financial benefit to the company.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, National Institute of General Medical Sciences or National Institute of Allergy and Infectious Diseases.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ma H, Galvin TA, Glasner DR, Shaheduzzaman S, Khan AS. Identification of a novel rhabdovirus in Spodoptera frugiperda cell lines. J Virol. 2014;88:6576–6585. doi: 10.1128/JVI.00780-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith GE, Summers MD, Fraser MJ. Production of human beta interferon in insect cells infected with a baculovirus expression vector. Mol Cell Biol. 1983;3:2156–2165. doi: 10.1128/mcb.3.12.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pennock GD, Shoemaker C, Miller LK. Strong and regulated expression of Escherichia coli beta-galactosidase in insect cells with a baculovirus vector. Mol Cell Biol. 1984;4:399–406. doi: 10.1128/mcb.4.3.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kost TA, Condreay JP, Jarvis DL. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat Biotechnol. 2005;23:567–575. doi: 10.1038/nbt1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Oers MM, Pijlman GP, Vlak JM. Thirty years of baculovirus-insect cell protein expression: from dark horse to mainstream technology. J Gen Virol. 2015;96:6–23. doi: 10.1099/vir.0.067108-0. [DOI] [PubMed] [Google Scholar]
- 6.Felberbaum RS. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors. Biotechnol J. 2015;10:702–714. doi: 10.1002/biot.201400438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaughn JL, Goodwin RH, Thompkins GJ, McCawley P. The establishment of two insect cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae) In Vitro. 1977;13:213–217. doi: 10.1007/BF02615077. [DOI] [PubMed] [Google Scholar]
- 8.Summers MD, Smith GE. A manual of methods for baculovirus vectors and insect cell culture procedures. Tx Ag Expt Stn Bull No. 1987;1555:5. [Google Scholar]
- 9.Marino JPJ, McAtee JJ, Washburn DG. PCT, editor. SEH and 11 β-HSD1 inhibitors and their use. SmithKline Beecham Corp; 2009. [Google Scholar]
- 10.Smith GE, Knell J, Vosnesensky A. USPTO, editor. Spodoptera frugiperda single cell suspension cell line in serum-free media, methods of producing and using. Protein Sciences Corporation; USA: 2011. [Google Scholar]
- 11.Haynes J. U.S.P.A.T. Office, editor. WO2015051255 A1. Methods of detection and removal of rhabdovirus from cell lines. 2015
- 12.Gilliland SM, Forrest L, Carre H, Jenkins A, Berry N, Martin J, Minor P, Schepelmann S. Investigation of porcine circovirus contamination in human vaccines. Biologicals. 2012;40:270–277. doi: 10.1016/j.biologicals.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Dubin G, Toussaint JF, Cassart JP, Howe B, Boyce D, Friedland L, Abu-Elyazeed R, Poncelet S, Han HH, Debrus S. Investigation of a regulatory agency enquiry into potential porcine circovirus type 1 contamination of the human rotavirus vaccine, Rotarix: approach and outcome. Hum Vacc Immunother. 2013;9:2398–2408. doi: 10.4161/hv.25973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puigbo P, Guzman E, Romeu A, Garcia-Vallve S. OPTIMIZER: a web server for optimizing the codon usage of DNA sequences. Nucl Acids Res. 2007;35:W126–131. doi: 10.1093/nar/gkm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toth AM, Geisler C, Aumiller JJ, Jarvis DL. Factors affecting recombinant Western equine encephalitis virus glycoprotein production in the baculovirus system. Prot Expr Purif. 2011;80:274–282. doi: 10.1016/j.pep.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarvis DL. Recombinant protein expression in baculovirus-infected insect cells. Meth Enzymol. 2014;536:149–163. doi: 10.1016/B978-0-12-420070-8.00013-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Reilly DR, Miller LK, Luckow VA. Baculovirus expression vectors. W.H. Freeman and Company; New York: 1992. [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blake MS, Johnston KH, Russell-Jones GJ, Gotschlich EC. A rapid, sensitive method for detection of alkaline phosphatase conjugated anti-antibody on western blot. Analyt Biochem. 1984;36:175–179. doi: 10.1016/0003-2697(84)90320-8. [DOI] [PubMed] [Google Scholar]
- 21.Miller JH. Experiments in Molecular Genetics. CSH Laboratory Press; Cold Spring Harbor, NY: 1972. Assay of β-galactosidase; pp. 352–355. [Google Scholar]
- 22.Lin CH, Jarvis DL. Utility of temporally distinct baculovirus promoters for constitutive and baculovirus-inducible transgene expression in transformed insect cells. J Biotechnol. 2013;165:11–17. doi: 10.1016/j.jbiotec.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geisler C, Mabashi-Asazuma H, Kuo CW, Khoo KH, Jarvis DL. Engineering beta1,4-galactosyltransferase I to reduce secretion and enhance N-glycan elongation in insect cells. J Biotechnol. 2015;193:52–65. doi: 10.1016/j.jbiotec.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liebman JM, LaSala D, Wang W, Steed PM. When less is more: enhanced baculovirus production of recombinant proteins at very low multiplicities of infection. Biotechniques. 1999;26:36–42. doi: 10.2144/99261bm05. [DOI] [PubMed] [Google Scholar]
- 25.Toth AM, Kuo CW, Khoo KH, Jarvis DL. A new insect cell glycoengineering approach provides baculovirus-inducible glycogene expression and increases human-type glycosylation efficiency. J Biotechnol. 2014;182–183:19–29. doi: 10.1016/j.jbiotec.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wagner RR. The Rhabdoviruses. Springer Sciences + Business Media; New York: 1987. [Google Scholar]
- 27.Rohrmann GF. Baculovirus Molecular Biology: Second Edition [Internet] National Center for Biotechnology Information (US); Bethesda (MD): 2011. [Google Scholar]
- 28.Kakumani PK, Malhotra P, Mukherjee SK, Bhatnagar RK. A draft genome assembly of the army worm, Spodoptera frugiperda. Genomics. 2014;104:134–143. doi: 10.1016/j.ygeno.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Kakumani PK, Shukla R, Todur VN, Malhotra P, Mukherjee SK, Bhatnagar RK. De novo transcriptome assembly and analysis of Sf21 cells using Illumina paired end sequencing. Biol Direct. 2015;10:44. doi: 10.1186/s13062-015-0072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fort P, Albertini A, Van-Hua A, Berthomieu A, Roche S, Delsuc F, Pasteur N, Capy P, Gaudin Y, Weill M. Fossil rhabdoviral sequences integrated into arthropod genomes: ontogeny, evolution, and potential functionality. Mol Biol Evol. 2012;29:381–390. doi: 10.1093/molbev/msr226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sf-rhabdovirus PCR product sequence. The diagnostic Sf-rhabdovirus PCR product obtained using primers Mono-1 and -2 and Sf9 cell cDNA as the template was directly sequenced, as described in Materials and methods, and the resulting DNA sequence was aligned with the Sf-rhabdovirus genome sequence originally published by Ma and coworkers [1].
Sf-RVN RPL-3 gene sequence. The Sf RPL-3 gene fragments amplified using Sf-RVN or Sf9 cell cDNA as the templates were sequenced and aligned with the Sf21, T. ni, and B. mori RPL-3 sequences from GenBank, as described in Materials and methods.
Sf-RVN COX-1 gene sequence. The Sf COX-1 gene fragments amplified using Sf-RVN or Sf9 cell cDNA as the templates were sequenced and aligned with the Sf21, T. ni, and B. mori COX-1 sequences from GenBank, as described in Materials and methods.
Sf-RVN GAPDH gene sequence. The Sf GAPDH gene fragments amplified using Sf-RVN or Sf9 cell cDNA as the templates were sequenced and aligned with the Sf21, T. ni, and B. mori GAPDH sequences from GenBank, as described in Materials and methods.