

Abstract
Background and Purpose
We tested the hypothesis that in resistance arteries from cardiovascular disease (CVD) patients, effects of an endothelium‐dependent vasodilator depend on the contractile stimulus.
Experimental Approach
Arteries dissected from parietal pericardium of cardiothoracic surgery patients were studied by myography and imaging techniques. Segments were sub‐maximally contracted by K+, the TxA2 analogue U46619 or endothelin‐1 (ET‐1).
Key Results
Relaxing effects of Na‐nitroprusside were comparable, but those of bradykinin (BK) were bigger in the presence of ET‐1 compared with K+ or U46619. BK‐induced relaxation was (i) abolished by L‐NAME in K+‐contracted arteries, (ii) partly inhibited by L‐NAME in the presence of U46619 and (iii) not altered by indomethacin, L‐NAME plus inhibitors of small and intermediate conductance calcium‐activated K+ channels, but attenuated by catalase, in ET‐1‐contracted arteries. This catalase‐sensitive relaxation was unaffected by inhibitors of NADPH oxidases or allopurinol. Exogenous H2O2 caused a larger relaxation of ET‐1‐induced contractions than those evoked by K+ or U46619 in the presence of inhibitors of other endothelium‐derived relaxing factors. Catalase‐sensitive staining of cellular ROS with CellROX Deep Red was significantly increased in the presence of both 1 μM BK and 2 nM ET‐1 but not either peptide alone.
Conclusions and Implications
In resistance arteries from patients with CVD, exogenous ET‐1 shifts the mediator of relaxing responses to the endothelium‐dependent vasodilator BK from NO to H2O2 and neither NADPH oxidases, xanthine oxidase nor NOS appear to be involved in this effect. This might have consequences for endothelial dysfunction in conditions where intra‐arterial levels of ET‐1 are enhanced.
Abbreviations
- A192621
(2R,3R,4S)‐4‐(1,3‐benzodioxol‐5‐yl)‐1‐[2‐(2,6‐diethylanilino)‐2‐oxoethyl]‐2‐(4‐propoxyphenyl)pyrrolidine‐3‐carboxylic acid
- BK
bradykinin
- BQ123
2‐[(3R,6R,9S,12R,15S)‐6‐(1H‐indol‐3‐ylmethyl)‐9‐(2‐methylpropyl)‐2,5,8,11,14‐pentaoxo‐12‐propan‐2‐yl‐1,4,7,10,13‐pentazabicyclo[13.3.0]octadecan‐3‐yl]acetic acid
- BQ788
sodium N‐{[(2R,6S)‐2,6‐dimethyl‐1‐piperidinyl]carbonyl}‐4‐methyl‐l‐leucyl‐N‐[(1R)‐1‐carboxylatopentyl]‐1‐(methoxycarbonyl)‐d‐tryptophanamide
- CVD
cardiovascular disease
- DPI
diphenyleneiodonium
- EDH
endothelium‐dependent hyperpolarization
- EDNO
endothelium‐derived NO
- EDRF
endothelium‐derived relaxing factor
- ET‐1
endothelin‐1
- L‐NAME
NΩ‐nitro l‐arginine methyl ester
- MEGJ
myo‐endothelial gap junction
- ODQ
1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one
- PD156707
α‐[2‐(4‐methoxyphenyl)‐2‐oxo‐1‐[(3,4,5‐trimethoxyphenyl)methyl]ethylidene]‐1,3‐benzodioxole‐5‐acetic acid
- PSS
physiological salt solution
- S6c
sarafotoxin S6c
- sGC
soluble guanylate cyclase
- SNP
Na‐nitroprusside
- TRAM‐34
1‐[(2‐chlorophenyl)diphenylmethyl]‐1H‐pyrazole
- TPEM
two‐photon excitation fluorescence microscopy
- U46619
(5Z)‐7‐[(1R,4S,5S,6R)‐6‐[(1E,3S)‐3‐hydroxy‐1‐octenyl]‐2‐oxabicyclo[2.2.1]hept‐5‐yl]‐5‐heptenoic acid
- UCL 1684
6,12,19,20,25,26‐hexahydro‐5,27:13,18:21,24‐trietheno‐11,7‐metheno‐7H‐dibenzo[b,n][1,5,12,16]tetraazacyclotricosine‐5,13‐diium dibromide
- XO
xanthine oxidase
Tables of Links
| TARGETS | |
|---|---|
| GPCRs a | Enzymes c |
| ETA receptor | COXs |
| ETB receptor | Endothelial NOS |
| TP receptor | Soluble guanylate cyclase |
| Voltage‐gated ion channels b | XO |
| KCa2.3 | |
| KCa3.1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,b,cAlexander et al., 2015a, 2015b, 2015c).
Introduction
The endothelium regulates vascular homeostasis and endothelial dysfunction is a marker of cardiovascular risk and contributes to the progression of atherosclerosis and cardiovascular events (Heitzer et al., 2001; Widlansky et al., 2003). Human endothelium‐dependent vasodilatations have been investigated in several vascular beds and in isolated blood vessels of different anatomical origin from volunteers and from a broad variety of patients. Studies were performed during basal vasomotor tone in vivo or during myogenic tone and constriction induced by different exogenously administered agonists in vitro. Vasodilatation was induced by different endothelium‐dependent stimuli such as acetylcholine, bradykinin (BK) or increased wall shear stress. Variations in the stimuli used may have contributed to different conclusions about the relative importance of specific endothelium‐derived relaxing factors (EDRFs) in different situations (Matoba et al., 2002; Morikawa et al., 2004; Phillips et al., 2007; Chadha et al., 2011; Liu et al., 2011; Freed et al., 2014). In coronary arterioles from cardiothoracic surgery patients contracted with endothelin‐1 (ET‐1), Gutterman and colleagues attributed relaxing effects of increased wall shear stress and of BK to endothelium‐derived hydrogen peroxide (H2O2) (Phillips et al., 2007; Liu et al., 2011). In mesenteric resistance arteries from gastrectomy patients, Shimokawa and colleagues observed a contribution of H2O2 to the relaxing effect of BK during contraction induced by ET‐1 (Morikawa et al., 2004) or prostaglandin F2α (Matoba et al., 2002). In mesenteric resistance arteries from intestinal surgery patients contracted with vasopressin, Chadha et al. attributed the effects of BK to endothelium‐derived NO (EDNO) and endothelium‐dependent hyperpolarization (EDH) conducted via myo‐endothelial gap junctions (MEGJ) (2011), with no notable effect of the H2O2 scavenger catalase. Recently, flow‐induced relaxation of adipose tissue arterioles constricted by ET‐1 was reported to involve EDNO in patients without coronary artery disease and H2O2 produced by endothelial mitochondria in patients with coronary artery disease (Freed et al., 2014).
Here, we investigated relaxations to one endothelium‐dependent vasodilator in one vascular bed, during contractions induced by different vasoconstrictors. We tested the hypothesis that in patients with cardiovascular diseases (CVD), the effects of an endothelium‐dependent vasodilator depend on the type of vasoconstrictor stimulus. We (i) studied resistance‐sized arteries isolated from a biopsy of the parietal pericardium obtained during elective cardiothoracic surgery, (ii) recorded relaxing effects of BK during contractions induced by elevated extracellular K+, the TxA2 analogue U46619 or exogenous ET‐1, (iii) used pharmacological inhibitors to investigate the mechanisms involved and (iv) used two‐photon excitation fluorescence microscopy (TPEM) to semi‐quantitatively estimate cellular levels of ROS. In the presence of ET‐1, the relaxing effects of BK persisted during blockade of cyclooxygenases, NOS, small‐conductance and intermediate‐conductance calcium‐activated K+ channels (KCa2.3 and KCa3.1) and soluble guanylate cyclase (sGC) but were inhibited by catalase and accompanied by a catalase‐sensitive increase in cellular ROS. This role of H2O2 in endothelium‐dependent vasodilatation in the presence of ET‐1 may contribute to microvascular complications in type 2 diabetes (Eringa et al., 2013), obesity (Campia et al., 2012) and pulmonary arterial hypertension (Kirkby et al., 2008; Miyagawa and Emoto, 2014) where arterial levels of this endothelium‐derived peptide are up‐regulated.
Methods
Patients and ethics
Biopsies from the parietal pericardium (approximately 2 × 2 cm) were collected from 121 patients at the initiation of elective cardiothoracic surgery (coronary artery bypass grafting and/or cardiac valve replacement). Patient characteristics are summarized in Table 1.
Table 1.
Clinical characteristics and reported pharmacotherapies of the study group
| N | 121 |
|---|---|
| Age (years) | 69 ± 1 |
| Male (%) | 82 |
| Surgery: CABG/valve/both (%) | 58/29/13 |
| Smoking (yes/former/no/unknown; %) | 19/45/33/3 |
| Body mass index (kg·m−2) | 27.0 ± 0.4 |
| Known hypertension (%) | 65 |
| Systolic blood pressure (mmHg) | 139 ± 2 |
| Diastolic blood pressure (mmHg) | 74 ± 1 |
| Type 2 diabetes mellitus (%) | 14 |
| Hyperlipidaemia (%) | 82 |
| Ejection fraction (%) | 54 ± 1 |
| HbA1c (mmol·mol−1) | 39 ± 1 |
| Plasma creatinine (μmol·L−1) | 95 ± 6 |
| Statin (%) | 70 |
| Low‐dose aspirin (%) | 66 |
| Warfarin/P2Y antagonist (%) | 7/12 |
| ACEI/ARB (%) | 24/21 |
| ≥2 anti‐hypertensive drugs (%) | 18 |
Data are shown as means ± SEM. ACEI, angiotensin‐converting enzyme inhibitor; ARB, angiotensin AT1 receptor antagonist; CABG, coronary artery bypass grafting (1–4); valve, aortic or mitral valve replacement.
All experiments were performed in accordance with institutional guidelines and were approved by the Medical Ethical Committee of the Region of Southern Denmark (S‐20100044). The experiments performed conformed to the principles outlined in the Declaration of Helsinki of the World Medical Association, and informed written consent was obtained from all patients prior to collection of the biopsy.
Recording of vasomotor responses
Pericardial biopsies were stored for 16 h in 30 mL PSS at 4°C to wash away anaesthetics and analgesics (Batenburg et al., 2004). One long resistance‐sized artery was then isolated from the parietal pericardial sheet in PSS at room temperature as recently described (Bloksgaard et al., 2015). The artery was dissected free from perivascular adipose tissue and cut into 2–9 segments for paired comparisons of effects of pharmacological inhibitors. Variations in group sizes can be explained by the fact that the number of different pharmacological interventions exceeded the number of arterial segments available that could be studied from one patient.
To record isometric tension development, 2‐mm‐long arterial segments were mounted in wire myograph chambers (DMT, Aarhus, Denmark) containing 5 mL PSS maintained at 37°C and aerated with 5% CO2 in air. Arterial segments were distended until a diameter and passive wall tension, which according to the law of Laplace, corresponded to a transmural pressure of 100 mmHg. The lumen diameter of 436 segments under these conditions averaged 187 ± 4 μm (mean of means ± SEM, n = 121), and their maximal contractile response to the combination of 16 nM ET‐1, 1 μM U46619 and 32 mM K+ had a median value of 1.4 N·m−1 with an interquartile range of 0.8 to 2.0 N·m−1.
Pharmacological protocols
From one long arterial segment (>1 cm), four 2‐mm‐long segments could be isolated and studied in parallel in a multichannel wire myograph. One control segment, without any pharmacological inhibitors, was studied and compared in parallel with segments that were incubated with inhibitors of endothelial‐derived relaxing factors, inhibitors of NADPH oxidases, an inhibitor of xanthine oxidase (XO) and/or catalase. This allowed us to compare the effects of different pharmacological interventions within the same artery from the same patient.
Cumulative concentration–response curves (CCRCs) to different vasoconstrictors and to BK, SNP or H2O2 were constructed in series, allowing at least 30 min between experimental steps. Effects of the vasodilators were studied during amplitude‐matched submaximal contractions. Segments were studied in the continuous presence of the selective inhibitors after incubation for at least 20 min. Because of the quasi‐irreversible nature of ET‐1‐induced contractions (De Mey et al., 2011), experiments with this contractile agonist were performed towards the end of the experiments.
Imaging of cellular H2O2 levels
ROS production was assessed by TPEM of arterial segments stained with CellROX Deep Red. Nine 1.5‐mm‐long arterial segments were isolated from one >2‐cm‐long artery from the same patient (n = 4) and studied in parallel in multichannel wire myographs. One segment was left untreated and not stained after the stretching procedure. The remaining eight segments were subjected to cumulative treatment with and without catalase (2000 U·mL−1 for 30 min), ET‐1 (2 nM for 10 min), BK (1 μM for 10 min) and combinations of the three. Staining was performed at the end of the experimental protocol using 5 μM CellROX Deep Red in PSS for 1 h. Finally, all nine segments were fixed in 4% paraformaldehyde for 1 h at 37°C. The staining protocol, fixation and subsequent imaging within 24 h from the staining and fixation were designed in accordance with the guidelines from the manufacturer. Imaging was conducted using a custom‐built two‐photon excitation fluorescence microscope on a Nikon TI (Ramcon A/S, Birerød, Denmark) eclipse platform (Brewer et al., 2013). The objective used was a 60× water immersion objective, NA 1.25. The laser was a Ti:Sa laser (HPeMaiTai DeepSee; Spectra Physics, Mountain View, CA, USA), and the excitation wavelength was 820 nm. Emission was split by a dichroic mirror (570DCXR; Chroma Technology Corp., Bellows Falls, VT, USA) and collected in two channels at 655/40 nm (D655/40M, Chroma Technology Corp., for CellROX Deep Red and spectral overlap from elastin autofluorescence) and 542/20 nm (Semrock Bright Line® filter, Semrock, Rochester, NY, USA, for elastin autofluorescence) using Becker and Hickl GmbH, Berlin, Germany HPM‐100‐40 hybrid detectors. Image analyses were performed using FIJI (Schindelin et al., 2012). Intensity (I) data representing the cellular ROS production within the samples were calculated as ICellROX − Iautofluorescence to eliminate pixels with a significant contribution of the autofluorescence to the data analyses. For each arterial segment, at least eight regions of interest (ROIs) without spatial overlap were imaged along the length of the arterial segment. Intensities in each calculated image (ICellROX − Iautofluorescence) were determined for 5–15 areas per ROI per sample without spatial overlap. Background fluorescence in the 655/40 nm channel, assessed using the ninth, unstained segment, was negligible for all four patients investigated.
Endothelium‐dependence
In arteries isolated from four patients (resting lumen diameter: 201 ± 22 μm), cannulated and pressurized largely as previously described (Hilgers et al., 2010; Bloksgaard et al., 2015), extra‐luminal application of 100 nM U46619 caused 69 ± 6% constriction that was reduced to 12 ± 5% by 1 μM BK. Following perfusion of the arterial lumen with 2 mL of air, no statistically significant BK‐induced dilatation could be observed, and endothelial cells, but not smooth muscle cells, had become permeable to the nuclear dye diamidino‐2‐phenylindole (DAPI), which did not stain nuclei in endothelial and smooth muscle cells of control arteries perfused with saline (data not shown).
Statistics
Results are expressed as contractile response (N·m−1) or as percentage reduction of the contraction (relaxations). Data are shown as mean ± SEM or as median (interquartile range) when not normally distributed. Individual CCRCs were fitted to a non‐linear regression curve with variable slope. Potency (pD2; −log10 EC50) and efficacy (E max) of agonists were compared either paired (when experiments were performed in arterial segments from the same patient) or unpaired (when experiments were performed in arteries from different patients) with two‐sided t‐test or extra sum‐of‐squares F‐test respectively. Differences in CellROX Deep Red intensities were analysed by one‐way ANOVA, followed by Bonferroni's post hoc test to compare multiple groups. P < 0.05 was considered statistically significant. Statistically significant differences in potency are indicated by asterisks (*) between curves; differences in efficacy are indicated by asterisks at the highest concentrations tested. All analyses were performed using graphpad prism 6.04 (GraphPad Software Inc., San Diego, CA, USA). pD2 and E max values are summarized in Supporting Information Table S1. Data and statistical analysis comply to the best of our abilities with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials
Physiological salt solution (PSS) contained (in mM) the following: NaCl, 115; NaHCO3, 25; K2HPO4, 2.5; MgSO4, 1.2; glucose, 5.5; HEPES, 10; and CaCl2, 1.3. High‐potassium PSS (KPSS) contained 20 mM NaCl and 95 mM KCl, instead of 115 mM NaCl. Solutions with intermediate K+ (and Na+) concentrations were prepared by mixing appropriate volumes of PSS and KPSS. Buffers were continuously aerated with 5% CO2 in air at 37°C. BQ123 (Bachem, Weil am Rhein, Germany), BQ788 (Bachem), CellROX® Deep Red (Molecular Probes™; Thermo Fisher Scientific, Waltham, MA, USA), diphenyleneiodonium (DPI) (Sigma‐Aldrich, Brøndby, Denmark), ODQ (Cayman Chemical, Ann Arbor, MI, USA), PD156707 (Tocris Bioscience, Bristol, UK), TRAM‐34 (Tocris Bioscience) and UCL 1684 (Tocris Bioscience) were dissolved in DMSO. Capsaicin (Sigma‐Aldrich), indomethacin (Sigma‐Aldrich), phenoxybenzamine (GlaxoSmithKline, Brøndby, Denmark) and U46619 (Tocris Bioscience) were dissolved in 100% ethanol. 5‐HT (serotonin; Sigma‐Aldrich), A192621 (Abbvie Labs, North Chicago, IL, USA), angiotensin II (Sigma‐Aldrich), apocynin (Sigma‐Aldrich), arginine vasopressin (Sigma‐Aldrich), BK (Sigma‐Aldrich), ET‐1 (Bachem), gp91 ds‐tat (AnaSpec, Seraing, Belgium), H2O2 (Sigma‐Aldrich), NΩ‐nitro l‐arginine methyl ester (L‐NAME) (Sigma‐Aldrich), Na‐nitroprusside (SNP) (Sigma‐Aldrich), noradrenaline (Sigma‐Aldrich), phenylephrine (Sigma‐Aldrich) and sarafotoxin‐6c (S6c) (Bachem) were dissolved in ddH2O. Allopurinol (Cayman Chemical) and uncontaminated catalase (C40, Sigma‐Aldrich) were dissolved in PSS. Actions and concentrations of the pharmacological inhibitors used are listed in Table 2.
Table 2.
Pharmacological inhibitors
| Chemical | Action | Concentration | Reference |
|---|---|---|---|
| A192621 | Selective ETB‐receptor antagonist | 30 nM | Davenport (2002) |
| Allopurinol | Inhibitor of xanthine oxidase | 0.5 mM | Lacy et al. (1998) |
| Apocynin | Inhibitor of NADPH oxidase | 10 μM | Stolk et al. (1994), Cifuentes‐Pagano et al. (2012) |
| BQ123 | Selective ETA‐receptor antagonist | 1 μM | Davenport (2002) |
| BQ788 | Selective ETB‐receptor antagonist | 1 μM | Davenport (2002) |
| Capsaicin | Irreversible desensitizer of sensorimotor nerves | 1 μM | Szallasi and Blumberg (1999) |
| Catalase (C40) | Scavenger of extracellular H2O2 | 2000 U·mL−1 | Matoba et al. (2000), Gauthier et al. (2011) |
| DPI | Inhibitor of NADPH oxidase | 1 μM | O'Donnell et al. (1993), Cifuentes‐Pagano et al. (2012) |
| gp91 ds‐tat | Selective inhibitor of NADPH oxidase 2 | 1 μM | Rey et al. (2001), Cifuentes‐Pagano et al. (2012) |
| Iberiotoxin | Inhibitor of large conductance Ca2 +‐activated K+ channels | 100 nM | Galvez et al. (1990) |
| Indomethacin | Inhibitor of cyclooxygenases 1 and 2 | 10 μM | Hart and Boardman (1963) |
| L‐NAME | Inhibitor of NOS | 100 μM | Rees et al. (1990) |
| ODQ | Inhibitor of soluble guanylate cyclase | 10 μM | Garthwaite et al. (1995) |
| PD156707 | Selective ETA‐receptor antagonist | 100 nM | Davenport (2002) |
| Phenoxybenzamine | Irreversible α‐adrenoceptor antagonist | 1 μM | Furchgott (1966) |
| TRAM‐34 | Inhibitor of intermediate conductance Ca2 +‐activated K+ channels | 1 μM | Hilgers et al. (2010) |
| UCL 1684 | Inhibitor of small conductance Ca2 +‐activated K+ channels | 1 μM | Rosa et al. (1998) |
Results
Contractile responses
All resistance arteries isolated from the parietal pericardium of patients with CVD that were included in this study contracted >0.1 N·m−1 in response to ET‐1, the TxA2 analogue U46619 and to increases in extracellular K+ concentration from 5 to 32 mM (Figure 1). The potency and efficacy of ET‐1 were significantly higher than those of U46619 and high K+. Contractile responses to 2 nM ET‐1, the EC50 of the peptide, to 1 μM U46619 and to 32 mM K+ were of comparable amplitude in arterial segments from the same patients (Figure 1) and were sustained for 30 min. Angiotensin II (0.1–100 nM) and vasopressin (1 pM–300 nM) caused contractions that were not sustained. Noradrenaline (0.1–10 μM) and phenylephrine (0.1 μM–1 mM) caused small contractions in arteries from less than 50% of the patients investigated. 5‐HT (0.1–10 μM) and S6c (selective ETB‐receptor agonist; 1 pM–1 nM) (Davenport, 2002) failed to induce contraction (Supporting Information Fig. S1).
Figure 1.
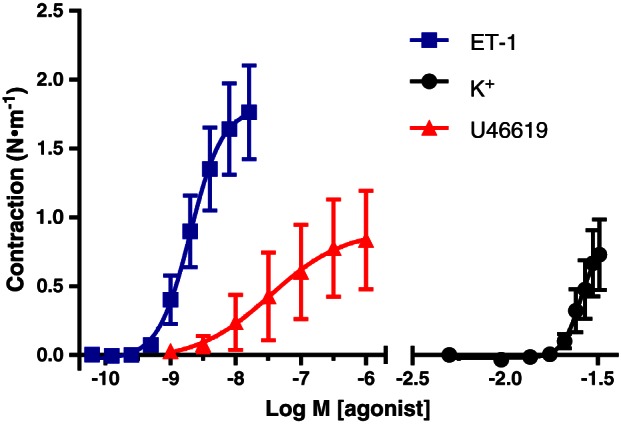
Contractile responses to cumulative concentrations of ET‐1, U46619 and increases in extracellular K+ concentration in pericardial resistance arteries from the same CVD patients. Means ± SEM (n = 13). Differences between the sensitivities to the stimuli and between the maximal responses to ET‐1 and those to the other two stimuli are statistically significant, analysed by comparing pD2 and E max values with paired two‐sided t‐test.
Most pharmacological inhibitors of EDRFs used in this study had no statistically significant effects on the sensitivity and maximal responses to the contractile stimuli, but there were notable exceptions. Maximal responses to U46619 (1.06 ± 0.17 vs. 0.84 ± 0.36 N·m−1) were increased by 1 μM UCL 1684 and 1 μM TRAM‐34 (inhibitors of KCa2.3 and KCa 3.1, respectively). Indomethacin and 2000 U·mL−1 catalase (scavenger of extracellular H2O2) each increased the potency of ET‐1 approximately threefold, and these effects were additive (Figure 2).
Figure 2.
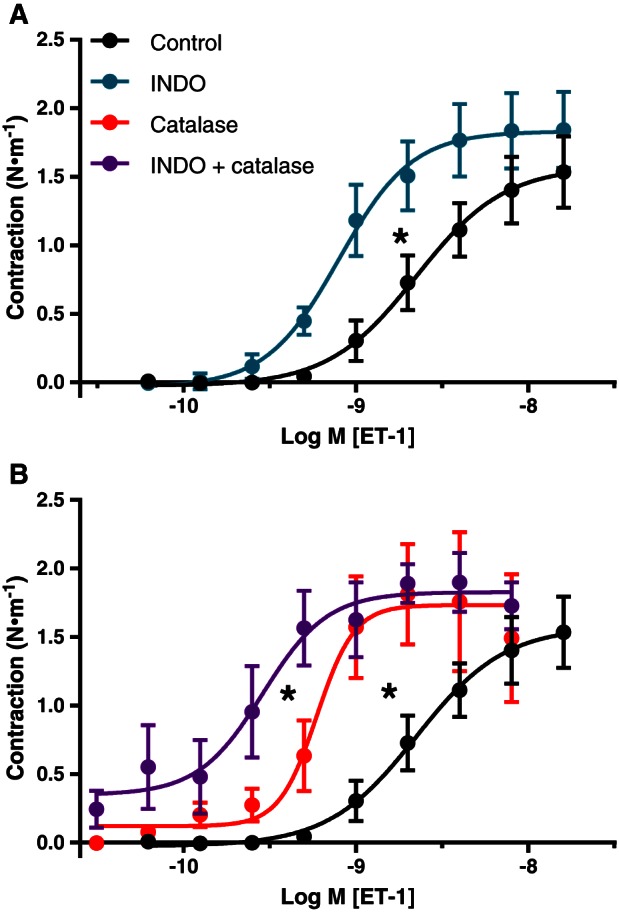
Effects of indomethacin (INDO) (A) and of catalase +/− INDO (B) on contractile responses to ET‐1 in pericardial resistance arteries from the same CVD patients. Means ± SEM (control and INDO: n = 11; catalase and catalase + INDO: n = 6). Effects on the sensitivity to ET‐1 are statistically significant, analysed by comparing pD2 and E max values with extra sum‐of‐squares F‐test.
In arteries from different patients, no statistically significant differences were observed between the contractile potency and efficacy of ET‐1 (i) during inhibition of several EDRFs by L‐NAME, UCL 1684 and TRAM‐34 and (ii) during inhibition of these pathways combined with irreversible blockade of α‐adrenoceptors with phenoxybenzamine, desensitization of sensorimotor nerves with capsaicin and presence of 30 nM A192621 or 1 μM BQ788 (selective ETB receptor antagonists) (Davenport, 2002) (Supporting Information Fig. S2). Absence of contractile responses to S6c was not modified by presence of L‐NAME, UCL 1684 and TRAM‐34, or their combination (Supporting Information Fig. S2). However, the contractile potency of ET‐1 was reduced in presence of 100 nM PD156707 (non‐peptide ETA antagonist; Supporting Information Fig. S3), and the efficacy of the peptide was reduced in presence of 1 μM BQ123 (peptidergic ETA antagonist; E max: 0.2 ± 0.1 vs. 1.4 ± 0.2 N·m−1). These indicate that ET‐1‐induced contractions were mediated by arterial smooth muscle ETA receptors.
Next, we used 2 nM ET‐1, 1 μM U46619 and 32 mM K+ to induce contractions of comparable amplitude in experiments investigating relaxing mechanisms. In the limited cases where inhibitors of relaxing mechanisms altered the sensitivity to contractile stimuli, the concentrations of these stimuli were modified accordingly.
Relaxing responses to BK and H2O2
BK (10 pM–1 μM) markedly relaxed sub‐maximally contracted pericardial resistance arteries from patients with CVD (Figure 3A). The potency of BK depended on the contractile stimulus. It was 10 times larger during contractions induced by 2 nM ET‐1 than during contractions in response to U46619 or K+. This effect was selective as the NO donor compound SNP relaxed the three types of contraction with comparable potency (Figure 3B).
Figure 3.
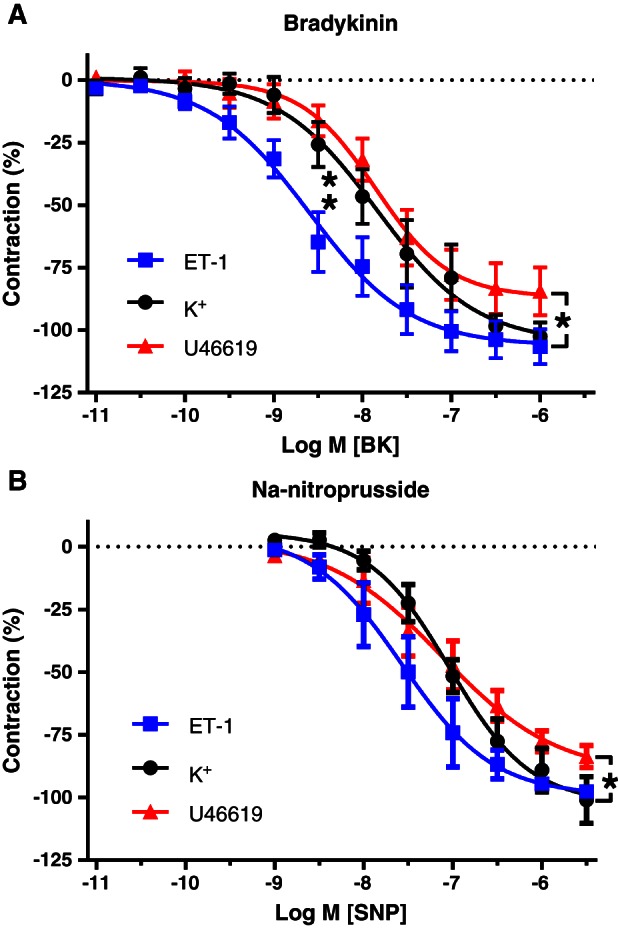
Relaxing responses to cumulative concentrations of bradykinin (BK; A) and Na‐nitroprusside (SNP; B) in pericardial resistance arteries from the same CVD patients during amplitude‐matched submaximal contractions induced by 2 nM ET‐1, 1 μM U46619 or 32 mM K+. Means ± SEM (n = 17, 8 and 9, respectively, for BK and 6 for SNP). The sensitivity to BK, but not the maximal relaxation, was significantly higher in the presence of ET‐1 than in the presence of U46619 or 32 mM K+, analysed by comparing pD2 and E max values with extra sum‐of‐squares F‐test (A, unpaired) or paired two‐sided t‐test (B, paired).
In K+‐contracted arteries, relaxing responses to BK were not modified by indomethacin but were strongly inhibited by L‐NAME. When, in addition to NOS, COXs were inhibited, relaxing responses to BK were abolished (Figure 4A). This indicates the involvement of NO in these responses.
Figure 4.
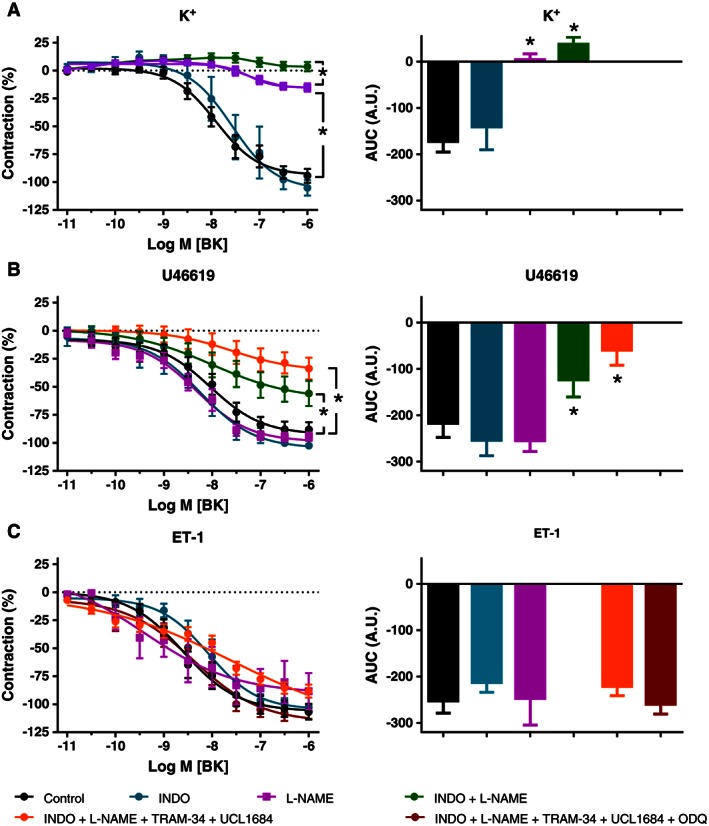
Effects of indomethacin (INDO), L‐NAME, TRAM‐34 and UCL 1684, and ODQ on relaxing responses to BK in pericardial resistance arteries made to contract with 32 mM K+ (A; n = 13, 6, 9, 13 and 6 for control, INDO, L‐NAME, INDO + L‐NAME and INDO + L‐NAME + TRAM‐34 + UCL 1684 respectively), 1 μM U46619 (B; n = 12, 7, 6, 14 and 8 for control, INDO, L‐NAME, INDO + L‐NAME and INDO + L‐NAME + TRAM‐34 + UCL 1684 respectively) and 2 nM ET‐1 (C; n = 17, 8, 9, 33 and 6 for control, INDO, L‐NAME, INDO + L‐NAME + TRAM‐34 + UCL 1684 and INDO + L‐NAME + TRAM‐34 + UCL 1684 + ODQ respectively). Means ± SEM, analysed by comparing pD2 and E max values and area under the curves (AUC; A.U., arbitrary units) with paired two‐sided t‐test.
During contraction induced by U46619, relaxing responses to BK were not significantly modified by indomethacin or L‐NAME but were reduced by approximately 50% when the two inhibitors were combined (Figure 4B). The remaining relaxations were reduced somewhat by the additional presence of 1 μM UCL 1684 and 1 μM TRAM‐34, but small resistant relaxations persisted (Figure 4B). These findings indicate that the effects of BK during TP receptor stimulation involve (i) NO when COXs are inhibited and (ii) a small component that cannot be attributed to COX, NOS or endothelial hyperpolarization via KCa2.3 and KCa3.1.
During contraction induced by ET‐1, the potent relaxing responses to BK were not modified by indomethacin, L‐NAME or by the combined blockade of COX, NOS, KCa2.3, KCa3.1 and inhibition of sGC with 10 μM ODQ (Figure 4C). These resistant relaxing effects of BK during ET‐1‐induced contraction were not significantly modified by 10 μM apocynin, 1 μM DPI or 1 μM gp91 ds‐tat (structurally different inhibitors of NADPH oxidases) (Cifuentes‐Pagano et al., 2012), or 0.5 mM allopurinol (inhibitor of XO) (Figure 5A) but were reduced in the presence of 0.1 μM iberiotoxin (inhibitor of BKCa) and were inhibited by 2000 U·mL−1 catalase (scavenger of H2O2) (Figure 5B). These findings indicate the involvement of H2O2 as a relaxing factor during contraction stimulated by ET‐1 and that this H2O2 is not derived from NADPH oxidases, XO, COX or NOS.
Figure 5.
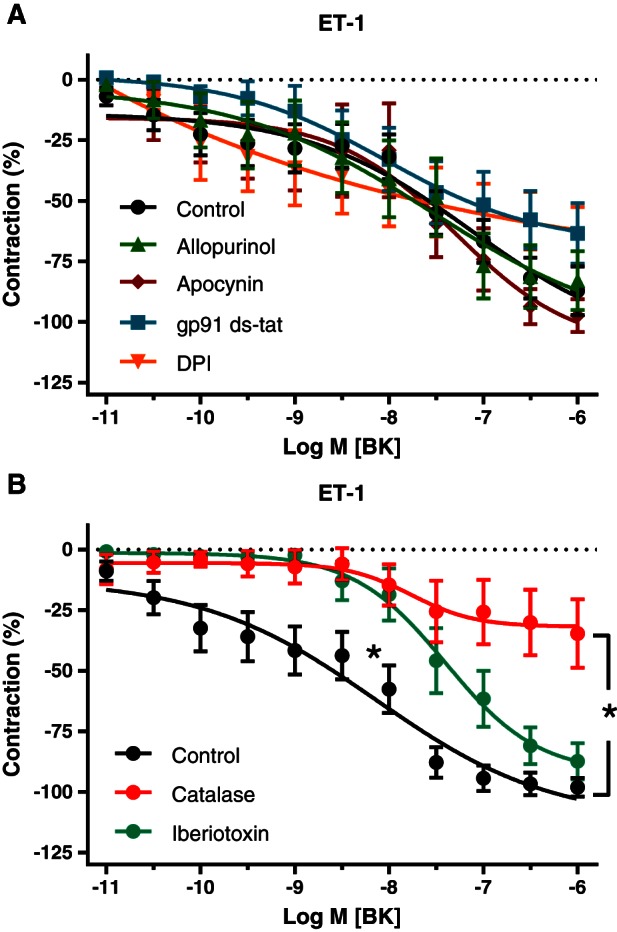
Effects of inhibitors of NADPH oxidases and xanthine oxidase (A) and of catalase and iberiotoxin (B) on relaxing responses to BK in pericardial resistance arteries made to contract with ET‐1 in the presence of indomethacin, L‐NAME, TRAM‐34 and UCL 1684 [control: n = 14 (A) and 12 (B)]. Means ± SEM. (A) Effects of allopurinol (n = 6), apocynin (n = 7), DPI (n = 5) and gp91 ds‐tat (n = 7) on sensitivity and maximal responses to BK in vessel segments from the same patients are not statistically significant. (B) The effect of catalase (n = 6) on the maximal relaxation and the effect of iberiotoxin (n = 7) on the potency of BK are statistically significant, analysed by comparing pD2 and E max values with unpaired two‐sided t‐test.
Exogenous H2O2 (100 nM–3.0 mM) caused concentration‐dependent relaxations in pericardial resistance arteries from CVD patients (Figure 6). In the presence of indomethacin, L‐NAME, UCL 1684 and TRAM‐34, the relaxing effect of H2O2 tended to be more potent (P = 0.06) and it was significantly more efficacious in arteries from the same individuals during ET‐1‐ than during U46619‐induced contraction. Compared with K+‐contracted arteries, the increased efficacy of exogenous H2O2 in the presence of ET‐1 was even more marked (Figure 6).
Figure 6.
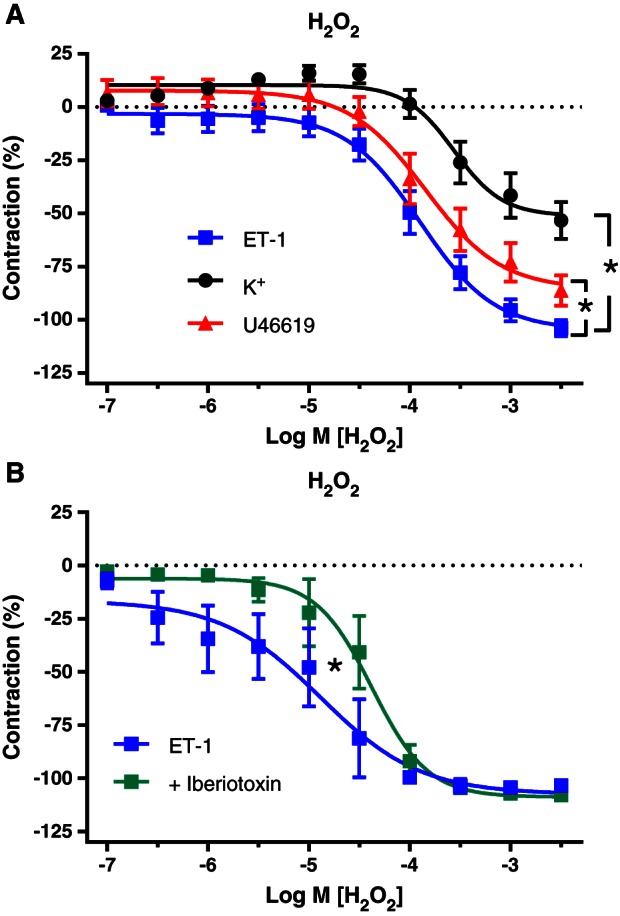
Relaxing responses to exogenous H2O2 in pericardial resistance arteries from the same patients during contraction induced by ET‐1 (n = 13), U46619 (n = 13) or K+ (n = 10) in presence of indomethacin, L‐NAME, TRAM‐34 and UCL 1684 (A) and during ET‐1‐induced contractions with the additional presence of iberiotoxin (B; n = 5). Means ± SEM. The relaxing effect of H2O2 is significantly larger in the presence of ET‐1 compared with U46619 and K+. Iberiotoxin significantly reduced the potency of H2O2 during contractions induced by ET‐1, analysed by comparing pD2 and E max values with paired two‐sided t‐test.
Production of H2O2
Cellular levels of ROS were investigated with CellROX Deep Red staining and TPEM. The intensity of the fluorescent staining was significantly more than the background fluorescence (determined in the absence of CellROX) but was not modified by catalase (2000 U·mL−1), ET‐1 (2 nM) or BK (1 μM). In the presence of both ET‐1 and BK, however, the staining intensity was significantly increased, and this was prevented by catalase (Figure 7). These findings indicate a significant production of ROS in the patients' resistance arteries under basal conditions and that the combined presence of the vasoconstrictor ET‐1 and the endothelium‐dependent vasodilator BK stimulate the production of H2O2.
Figure 7.
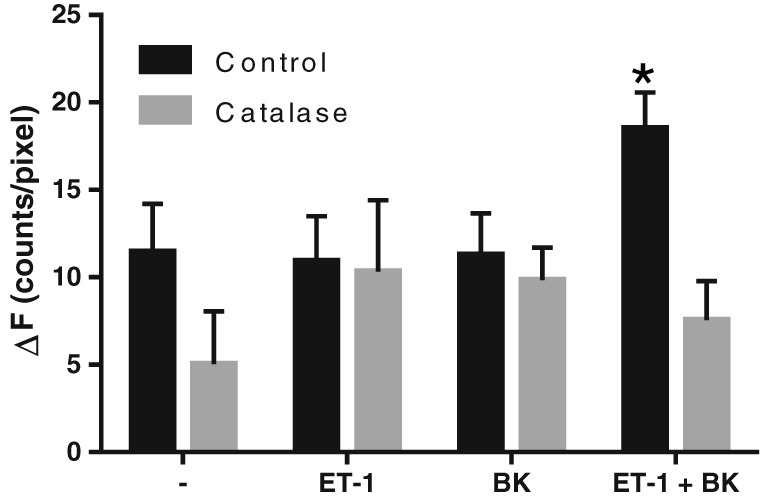
Effects of catalase (2000 U·mL−1), ET‐1 (2 nM) and BK (1 μM) on cellular ROS levels. Fluorescence intensity above background autofluorescence (ΔF) is shown for arterial segments stained with CellROX Deep Red after incubation in the absence or presence of catalase with or without ET‐1 and/or BK. Means ± SEM. *The difference is statistically significant, analysed by Bonferroni's post hoc test, n = 4.
Discussion and conclusions
The main findings of this investigation of patient resistance artery responses to an endothelium‐dependent vasodilator are (i) more potent relaxation responses occur during ET‐1‐ than during K+‐ or U46619‐induced contractions, (ii) the responses are significantly reduced by NOS blockade when the contraction is induced by depolarization and by combined NOS and COX blockade when mediated by TP receptors but not ETA receptors, (iii) this resistant relaxation persists during inhibition of KCa2.3 and KCa3.1 and (iv) it can be subsequently blocked by catalase but not inhibitors of NADPH oxidases or XO. In this study, we demonstrated that ET‐1 induces a shift from NO‐ to H2O2‐mediated relaxation and that NOS, NADPH oxidase stimulation and XO activation are not involved in this effect.
In conduit arteries, NO is the main EDRF and impairment of its production and scavenging by superoxide anions are responsible for endothelial dysfunction (Heitzer et al., 2001). In resistance arteries, additional mechanisms have been identified, such as endothelium‐derived K+ (Edwards et al., 1998), epoxyeicosatrienoic acids (Fisslthaler et al., 1999), H2O2 (Matoba et al., 2002; Morikawa et al., 2004; Phillips et al., 2007; Liu et al., 2011) and hyperpolarization of smooth muscle via MEGJ (Feletou and Vanhoutte, 2006). The contributions of each of these were observed to be different in animal models of CVD (Yada et al., 2006; Feletou and Vanhoutte, 2009). Studies of endothelium‐dependent responses in human resistance arteries have used vessels isolated from different vascular beds from volunteers and various patient groups, and a variety of endothelial stimuli have been used to elicit the responses during different agonist‐induced contractions (Matoba et al., 2002; Morikawa et al., 2004; Phillips et al., 2007; Chadha et al., 2011; Liu et al., 2011; Freed et al., 2014). These discrepancies in the experimental conditions may have contributed to different conclusions. Hence, in the present study we investigated whether the contractile stimulus influences the responses of resistance arteries from patients with CVD to an endothelium‐dependent vasodilator.
Large numbers of CVD patients have to undergo life‐saving cardiothoracic surgery despite chronic antithrombotic, cholesterol‐lowering, antidiabetic and antihypertensive treatments (Table 1). These patients are in our opinion important targets for the development of new drug concepts. During cardiothoracic surgery, the pericardium must be opened to access the heart. From biopsies of this structure, one long resistance artery can be isolated and divided into several segments (Bloksgaard et al., 2015). This facilitated characterization of mechanisms of vasodilatation in CVD patients with various pharmacological inhibitors during ex vivo contractions.
We identified three stimuli that induce sustained contractions in these resistance arteries. Maximal contractility varied considerably between patients (0.1–5.4 N·m−1), but we took special care to compare relaxing responses during amplitude‐matched contractions in vessels from each individual. The contractile effects of ET‐1 were not mimicked or modified by a selective agonist and antagonist of ETB receptors, alkylation of α‐adrenoceptors, desensitization of sensorimotor nerves, inhibition of NOS and of KCa2.3 and KCa3.1. They were thus mediated by smooth muscle ETA receptors and not modulated by ETB receptors, endogenous NO or perivascular nerves, which can be affected by ET‐1 (Wang and Wang, 2004; Meens et al., 2009; De Mey et al., 2011). They were, however, enhanced by COX inhibition and catalase, which suggests that basal or stimulated prostaglandin and H2O2 production are involved, and this remains to be established. ET‐1 was not only more potent but also more efficacious than U46619 and depolarization at inducing a contraction, which is in line with the broader signal transduction stimulated by ETA receptors than by TP receptors and depolarization in arterial smooth muscle (Momotani et al., 2011; Takeya et al., 2015).
High‐potassium (low‐sodium) buffer causes depolarization, contracts smooth muscle, reduces endothelial activation and inhibits endothelium‐dependent hyperpolarizations (Feletou and Vanhoutte, 2006; Feletou and Vanhoutte, 2009). Under this contractile condition, the relaxing response to BK was blocked by L‐NAME, indicating that it was due to NO. During contractions induced by the TP receptor stimulus U46619, NO only played a major role when COX was inhibited. An additional component persisted in the presence of COX and NOS inhibitors, which was not observed in depolarized arteries. This persistent relaxation response was not significantly affected by TRAM‐34 and UCL 1684 and thus differs from the classical EDH mechanisms that are initiated by activation of endothelial KCa2.3 and KCa3.1 (Feletou and Vanhoutte, 2006; Feletou and Vanhoutte, 2009). This is in line with observations from animal studies indicating that TP receptor stimulation inhibits calcium‐activated potassium channels on endothelial and arterial smooth muscle cells (Crane and Garland, 2004; Ellinsworth et al., 2014). A different pattern was observed when the same vessels from the same patients were made to contract with ET‐1. Here, BK was significantly more potent, and the relaxations were not modified by inhibition of COX, NOS, KCa2.3, KCa3.1 or sGC. Because an NO donor was equally potent in relaxing contractions induced by depolarization, U46619 and ET‐1, resistance to L‐NAME indicates inhibited production of NO in the presence of ET‐1. Catalase blocked the resistant BK‐induced relaxing effects. Inhibition by this selective large molecular weight scavenger indicates the involvement of H2O2 as a relaxing factor in the effects of BK in the presence of ET‐1. In addition to these pharmacological findings, staining with CellROX Deep Red revealed increased cellular levels of H2O2 in the presence of both ET‐1 and BK. Taken together, our findings indicate that ET‐1 stimulates the transition from an NO‐mediated to an H2O2‐mediated effect of an endothelium‐dependent vasodilator.
Uncoupled endothelial NOS generates superoxide anions instead of NO, and the oxygen free radicals can be dismutated into H2O2 (Heitzer et al., 2001; Forstermann and Munzel, 2006). In line with this, endothelium‐derived H2O2 was reported to be absent in NOS‐deficient mice (Matoba et al., 2000; Satoh et al., 2014). From our observations, uncoupled NOS and cyclooxygenases, another source of superoxide anions, are, however, not involved because catalase was observed to have an inhibitory effect in the presence of L‐NAME and indomethacin. We considered that stimulation of superoxide‐generating NADPH oxidases by ET‐1 (Li et al., 2003; Loomis et al., 2005) might reduce the bioavailability of NO and provide a source of H2O2 (Heitzer et al., 2001; Forstermann and Munzel, 2006). Yet, three inhibitors of NADPH oxidases with different selectivity and specificity for different enzyme subtypes (Cifuentes‐Pagano et al., 2012) did not significantly modify the catalase‐sensitive effects. Furthermore, inhibition of XO by allopurinol (Lacy et al., 1998) did not affect BK‐induced relaxation during ET‐1 contractions either. This leaves lipoxygenases and, in particular, the electron transport chain of mitochondria as potential sources of superoxide anion and H2O2 (Freed et al., 2014) to seriously consider in future research.
In addition to promoting the transition of NO to H2O2, ET‐1‐stimulated contractions were particularly sensitive to relaxation by H2O2. Catalase increased the sensitivity to the contractile effect of ET‐1, indicating inhibition by endogenous H2O2. Moreover, exogenous H2O2 was more efficacious in relaxing responses to ET‐1 than to U46619 and K+. This suggests that the contractile mechanisms stimulated by arterial smooth muscle ETA receptors but not TP receptors or depolarization (Momotani et al., 2011; Takeya et al., 2015) are particularly sensitive to inhibition by H2O2.
Although this study comes with important perspectives, several limitations need to be addressed as well. We introduced the use of human pericardial resistance arteries for pharmacological studies. They can frequently be obtained from patients, with and without coronary artery disease (bypass and valve replacement), who are undergoing of cardiothoracic surgery. They are then cut into several segments, which is helpful for paired investigation of drug effects in a diverse population. Whether pericardial arteries are representative of other vascular beds remains to be established. The observed dependence of BK‐induced relaxation on the contractile stimulus advises caution when comparing studies of endothelial function in vitro with the in vivo situation, where the nature of the underlying vasomotor tone is unknown. The dependence on the contractile stimulus was deduced from the discrepancies between observations with L‐NAME and with BK. Conclusions about endothelial (dys)function can thus depend on the use of a particular inhibitor or stimulus.
Furthermore, the concentration of catalase used in the present study (2000 U·mL−1) might be on the high side, as it was previously demonstrated that a concentration of 140 U·mL−1 was sufficient to abolish the effects of exogenous H2O2 on vascular tone (You et al., 2005). We used catalase at 2000 U·mL−1 so that are results are comparable with the earlier findings of Gutterman and Shimokawa.
It remains to be established whether, in addition to exogenous ET‐1, endogenous ET‐1 can also induce the NO to H2O2 transition. In that case, our findings are particularly relevant for obesity, insulin resistance and pulmonary hypertension, where intra‐arterial levels of ET‐1 are up‐regulated (Kirkby et al., 2008; Campia et al., 2012; Eringa et al., 2013; Miyagawa and Emoto, 2014). The molecular mechanisms of the transition have not yet been addressed. The roles of the various ET receptor subtypes (De Mey et al., 2011), sphingolipids (Spijkers et al., 2011; Freed et al., 2014) and mitochondria (Freed et al., 2014) should also be considered.
From this study, we conclude that in resistance arteries from patients with CVD, exogenous ET‐1 shifts the mediator of relaxing responses to the endothelium‐dependent vasodilator BK from NO to H2O2.
Author contributions
T.M.L., M.B., J.R.B., L.M.R., and J.G.R.D.M. participated in study design. L.A.B., M.L.H., L.M.R., and A.I. provided access to essential methods and materials. T.M.L., M.B., J.R.B., M.H.F., and K.R. conducted the experiments. T.M.L., M.B., J.R.B., M.H.F., M.L.H., L.M.R., and J.G.R.D.M. performed data analysis. T.M.L., M.B., J.R.B., M.H.F., and J.G.R.D.M. wrote or contributed to the writing of the manuscript. All authors have read and approved the final manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1 Overview of pD2 and Emax values from all figures
Figure S1 Contractile responses to cumulative concentrations of angiotensin II (Ang II; n = 27), vasopressin (n = 5), noradrenaline (n = 13), phenylephrine (n = 7), 5‐hydroxytryptamine (5‐HT, serotonin; n = 5) and sarafotoxin 6c (n = 7). Means ± SEM.
Figure S2 Contractile responses to cumulative concentrations of ET‐1 (A) and sarafotoxin 6c (B) in the absence and presence of 100 μM L‐NAME, 1 μM TRAM‐34 and 1 μM UCL 1684 and their combination. All responses to sarafotoxin 6c in presence of 10 μM indomethacin. Means ± SEM (A: n = 14, 11, and 10 for control, L‐NAME and TRAM‐34 + UCL 1684, respectively. B: n = 7, 7, 7, and 6 for control, L‐NAME, TRAM‐34 + UCL 1684 and L‐NAME + TRAM‐34 + UCL 1684, repectively).
Figure S3 Contractile responses to cumulative concentrations of ET‐1 in the absence and presence of the non‐peptide ETA receptor antagonist PD156707 (100 nM). Means ± SEM (n = 7). The difference in potency is statistically significant, analysed by paired two‐sided T‐test.
Supporting info item
Supporting info item
Acknowledgements
This work was supported by Odense University Hospital (grant number A735), the University of Southern Denmark and the Danish Council for Independent Research (grant number DFF‐1333‐00038) and was conducted within the frame of the elite research centre CIMA – Center for Individualized Medicine in Arterial Diseases of the Odense University Hospital.
The authors thank Anne‐Marie Jakobsen, Pia Søndergaard Jensen and Simone Rørdam‐Preil from the Department of Clinical Biochemistry and Pharmacology (OUH) for their assistance with the collection and handling of patient biopsies and characteristics in the project. The Danish Molecular Biomedical Imaging Center (DaMBIC) is acknowledged for the use of equipment.
Leurgans, T. M. , Bloksgaard, M. , Brewer, J. R. , Bagatolli, L. A. , Fredgart, M. H. , Rosenstand, K. , Hansen, M. L. , Rasmussen, L. M. , Irmukhamedov, A. , and De Mey, J. GR. (2016) Endothelin‐1 shifts the mediator of bradykinin‐induced relaxation from NO to H2O2 in resistance arteries from patients with cardiovascular disease. British Journal of Pharmacology, 173: 1653–1664. doi: 10.1111/bph.13467.
References
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015c). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batenburg WW, Garrelds IM, van Kats JP, Saxena PR, Danser AH (2004). Mediators of bradykinin‐induced vasorelaxation in human coronary microarteries. Hypertension 43: 488–492. [DOI] [PubMed] [Google Scholar]
- Bloksgaard M, Leurgans TM, Nissen I, Jensen PS, Hansen ML, Brewer JR et al. (2015). Elastin organization in pig and cardiovascular disease patients' pericardial resistance arteries. J Vasc Res 52: 1–11. [DOI] [PubMed] [Google Scholar]
- Brewer J, Bloksgaard M, Kubiak J, Sorensen JA, Bagatolli LA (2013). Spatially resolved two‐color diffusion measurements in human skin applied to transdermal liposome penetration. J Invest Dermatol 133: 1260–1268. [DOI] [PubMed] [Google Scholar]
- Campia U, Tesauro M, Cardillo C (2012). Human obesity and endothelium‐dependent responsiveness. Br J Pharmacol 165: 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadha PS, Liu L, Rikard‐Bell M, Senadheera S, Howitt L, Bertrand RL et al. (2011). Endothelium‐dependent vasodilation in human mesenteric artery is primarily mediated by myoendothelial gap junctions intermediate conductance calcium‐activated K+ channel and nitric oxide. J Pharmacol Exp Ther 336: 701–708. [DOI] [PubMed] [Google Scholar]
- Cifuentes‐Pagano E, Csanyi G, Pagano PJ (2012). NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol Life Sci 69: 2315–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane GJ, Garland CJ (2004). Thromboxane receptor stimulation associated with loss of SKCa activity and reduced EDHF responses in the rat isolated mesenteric artery. Br J Pharmacol 142: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AP (2002). International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharmacol Rev 54: 219–226. [DOI] [PubMed] [Google Scholar]
- De Mey JG, Compeer MG, Lemkens P, Meens MJ (2011). ETA‐receptor antagonists or allosteric modulators? Trends Pharmacol Sci 32: 345–351. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396: 269–272. [DOI] [PubMed] [Google Scholar]
- Ellinsworth DC, Shukla N, Fleming I, Jeremy JY (2014). Interactions between thromboxane A(2), thromboxane/prostaglandin (TP) receptors, and endothelium‐derived hyperpolarization. Cardiovasc Res 102: 9–16. [DOI] [PubMed] [Google Scholar]
- Eringa EC, Serne EH, Meijer RI, Schalkwijk CG, Houben AJ, Stehouwer CD et al. (2013). Endothelial dysfunction in (pre)diabetes: characteristics, causative mechanisms and pathogenic role in type 2 diabetes. Rev Endocr Metab Disord 14: 39–48. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM (2006). Endothelium‐derived hyperpolarizing factor: where are we now? Arterioscler Thromb Vasc Biol 26: 1215–1225. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM (2009). EDHF: an update. Clin Sci (Lond) 117: 139–155. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I et al. (1999). Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401: 493–497. [DOI] [PubMed] [Google Scholar]
- Forstermann U, Munzel T (2006). Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113: 1708–1714. [DOI] [PubMed] [Google Scholar]
- Freed JK, Beyer AM, LoGiudice JA, Hockenberry JC, Gutterman DD (2014). Ceramide changes the mediator of flow‐induced vasodilation from nitric oxide to hydrogen peroxide in the human microcirculation. Circ Res 115: 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF (1966). The use of β‐haloalkylamines in the differentiation of receptors and in determination of dissociation constants of receptor‐agonist complexes In: Harper NJ, Simmonds AB. (eds). Advances in Drug Research, Vol. 3 Academic Press, pp. 21–55. [Google Scholar]
- Galvez A, Gimenez‐Gallego G, Reuben JP, Roy‐Contancin L, Feigenbaum P, Kaczorowski GJ et al. (1990). Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium‐activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem 265: 11083–11090. [PubMed] [Google Scholar]
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B (1995). Potent and selective inhibition of nitric oxide‐sensitive guanylyl cyclase by 1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one. Mol Pharmacol 48: 184–188. [PubMed] [Google Scholar]
- Gauthier KM, Olson L, Harder A, Isbell M, Imig JD, Gutterman DD et al. (2011). Soluble epoxide hydrolase contamination of specific catalase preparations inhibits epoxyeicosatrienoic acid vasodilation of rat renal arterioles. Am J Physiol Renal Physiol 301: F765–F772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart FD, Boardman PL (1963). Indomethacin: a new non‐steroid anti‐inflammatory agent. Br Med J 2: 965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T (2001). Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104: 2673–2678. [DOI] [PubMed] [Google Scholar]
- Hilgers RH, Janssen GM, Fazzi GE, De Mey JG (2010). Twenty‐four‐hour exposure to altered blood flow modifies endothelial Ca2+‐activated K+ channels in rat mesenteric arteries. J Pharmacol Exp Ther 333: 210–217. [DOI] [PubMed] [Google Scholar]
- Kirkby NS, Hadoke PW, Bagnall AJ, Webb DJ (2008). The endothelin system as a therapeutic target in cardiovascular disease: great expectations or bleak house? Br J Pharmacol 153: 1105–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy F, Gough DA, Schmid‐Schonbein GW (1998). Role of xanthine oxidase in hydrogen peroxide production. Free Radic Biol Med 25: 720–727. [DOI] [PubMed] [Google Scholar]
- Li L, Fink GD, Watts SW, Northcott CA, Galligan JJ, Pagano PJ et al. (2003). Endothelin‐1 increases vascular superoxide via endothelin(A)‐NADPH oxidase pathway in low‐renin hypertension. Circulation 107: 1053–1058. [DOI] [PubMed] [Google Scholar]
- Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD (2011). H2O2 is the transferrable factor mediating flow‐induced dilation in human coronary arterioles. Circ Res 108: 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomis ED, Sullivan JC, Osmond DA, Pollock DM, Pollock JS (2005). Endothelin mediates superoxide production and vasoconstriction through activation of NADPH oxidase and uncoupled nitric‐oxide synthase in the rat aorta. J Pharmacol Exp Ther 315: 1058–1064. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Nakashima M, Hirakawa Y, Mukai Y, Hirano K et al. (2000). Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest 106: 1521–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H, Kubota H, Morikawa K, Fujiki T, Kunihiro I et al. (2002). Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in human mesenteric arteries. Biochem Biophys Res Commun 290: 909–913. [DOI] [PubMed] [Google Scholar]
- Meens MJ, Fazzi GE, van Zandvoort MA, De Mey JG (2009). Calcitonin gene‐related peptide selectively relaxes contractile responses to endothelin‐1 in rat mesenteric resistance arteries. J Pharmacol Exp Ther 331: 87–95. [DOI] [PubMed] [Google Scholar]
- Miyagawa K, Emoto N (2014). Current state of endothelin receptor antagonism in hypertension and pulmonary hypertension. Ther Adv Cardiovasc Dis 8: 202–216. [DOI] [PubMed] [Google Scholar]
- Momotani K, Artamonov MV, Utepbergenov D, Derewenda U, Derewenda ZS, Somlyo AV (2011). p63RhoGEF couples Galpha(q/11)‐mediated signaling to Ca2+ sensitization of vascular smooth muscle contractility. Circ Res 109: 993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morikawa K, Fujiki T, Matoba T, Kubota H, Hatanaka M, Takahashi S et al. (2004). Important role of superoxide dismutase in EDHF‐mediated responses of human mesenteric arteries. J Cardiovasc Pharmacol 44: 552–556. [DOI] [PubMed] [Google Scholar]
- O'Donnell BV, Tew DG, Jones OT, England PJ (1993). Studies on the inhibitory mechanism of iodonium compounds with special reference to neutrophil NADPH oxidase. Biochem J 290 (Pt 1): 41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips SA, Hatoum OA, Gutterman DD (2007). The mechanism of flow‐induced dilation in human adipose arterioles involves hydrogen peroxide during CAD. Am J Physiol Heart Circ Physiol 292: H93–100. [DOI] [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S (1990). Characterization of three inhibitors of endothelial nitric oxide synthase in vitro and in vivo . Br J Pharmacol 101: 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Cifuentes ME, Kiarash A, Quinn MT, Pagano PJ (2001). Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(‐) and systolic blood pressure in mice. Circ Res 89: 408–414. [DOI] [PubMed] [Google Scholar]
- Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH (1998). Bis‐quinolinium cyclophanes: 6,10‐diaza‐3(1,3),8(1,4)‐dibenzena‐1,5(1,4)‐ diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non‐peptidic blocker of the apamin‐sensitive Ca(2+)‐activated K+ channel. J Med Chem 41: 2–5. [DOI] [PubMed] [Google Scholar]
- Satoh K, Godo S, Saito H, Enkhjargal B, Shimokawa H (2014). Dual roles of vascular‐derived reactive oxygen species – with a special reference to hydrogen peroxide and cyclophilin A. J Mol Cell Cardiol 73: 50–56. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al. (2012). Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spijkers LJ, van den Akker RF, Janssen BJ, Debets JJ, De Mey JG, Stroes ES et al. (2011). Hypertension is associated with marked alterations in sphingolipid biology: a potential role for ceramide. PLoS One 6: e21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ (1994). Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy‐substituted catechol. Am J Respir Cell Mol Biol 11: 95–102. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM (1999). Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol Rev 51: 159–212. [PubMed] [Google Scholar]
- Takeya K, Wang X, Kathol I, Loutzenhiser K, Loutzenhiser R, Walsh MP (2015). Endothelin‐1, but not angiotensin II, induces afferent arteriolar myosin diphosphorylation as a potential contributor to prolonged vasoconstriction. Kidney Int 87: 370–381. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang DH (2004). Prevention of endothelin‐1‐induced increases in blood pressure: role of endogenous CGRP. Am J Physiol Heart Circ Physiol 287: H1868–H1874. [DOI] [PubMed] [Google Scholar]
- Widlansky ME, Gokce N, Keaney JF Jr, Vita JA (2003). The clinical implications of endothelial dysfunction. J Am Coll Cardiol 42: 1149–1160. [DOI] [PubMed] [Google Scholar]
- Yada T, Shimokawa H, Hiramatsu O, Haruna Y, Morita Y, Kashihara N et al. (2006). Cardioprotective role of endogenous hydrogen peroxide during ischemia‐reperfusion injury in canine coronary microcirculation in vivo. Am J Physiol Heart Circ Physiol 291: H1138–H1146. [DOI] [PubMed] [Google Scholar]
- You J, Golding EM, Bryan RM Jr (2005). Arachidonic acid metabolites, hydrogen peroxide, and EDHF in cerebral arteries. Am J Physiol Heart Circ Physiol 289: H1077–H1083. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Overview of pD2 and Emax values from all figures
Figure S1 Contractile responses to cumulative concentrations of angiotensin II (Ang II; n = 27), vasopressin (n = 5), noradrenaline (n = 13), phenylephrine (n = 7), 5‐hydroxytryptamine (5‐HT, serotonin; n = 5) and sarafotoxin 6c (n = 7). Means ± SEM.
Figure S2 Contractile responses to cumulative concentrations of ET‐1 (A) and sarafotoxin 6c (B) in the absence and presence of 100 μM L‐NAME, 1 μM TRAM‐34 and 1 μM UCL 1684 and their combination. All responses to sarafotoxin 6c in presence of 10 μM indomethacin. Means ± SEM (A: n = 14, 11, and 10 for control, L‐NAME and TRAM‐34 + UCL 1684, respectively. B: n = 7, 7, 7, and 6 for control, L‐NAME, TRAM‐34 + UCL 1684 and L‐NAME + TRAM‐34 + UCL 1684, repectively).
Figure S3 Contractile responses to cumulative concentrations of ET‐1 in the absence and presence of the non‐peptide ETA receptor antagonist PD156707 (100 nM). Means ± SEM (n = 7). The difference in potency is statistically significant, analysed by paired two‐sided T‐test.
Supporting info item
Supporting info item