

Abstract
Background and Purpose
The organic cation transporter 1 (OCT1) transports cationic drugs into hepatocytes. The high hepatic expression of OCT1 is controlled by the HNF4α and USF transcription factors. Pregnane X receptor (PXR) mediates induction of the principal xenobiotic metabolizing enzymes and transporters in the liver. Here, we have assessed the down‐regulation of OCT1 expression by PXR activation.
Experimental Approach
We used primary human hepatocytes and related cell lines to measure OCT1 expression and activity, by assaying MPP+ accumulation. Western blotting, qRT‐PCR, the OCT1 promoter gene reporter constructs and chromatin immunoprecipitation assays were also used.
Key Results
OCT1 mRNA in human hepatocytes was down‐regulated along with reduced [3H]MPP+ accumulation in differentiated HepaRG cells after treatment with rifampicin. Rifampicin and hyperforin as well as the constitutively active PXR mutant T248D suppressed activity of the 1.8 kb OCT1 promoter construct in gene reporter assays. Silencing of both PXR and HNF4α in HepaRG cells blocked the PXR ligand‐mediated down‐regulation of OCT1 expression. The mutation of HNF4α and USF1 (E‐box) responsive elements reversed the PXR‐mediated inhibition in gene reporter assays. Chromatin immunoprecipitation assays indicated that PXR activation sequestrates the SRC‐1 coactivator from the HNF4α response element and E‐box of the OCT1 promoter. Consistent with these findings, exogenous overexpression of the SRC‐1, but not the PGC1α coactivator, relieved the PXR‐mediated repression of OCT1 transactivation.
Conclusions and Implications
PXR ligands reduced the HNF4α‐mediated and USF‐mediated transactivation of OCT1 gene expression by competing for SRC‐1 and decreased delivery of a model OCT1 substrate into hepatocytes.
Abbreviations
- CAR
constitutive androstane receptor
- ChiP
chromatin immunoprecipitation assay
- CML
chronic myeloid leukaemia
- DR‐2
direct repeat separated by two nucleotides
- HNF4α
hepatocyte nuclear factor‐4‐α
- OCT1
organic cation transporter 1
- PXR
pregnane X receptor
- SRC‐1
steroid receptor coactivator
- PGC1α
PPARγ coactivator 1α
- USFs
upstream stimulating factors
Tables of Links
| TARGETS |
|---|
| Transporters a |
| OCT‐1, organic cation transporter 1, SLC22A1 |
| Nuclear hormone receptors b |
| CAR, constitutive androstane receptor |
| GRIP‐1, NCOA2 |
| HNF4α, hepatocyte nuclear factor‐4‐α, NR2A1 |
| PGC1α, PPARGC1A |
| PXR, pregnane X receptor |
| SRC‐1, steroid receptor coactivator, NCOA1 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a, 2015b).
Introduction
The human organic cation transporter 1 (OCT1), encoded by the SLC22A1 gene, is responsible for the drug delivery of various cationic drugs (the antiviral drugs lamivudin, zalcitabine, ganciclovir and acyclovir; the anticancer drug oxaliplatin; the anti‐hyperglycaemic drug metformin) and endogenous substrates such as dopamine, serotonin and choline into the hepatocyte from sinusoidal blood (Jonker and Schinkel, 2004; Koepsell et al., 2007; Nies et al., 2009; Boxberger et al., 2014). Notably, hepatocytes are the pharmacological target of some OCT1 substrate drugs including metformin and lamivudine, and they are the site of the biotransformation and clearance of most drugs in general.
Studies with Oct1‐null mice have clearly shown that Oct1 is the major physiological hepatic uptake system for small organic cations (Jonker et al., 2003). In Oct1−/− mice, the blood glucose‐lowering effect of metformin was completely abolished (Shu et al., 2007), and the liver concentration of metformin was approximately 30 times higher in wild‐type mice than in Oct1−/− mice (Wang et al., 2002; Wang et al., 2003). In addition, genetic variation rs622342 of the human SLC22A1 gene has been associated with the glucose‐lowering effect of metformin in patients with diabetes mellitus (Becker et al., 2009).
The hepatocyte‐specific expression of OCT1 is strongly controlled by the liver‐enriched transcription factor hepatocyte nuclear factor‐4‐α (HNF4α, NR2A1) (Saborowski et al., 2006; Kamiyama et al., 2007; Rulcova et al., 2013). Saborowski et al. (2006) identified two interacting HNF4α response elements of the direct repeat (DR‐2) format in the OCT1 promoter at −1479 to −1441 bp of 5′‐flanking region of the OCT1 (SLC22A1) gene. In addition, a functional E‐box (CACGTG) has been identified in the core proximal promoter region controlling the initiation of transcription. The E‐box binds the upstream stimulating factors (USFs) USF1 and USF2 that further stimulate the HNF4α‐mediated transactivation of the OCT1 gene (Kajiwara et al., 2008). OCT1 mRNA and protein expression varies between 113‐fold and 83‐fold, respectively, in human livers population (Nies et al., 2009).
Recently, however, contradictory data have also been published regarding OCT1 gene regulation by pregnane X receptors (PXR, NR1I2). PXR, the ligand‐activated nuclear receptor of nuclear receptor subfamily NR1I, is the xenobiotic receptor governing the inducible expressions of a broad spectrum of target genes that encode phase‐I and phase‐II xenobiotic‐metabolizing enzymes and drug transporters. Thus, PXR mediates a detoxification response to potentially toxic xenobiotics and also to toxic endogenous compounds (Chen et al., 2012).
Cho and co‐workers have reported that rifampicin increases the glucose‐lowering effect of metformin, an OCT1 substrate, by 54% and that rifampicin significantly induces OCT1 mRNA in peripheral blood cells. Therefore, the authors hypothesize that rifampicin induces OCT1 in hepatocytes, which subsequently increases the uptake of metformin into hepatocytes, resulting in the increased effect of metformin (Cho et al., 2011). Similarly, agonists of PXR induced OCT1 mRNA in the chronic myeloid leukaemia (CML) cell line and primary CML cells (Austin et al., 2015). However, contradictory data have been also published indicating the down‐regulation of OCT1 mRNA in human hepatocytes after treatment with rifampicin (Jigorel et al., 2006; Badolo et al., 2013). As yet, the underlying molecular mechanism of the rifampicin‐mediated changes in OCT1 expression has not been determined.
Therefore, in the current work, we aimed to examine whether OCT1 is regulated by the PXR nuclear receptor and to elucidate the mechanism of this regulation in primary human hepatocytes and in hepatocyte‐derived cellular models.
Methods
Cellular models
The human Caucasian hepatocellular carcinoma (HepG2) and human hepatocellular carcinoma HuH7 (D12) cell lines were purchased from the European Collection of Cell Cultures (Salisbury, UK) or Japanese Collection of Research Bioresources Cell Bank, Osaka, Japan. Because the proliferating tumour hepatic cell lines lack appropriate hepatic phenotype of differentiated hepatocytes and do not express enough functional endogenous PXR under normal conditions, we also used differentiated HepaRG cells and primary human hepatocyte models.
Cryopreserved HepaRG™ (GIBCO®) cells and media were purchased from Life Technologies (Carlsbad, CA, USA). The HepaRG™ cell line is an immortalized and terminally differentiated hepatic cell line that retains many liver‐specific characteristics of primary human hepatocytes. The HepaRG cells were initially isolated from a liver tumour of a female patient suffering from hepatocarcinoma (Gripon et al., 2002). A substantial expression of OCT1 mRNAs reaching 50% of those found in primary human hepatocytes was detected in confluent and DMSO‐treated HepaRG cells cultured in conditions promoting their differentiation (Le Vee et al., 2006). In contrast, HepG2 and Huh7 cells express low OCT1 mRNA levels (Hilgendorf et al., 2007); unpublished data of present authors).
The primary human hepatocytes were prepared from lobectomy segments resected from adult patients for medical reasons unrelated to our research programme (for details see Supporting Information). The human tissue acquisition was undertaken according to procedures complying with the current Czech and French legislation. In addition, five commercial cultures of long‐term human hepatocytes, as monolayers, were used (Biopredic International, Rennes, France or Primcyt, Scwerin, Germany)(Supporting Information).
Cell lines, HepaRG and primary human hepatocyte cells cultivation protocols are in Supporting Information.
Plasmids and siRNA
To produce a 1.8 kb OCT1 reporter construct with two DR‐2 HNF4a binding sites (Saborowski et al., 2006), the promoter sequence from −1812 to +102 was synthesized and inserted into the pGL4.10 vector (Promega) using KpnI and XhoI restriction enzymes (pOCT1 1.8 kb‐luc). Sequences −1649 to +102, −1458 to +102, −430 to +102 and −99 to +102 were PCR‐amplified and inserted into the pGL4.10 vector (Promega) using KpnI and XhoI restriction enzymes (Rulcova et al., 2013). Construct pOCT1(−1649/+102)‐luc contains both DR‐2 elements binding HNF4α, whereas construct pOCT1(−1458/+102)‐luc lacks the first HNF4α response element critical for OCT1 promoter transactivation with HNF4α. Construct OCT1(−430/+102)‐luc lacks both DR‐2 elements. Generation of mutant plasmids and description of additional constructs is mentioned in Supporting Information.
Transient transfection assays
The transient transfection assays were carried out using TransFectin transfection reagent (BioRad, Hercules, CA, USA) in the case of the HepG2 cells and using JetPEI (Polyplus‐transfection SA, Illkirch, France) to transfect the HuH‐7 cell line according to the manufacturer's protocol as described elsewhere (Smutny et al., 2014)(Supporting Information).
qRT‐PCR
In these series of experiments, the HepG2 cells were seeded into 12‐well plates and 24 h later were transfected with PXR expression vector (300 ng per well) or with empty control plasmid as described above. The HepaRG cells were seeded into 12‐well plates without exogenous PXR transfection. Both cell lines were then maintained in cultivating medium supplemented with the tested compounds at the indicated concentrations for 24 hours. Total RNA isolation and qRT‐PCR is described in Supporting Information.
Uptake studies in HepaRG cells
Uptake assays were carried out in 12‐well plates as described previously (Mandikova et al., 2013). The transport assays were performed in HepaRG cells differentiated for 14 days under confluent conditions and pre‐treated with 10 μM rifampicin for 24 or 48 h. Before the accumulation experiment, the HepaRG cultivation medium was removed, and the cells were washed with transport solution (NaCl 130 mM, KCl 4 mM, CaCl2 1 mM, MgCl2 1 mM, glucose 5 mM and HEPES 10 mM, pH 7.4) and pre‐incubated for 10 min at 37°C. The standard radiolabelled substrate for the OCT1 transporter, [3H]MPP+, was used at 1 μM. After a 2 min incubation interval at 37°C, the incubation was stopped by washing the cells twice with ice‐cold solution containing 137 mM NaCl and 10 mM HEPES, pH 7.4. The cells were lysed with 0.1 mL of 0.5% Triton X‐100 in 100 mM NaOH for 30 min. Radioactivity of the samples in scintillation solution (Sigma‐Aldrich) was measured with a β counter (Tri‐Carb 2900TR; Perkin Elmer, Shelton, CT, USA).
ChIP assay
Steroid receptor coactivator (SRC)‐1 recruitment to the HNF4α responsive elements (DR‐2) and E‐box in the OCT1 gene promoter was determined using a chromatin immunoprecipitation (ChIP) assay in the HepG2 cells, which were transfected with pSG5‐hPXR and pSG5‐FLAG hSRC‐1 (3.2 μg in 25 cm2 flask) expression plasmids after one day of cultivation. Cells were then treated using 10 μM rifampicin or 1‰ DMSO as control sample for 24 h. After the interval, the cells were harvested, and immunoprecipitation was conducted with the Imprint® Chromatin Immunoprecipitation Kit (Sigma‐Aldrich) according to the manufacturer's protocol. Anti‐FLAG M2 antibody (Sigma‐Aldrich), normal mouse IgG antibody and anti‐RNA polymerase II (Pol II) antibody were used for precipitation. Levels of SRC‐1 recruitment to the HNF4α response elements and into the E‐box were analysed by qRT‐PCR with primers and probes specifically designed to amplify indicated regions (Figure 6, upper panel, arrows). Data are presented as a relative binding to control vehicle (DMSO)‐treated cell samples (100%) immunoprecipitated with the same antibody. Non‐specific immunoprecipitation with mouse IgG antibody was 1% lower than with anti‐FLAG or the anti‐Pol II antibodies. Primers for ChiP experiments are listed in Supporting Information Table S2.
Figure 6.
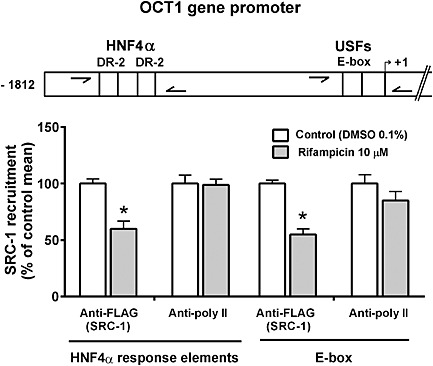
Recruitment of SRC‐1 into the OCT1 gene promoter regions in response to rifampicin treatment in HepG2 cells. Chromatin immunoprecipitation (ChIP) assay was performed to analyse the association of SRC‐1 within the HNF4α response elements (DR‐2 motifs) and E‐box of the OCT1 gene. HepG2 were transfected with pSG5‐FLAG hSRC‐1 and pSG5‐hPXR constructs and then treated with rifampicin (10 μM) or vehicle (DMSO, 0.1%) for 24 h. After this, immunoprecipitated DNA fragments extracted from sonicated HepG2 cell lysates with anti‐FLAG antibody (M2 antibody, Sigma‐Aldrich) or anti RNA polymerase II (poly II) antibody were analysed using qPCR with specific TaqMan probes that detect the individual regions of the OCT1 gene promoter (arrows shown in upper panel). All vehicle‐treated control and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis so the control group value is 100%. The effects of PXR activation on SRC‐1 recruitment are presented as % of the control group's mean value (n = 5).*P < 0.05, significantly different from vehicle‐treated cells; Student's unpaired two‐tailed t‐test.
Immunoblotting analysis of OCT1 protein
A Western blotting analysis of OCT1 protein expression in differentiated HepaRG cells, in primary hepatocytes LH42 (48 h treatment) or in human hepatocytes in monolayer‐long term cultures (Biopredic, Rennes, France, Batch No. HEP220879, female, Caucasian, 65 years old, liver metastases, 96 h treatment) was performed as described previously (Rulcova et al., 2013). To determine the protein level of OCT1 transporter, the primary hepatocyte cultures or HepaRG cells (differentiated as described before for 3 weeks) were treated with 10 μM rifampicin, 10 μM hyperforin or 1‰ DMSO (control) for 48 or 96 h. Total cellular fractions were prepared, denatured for 5 min at 95°C in 2× Laemmli sample buffer and separated (50 μg of protein per well) on 7.5% SDS/polyacrylamide gels. Membranes were then incubated for 24 h at 4°C with anti‐OCT1 antibody (ab55916; Abcam, Cambridge, UK) (1:1000 dilution), anti‐HNF‐4‐α antibody (ab139420; Abcam), anti‐PXR antibody (ab85451; Abcam) or anti‐GADPH polyclonal antibody (PA1‐987; Thermo Fisher, Waltham, MA, USA; 1:5000 dilution). After washing with Tris‐buffered saline with Tween 20 (0,1%v/v), pH 7.4, membranes were incubated with the corresponding goat anti‐rabbit HRP‐conjugated ab136636 (Abcam) or sc‐2004 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies (1:5000 dilution) for 60 min at room temperature. Chemiluminescence was detected using the Image Quant 400 CCD camera (GE Healthcare, Little Chalfont, UK). Digital image densitometry analysis has been performed with labimage 2.7 software (Kapelan, Leipzig, Germany).
Study design and statistical analysis
The study design conforms to the recent guidance on experimental design and analysis (Curtis et al., 2015). In all experiments, data subjected to statistical analysis are from at least 5 independent values or samples). Groups were of equal sizes. In Figure 3A and D, data have been obtained from three independent experiments (n = 3) and have not been subjected to statistical analysis. Justification is given in the Results section. The order of treatment in cellular assays were randomized (i.e. vehicle‐treated controls were not systematically treated first). Data analysis was blinded to the analyst who helped with statistical analysis.
Figure 3.
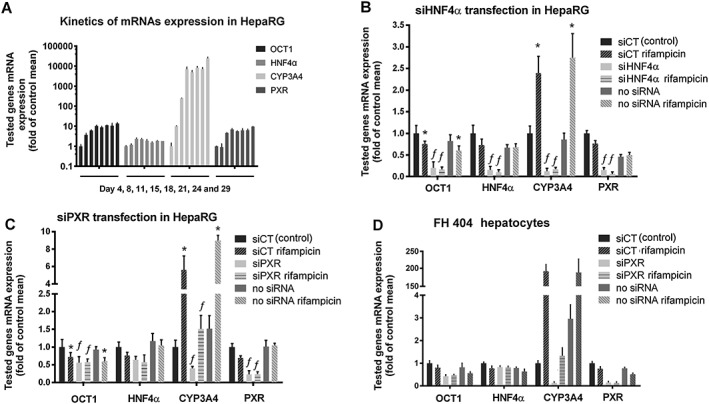
Silencing of PXR and HNF4α prevents PXR‐mediated down‐regulation of OCT1 mRNA. (A) Optimization of HepaRG cultivation and kinetics of OCT1, HNF4α, PXR and CYP3A4 mRNAs expression in HepaRG cells during 4 weeks of differentiation in DMSO‐free medium. Levels of tested genes were assessed twice a week (on days 4, 8, 11, 15, 18, 21, 24 and 29, respectively); total mRNA was extracted, and tested genes mRNA were quantified using qRT‐PCR. Data are presented as the mean ± SD from three experiments performed in duplicate and are expressed as fold of the control group's mean value (day 4, the control group value is 1. (B–C) HepaRG cells or representative primary human hepatocyte culture FH 404 (D) were transfected with siHNF4α (20 nM in medium) (B) or siPXR (10 nM) (C, D). Total RNA was then isolated and OCT1, HNF4α, CYP3A4 and PXR mRNA expression was analysed using qRT‐PCR. All control and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis so the control group value is 1. The effects of tested combinations on the normalized tested genes mRNA expression are presented as fold of the control group's mean value (n = 5).*P < 0.05, significant effect of rifampicin (10 μM) on OCT1, CYP3A4 or PXR mRNA expression in samples transfected with the same siRNAs; †P < 0.05, significant effect of siPXR or siHNF4α on tested genes expression compared with siCT (scramble, control) transfected sample counterparts; ANOVA with Dunnett's post hoc test.
The absolute effect of PXR activation on OCT1 expression is useless and we therefore, normalized these data. In all figures, control and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis, so the control group value is 1 or 100%. Data for parametric statistical analysis were not normalized.
All results are presented as the mean ± SD. Differences between the groups were compared using a Student's unpaired two‐tailed t‐test. One‐way analysis of variance with a Dunnett's post hoc test was applied to the data if more than two groups were analysed, only if F achieved the level of significance P < 0.05 and no significant variance inhomogeneity was observed. A nonparametric Mann–Whitney U‐test (also called Wilcoxon rank‐sum test) was used to compare OCT1 mRNA expression in two sets of samples of primary human hepatocytes or HepaRG cells. All of the statistical analyses were performed using graph‐pad prism 6 software (GraphPad Software Inc., San Diego, CA, USA). A P‐value of <0.05 was considered to be statistically significant.
Materials
Rifampicin and hyperforin, non‐essential amino acids (NEAA), DMSO and DMEM medium were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Drug stock solutions (1000× or 5000×) were prepared in DMSO. Radio‐labelled methyl‐4‐phenylpyridinium acetate ([3H]MPP+), the substrate for OCT1, was obtained from American Radiolabeled Chemicals (St. Louis, MO, USA). Ultima Gold™ LSC cocktail was purchased from Perkin Elmer (Waltham, MT, USA) and MPP+ from Sigma‐Aldrich.
Results
Ligand‐mediated activation of PXR resulted in the down‐regulation of OCT1 mRNA in hepatocyte cellular models
Firstly, we determined the effects of activation of the PXR nuclear receptor on the OCT1 transporter at mRNA and protein level in primary human hepatocytes and in differentiated HepaRG cells. In a group of 15 primary human hepatocyte preparations from different donors, rifampicin (10 μM), a model agonist of PXR, significantly (P < 0.0054, Mann–Whitney U‐test) suppressed OCT1 mRNA expression after 24 h treatment. In these experiments, OCT1 mRNA levels in rifampicin‐treated samples were consistently lower than in vehicle‐treated samples in all cases examined, with the exception of two preparations (Figure 1A).
Figure 1.
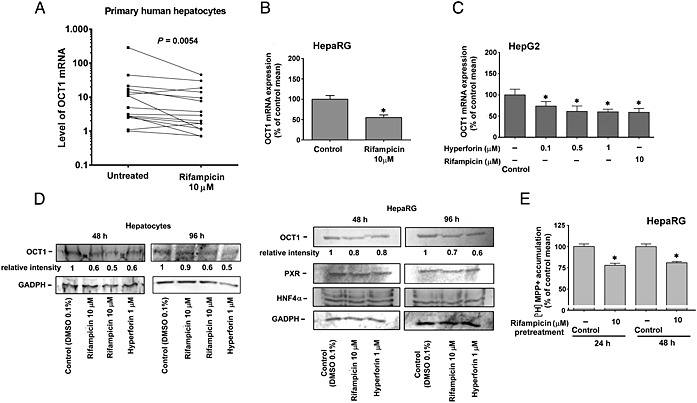
Rifampicin down‐regulates OCT1 mRNA. (A) Rifampicin down‐regulates OCT1 mRNA in a set of primary human hepatocytes. Different primary human hepatocyte preparations were isolated and cultivated according to standard protocol and then either treated with rifampicin (10 μM) for 24 h or treated with vehicle (0.1% DMSO, control). The expression of OCT1 mRNA normalized to the housekeeping gene is shown, and data have been analysed using the Mann–Whitney U‐test. Rifampicin and hyperforin suppress the expression of OCT1 mRNA in differentiated HepaRG (B) and HepG2 (C) cells. HepG2, but not HepaRG, cells were transfected with pSG5‐hPXR and treated with the indicated concentrations of PXR ligands, rifampicin and hyperforin, or with vehicle (DMSO 0.1%, control), for 24 h. After this, total RNA was isolated, and OCT1 mRNA levels were analysed employing qRT‐PCR. All control and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis, so the control group value is 100%. The effects of compounds on the normalized OCT1 mRNA levels are presented as % of the control group's mean value (n = 5).*P < 0.05, significant effect of rifampicin or hyperforin on OCT1 mRNA expression, compared with vehicle‐treated controls; ANOVA with Dunnett's post hoc test. (D) PXR ligands down‐regulate OCT1 protein expression in differentiated primary human hepatocytes and HepaRG cells. Western blots were performed with anti‐OCT1 polyclonal antibody, anti‐PXR and anti‐HNF4α antibodies after 48 or 96 h of treatment with rifampicin (10 μM) and hyperforin (10 μM). Total cellular lysates were used for immunoblotting experiments. Densitometry analyses were performed to evaluate relative intensity of OCT1 bands. (E) Relative accumulation of OCT1 substrate [3H]MPP+ in HepaRG cells. Accumulation of [3H]MPP+ was reduced after 24 and 48 h of treatment with 10 μM rifampicin in differentiated HepaRG cells. *P < 0.05. Accumulation of [3H]MPP+ in control cells was set as 100%, which represent about 1.1% of [3H]MPP+ radioactivity in the medium. All control and test values were normalized to the mean value of the experimental control group. The effects of rifampicin on the normalized [3H]MPP+ radioactivity levels are presented as % of the control group's mean value (n = 5). *P < 0.05, significant effect of rifampicin on [3H]MPP+ accumulation in HepaRG cells, compared with vehicle‐treated controls; Student's unpaired two‐tailed t‐test.
In HepaRG cells differentiated for 14 days in medium without 1.5% DMSO, rifampicin down‐regulated OCT1 mRNA by 50% after 24 h treatment (Figure 1B). Moreover, in HepG2 cells transiently transfected with exogenous PXR, a consistent and concentration‐dependent down‐regulation of OCT1 mRNA level was observed after 24 h treatment with rifampicin or hyperforin, activators of PXR (Figure 1C). The suppressive effect of these PXR ligands was apparent only with exogenous PXR cDNA transfected into cells, a finding, which is in accordance with the low functional expression of PXR in hepatic HepG2 cells (Smutny et al., 2014) and clearly indicates involvement of PXR in OCT1 gene down‐regulation.
Using Western blotting, we confirmed down‐regulation of OCT1 protein in differentiated HepaRG cells and in primary human hepatocytes by the PXR ligands, rifampicin and hyperforin (Figure 1D), after 48 and 96 h of treatment.
We also performed accumulation experiments to confirm the influence of PXR activation on OCT1 transporter activity in HepaRG cells pre‐treated with rifampicin. A decreased accumulation of the model substrate [3H]MPP+ by the OCT1 transporter was decreased in cells pre‐treated with 10 μM rifampicin for 24 or 48 h when compared with control DMSO‐treated cells (Figure 1E). These data confirm that the down‐regulation of OCT1 gene expression correlates with the reduced delivery of OCT1 substrates into hepatic cells.
Transactivation of OCT1 gene is suppressed by activated PXR in hepatocyte‐derived cell lines
First, HepG2 and HuH7 cells were transfected with a pOCT1 1.8 kb‐luc reporter construct harbouring 1.8 kb of the OCT1 promoter sequence upstream of the transcription start together with PXR and/or HNF4α expression plasmids. We observed a significant suppression of OCT1 gene reporter construct activity after co‐transfection with the PXR expression construct. These observations are consistent with a degree of ligand‐independent activity of PXR in HepG2 cells, a finding which is likely to be the consequence of an endogenous ligand. Increasing amounts of exogenous PXR progressively repressed OCT1 transactivation both alone and in the presence of rifampicin (data not shown). Treatment with rifampicin or hyperforin further potentiated PXR‐mediated transcriptional repression (Figure 2A, B and C). In experiments with cells transfected with only HNF4α (without PXR), basal activity of OCT1 gene reporter was increased as expected, and rifampicin had no suppressive effect on OCT1 promoter construct activity (Figure 2A).
Figure 2.
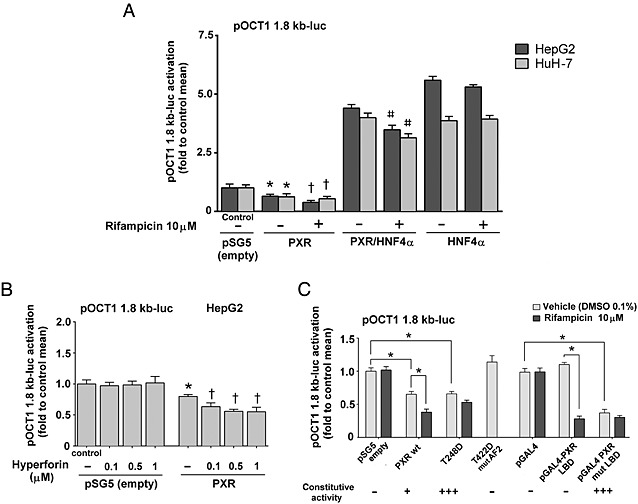
PXR mediates the inhibition of OCT1 promoter transactivation in gene reporter assays. Hepatic HepG2 and HuH‐7 (A) were transfected with the pOCT1 1.8 kb‐luc reporter construct and cotransfected with PXR, HNF4α or their combination as indicated or only with PXR expression construct. (B) Cells were then treated with rifampicin (10 μM), hyperforin (0.1, 0.5, 1 μM) or with vehicle (−) as a control for 24 h. Activity of OCT1 gene reporter construct was detected with the use of Dual‐Luciferase Reporter Assay (Promega). All control and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis so the control group value is 1. The effects of tested combinations on the normalized pOCT1 1.8‐luc activation levels are presented as fold of the control group's mean value (n = 5).*P < 0.05 indicates a statistically significant effect of exogenous PXR expression on basal OCT1 transactivation compared with the mock (empty expression vector) transfected cells. †P < 0.05, significant effect of a PXR ligand (rifampicin or hyperforin) on OCT1 transactivation in PXR expressing cells; ANOVA with Dunnett's post hoc test. # P <0.05, significant effect of rifampicin on OCT1 transactivation in PXR and HNF4 expressing cells. (C.) HepG2 cells were transfected with expression constructs encoding either wild‐type human PXR, constitutively active PXR T248D mutant, inactive PXR T422D mutant with mutated AF2 domain, pGAL4‐PXR LBD construct, constitutively active pGAL4‐PXR LBD S247W/C284W mutant or empty pSG5 or GAL4 vectors (100 ng per well), respectively, and treated with rifampicin (10 μM) or vehicle (DMSO 1 ‰, v/v) for 24 h. Activity of OCT1 gene reporter construct (150 ng per well) was detected with the use of Dual‐Luciferase Reporter Assay. All control (empty expression constructs with vehicle) and test values were normalized to the mean value of the experimental control group, and the data are presented as fold of the control group's mean value (n = 5).*P < 0.05, significant effect; ANOVA with Dunnett's post hoc test.
Finally, gene reporter assays were performed with pOCT1 1.8 kb‐luc reporter vector together with a series of modified PXR expression constructs. The constitutively active PXR T248D construct, but not the inactive PXR T422D construct with a mutation in the AF2 domain, suppressed OCT1 transactivation (Figure 2C). Importantly, the pGAL4‐PXR LBD construct, lacking the DNA binding domain, after rifampicin treatment and its constitutively active variant pGAL4‐PXR LBD (S247W/C248W) with a mutated LBD also diminished OCT1 promoter construct activation (Figure 2C). The effects of the T248D mutant was only similar to but not stronger than, unliganded wild‐type PXR, which may reflect a promoter‐specific effect of the mutant, in comparison with CYP3A4 regulation (Doricakova et al., 2013).
These results indicate that (i) PXR‐mediated suppression of OCT1 gene transactivation is mediated by PXR‐HNF4α crosstalk; (ii) the activation domain AF2 of the PXR LBD is critical for the phenomenon; (iii) the phenomenon is independent of the PXR DNA‐binding domain; and (iv) the phenomenon is likely to be hepatocyte‐cell dependent and HNF4α dependent.
Silencing of PXR and HNF4α in HepaRG cells blocks the PXR‐induced suppression of OCT1 gene expression
HepaRG is the only cell line that, under differentiation conditions, has hepatic phenotype and expresses key nuclear receptors and drug‐metabolizing enzymes. In our initial experiments, the mRNA levels of OCT1, HNF4α, CYP3A4 and PXR (NR1I2) genes were analysed in HepaRG cells, cultivated under the standard differentiation protocol with 1.5% DMSO. We observed that the level of OCT1 and HNF4α was decreased after DMSO addition (data not shown). Therefore, it was decided to perform a kinetic experiment in the confluent cells, 2 weeks after their seeding. For this purpose, HepaRGs were differentiated, without addition of DMSO to the regular culture medium, for another two weeks (Figure 3A). mRNA levels of OCT1, HNF4α, CYP3A4 and PXR (NR1I2) genes were assessed twice a week (on days 4, 8, 11, 15, 18, 21, 24 and 29). All four tested genes were stably expressed after 15 days of the DMSO‐free differentiation period (n = 3, Figure 3A). Therefore, based on these preliminary experiments, we chose a 2 week (15 days) differentiation protocol without DMSO as providing the most suitable and reliable experimental conditions.
At this time, the silencing of HNF4α in HepaRG cells was tested. Transfection of siRNA targeting HNF4α was performed twice in 2 days to improve silencing efficiency. HNF4α mRNA levels and the relevant target genes OCT1, CYP3A4 and PXR were decreased (Figure 3B). Importantly, the silencing of HNF4α abolished the suppressive PXR‐mediated effect on OCT1 gene expression.
In the next series of experiments, HepaRG cells were transfected three times with siRNA against PXR to obtain a greater efficiency of PXR silencing. Levels of both PXR and its main target gene CYP3A4 were significantly decreased. We observed slight suppressive effects of siPXR on OCT1 and HNF4α (Figure 3C). Also of note, siPXR again blocked the rifampicin‐induced PXR‐mediated suppression of OCT1 expression (Figure 3C).
Finally, the effect of PXR silencing in primary human hepatocytes (Figure 3D) was also determined. We analysed levels of OCT1, HNF4α, PXR and CYP3A4 mRNAs in three different batches of human hepatocytes (n = 3) after transfection with siRNA targeting PXR and treatment with 10 μM rifampicin. In concordance with our previous findings in HepaRG cells, there were both a decrease in OCT1 mRNA levels and the prevention of PXR‐mediated repression of OCT1 expression, after the silencing of PXR in primary human hepatocytes (Figure 3D, a representative data from FH 404 preparation are shown). We observed similar profiles in all the primary human hepatocyte preparations we used, even though there was typically high inter‐individual variation.
Interestingly, the silencing of PXR in both HepaRG and in primary human hepatocytes did not lead to up‐regulation of OCT1 mRNA but to the opposite phenomenon, which was consistent with the results obtained using the inactive PXR T422D mutant (Figure 2C).
These results demonstrate that human PXR is responsible for rifampicin‐mediated OCT1 repression in hepatic cells and that HNF4α, a key regulator of OCT1 expression, is necessary for the underlying molecular mechanism. PXR is unlikely to transrepress OCT1 gene expression, because the silencing or inactivation of PXR does not up‐regulate OCT1 mRNA expression.
Cotransfection of SRC‐1, but not PGC1α coactivator, blocks PXR‐mediated down‐regulation of OCT1 gene transactivation
In subsequent experiments, we tested to see whether the forced expression of SRC‐1 or the PPARγ coactivator 1α, (PGC1α) in HepG2 cells could reverse the rifampicin‐induced repression of OCT1 gene expression. In transient transfection gene reporter assays with the 1.8 kb OCT1‐luc construct, incremental amounts of SRC‐1 elicited a progressive reduction of the PXR‐mediated repression of OCT1 transactivation (Figure 4A). When the SRC‐1 expression construct (400 ng per well) was co‐transfected, we even observed the reversal of the phenomenon and induction of OCT1 transactivation. In other experiments, increasing amounts of SRC‐1 significantly stimulated PXR‐responsive p3A4‐luc luciferase reporter construct with critical CYP3A4 gene promoter regulatory elements (Figure in Supporting Information). In contrast, increasing amounts of exogenous PGC1α did not reverse the suppression of OCT1 by activated PXR (Figure 4B).
Figure 4.
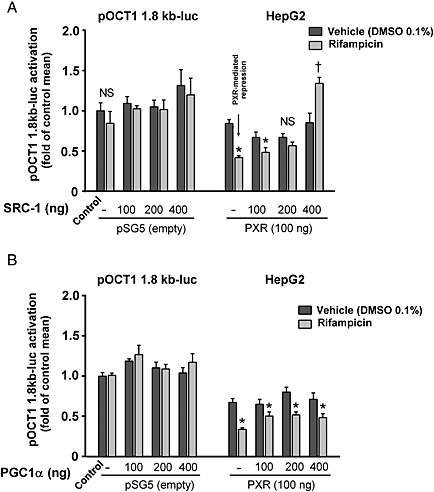
Cotransfection of SRC‐1, but not PGC1α coactivator, blocks PXR‐mediated down‐regulation of OCT1 gene transactivation. HepG2 cells were co‐transfected with the 1.8 kb OCT1 reporter construct, pSG5‐hPXR expression construct and with increasing amount (100, 200 or 400 ng per well) of SCR‐1 (A) or PGC1α (B) expression vectors. Cells were then treated with rifampicin (10 μM) or with vehicle (−) for 24 h. Activity of OCT1 gene reporter construct was detected with the use of Dual‐Luciferase Reporter Assay (Promega). All vehicle‐treated empty construct‐transfected control, and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis so the control group value is 1. The effects of tested combinations on the normalized pOCT1 1.8 kb‐luc activation are presented as fold of the control group's mean value (n = 5).*P < 0.05, significant suppression of OCT1 construct transactivation by rifampicin in comparison with samples transfected with the same expression constructs and treated with vehicle; †P < 0.05, significant augmentation of OCT1 promoter construct transactivation by rifampicin in comparison with samples transfected with the same expression constructs and treated with vehicle; ANOVA with Dunnett's post hoc test was used. NS – non‐significant effect of rifampicin.
These data further support the idea that the common coactivator SRC‐1 is depleted from HNF4α in the OCT1 promoter, after rifampicin‐treatment, by activated PXR and that this depletion results in the repression of the OCT1 gene. However, if SRC‐1 is overexpressed in cells, depletion from HNF4α does not occur, nor is transactivation OCT1 expression by HNF4α substantially attenuated.
HNF4α and E‐box response elements are involved in PXR‐mediated suppression of OCT1 expression
In order to further confirm and evaluate the involvement of the HNF4α and USF factors in OCT1 down‐regulation, we generated a series of OCT1 gene promoter luciferase reporter constructs (Figure 5).
Figure 5.
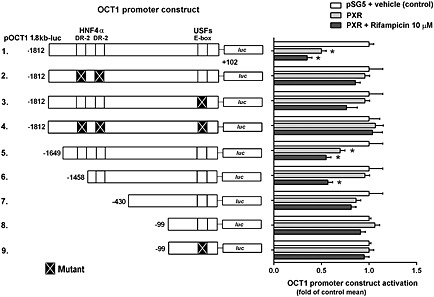
Involvement of HNF4α response elements (DR‐2) and E‐box in transcriptional regulation of the OCT1 gene through PXR nuclear receptors in the HepG2 cell line. Transactivation of the wild‐type pOCT1 1.8 kb‐luc luciferase reporter gene construct, its truncated forms and HNF4α RE or E‐box mutants (150 ng/well) were examined in HepG2 cells transiently co‐transfected with PXR or empty expression pSG5 vectors (100 ng per well). pRL‐TK control plasmid was used for transfection normalization (30 ng per well). Transfected cells were maintained in a medium containing rifampicin (10 μM) for 24 h. After incubation, the cells were lysed and analysed for both firefly and Renilla luciferase activities. Firefly luciferase activity was normalized to Renilla activity. All control and test values were normalized to the mean value of the experimental control group in order to set the Y‐axis so the control group value is 1. The effects of PXR on the normalized OCT1 constructs activation are presented as fold of the control group's mean value (n = 5).*P < 0.05, significantly different from the vehicle‐treated cells transfected with the same luciferase reporter construct and an empty expression vector; ANOVA with Dunnett's post hoc test.
The USF1 and USF2 basal transcription factors, members of the eukaryotic basic helix‐loop‐helix leucine zipper transcription factor family and their E‐box response element act in cooperation with HNF4α, in hepatocyte‐specific OCT1 expression (Kajiwara et al., 2008). Importantly, USF1 has been found to be co‐activated by SRC‐1 (Huang et al., 2007). Therefore, we generated a series of the E‐box and HNF4α response elements mutants of the OCT1 promoter.
We observed statistically significant PXR‐mediated suppressive effects on OCT1 reporter constructs with −1812/+102 promoter sequence (lane 1), truncated −1692/+102 sequence (lane 5) and in −1458/+102 construct, which lacks the first HNF4α response element (lane 6). However, mutation of either HNF4α response elements or USFs response element (E‐box) significantly decreased the PXR‐mediated suppression of the OCT1 promoter activation in these experiments.
These results indicate that both HNF4α response elements and the USFs response element (E‐box) are together critical for the PXR‐mediated transrepression of OCT1 promoter constructs in the gene reporter assays.
Chromatin immunoprecipitation studies to monitor SRC‐1 recruitment to HNF4α and E‐box elements after treatment with rifampicin
To demonstrate that competition for binding to the SRC‐1 coactivator may be the underlying mechanism of the PXR‐mediated suppression of OCT1 expression, chromatin immunoprecipitation experiments with probes designed to amplify both HNF4α response elements and E‐box elements using qPCR were performed (Figure 6, arrows in upper panel). Rifampicin treatment resulted in the dissociation of SRC‐1 from HNF4α response elements and the E‐box, as indicated by a decreased amount of DNA fragments immunoprecipitated with the anti‐FLAG SRC‐1 antibody (Figure 6).
These results are again consistent with our hypothesis that ligand‐activated PXR interacts with HNF4α (and USFs) signalling in OCT1 regulation by targeting the common coactivator SRC‐1. In other words, these data suggest that after PXR activation, PXR, HNF4α and USF all compete with each other to bind to the common co‐activator SRC‐1.
Discussion
Drug delivery into hepatocytes is critical for hepatic metabolism and the biliary elimination of most drugs. Drug transporters determine entry into hepatocytes via the sinusoidal (basolateral) membrane. These membrane transporters are also critical for responses to drugs, in those cases in which hepatocytes are the pharmacological target site. The OCT1 transporter ranks among the most important liver uptake carriers, which transport positively charged substances. At physiological pH, approximately 40% of drugs are organic cations or weak bases, and thus, many of them are potential substrates for OCT1 (Nies et al., 2009). As variable OCT1 expression and gene regulation contributes to intra‐individual and inter‐individual variation in the hepatic disposition of some cationic drugs, further inquiries into OCT1 gene regulation are necessary to predict cationic drug delivery and response in hepatocytes, in the context of clinical consequences of the interaction.
In the current study, the effect of PXR activation on OCT1 expression was examined in several validated hepatocyte cellular models including primary human hepatocytes and differentiated HepaRG cells. In contrast to tumour hepatic cell lines, the differentiated HepaRG cells express functional endogenous PXR and OCT1. All our data indicate that OCT1 hepatic expression is down‐regulated by PXR agonists at the transcriptional level and that the accumulation of the model OCT1 substrate MPP+ is reduced in HepaRG cells pre‐treated with rifampicin. Moreover, we propose the mechanism of the down‐regulation and postulate that PXR reduces OCT1 transactivation by competing for the SRC‐1 coactivator, with the HNF4α and USF factors which control the high constitutive hepatic expression of the OCT1 gene. Such competition for coactivators is called squelching (Cahill et al., 1994; Min et al., 2002) and we shall use this term from now on. For our experiments, a squelching mechanism is based on the results of experiments involving the over‐expression of SRC‐1 coactivator or various PXR mutants, silencing of PXR and HNF4α in HepaRG cells and primary human hepatocytes, mutagenesis of the HNF4α responsive DR‐2 and E‐box response elements and chromatin immunoprecipitation assays.
Recently, Cho and co‐workers hypothesized that rifampicin induced the OCT1 transporter in hepatocytes and thus increased uptake of metformin (Cho et al., 2011). In contrast to this speculation, two published studies showed that rifampicin, via PXR activation, actually down‐regulated OCT1 mRNA expression in primary human hepatocytes (Jigorel et al., 2006; Badolo et al., 2013). Consistent with the latter reports, we observed OCT1 mRNA down‐regulation in most of our tested samples of 15 primary human hepatocytes derived from different donors (Figure 1A) and in differentiated HepaRG cells (Figure 1B). In addition, employing a gene reporter assay, the PXR‐mediated inhibition of OCT1 gene reporter construct was observed in hepatocyte‐derived HuH‐7 and HepG2 cell lines (Figure 2).
The hepatocyte‐specific expression of OCT1 is tightly controlled by the liver‐enriched HNF4α (NR2A1) (Saborowski et al., 2006; Kamiyama et al., 2007; Rulcova et al., 2013). HNF4α is an orphan member of the nuclear receptor superfamily, which has a critical role as a transcription factor in the regulation of a wide variety of liver‐specific genes, involved in lipid and glucose metabolism, hepatocyte differentiation and the maintenance of the hepatic gene expression profile (Watt et al., 2003). Two cooperating DR‐2 HNF4α response elements have been identified between nucleotides −1479 and −1441 of the 5′‐flanking region of the OCT1 gene upstream of the transcription initiation site, which contains directly repeated hexamers separated by two bases (Saborowski et al., 2006). This regulation explains well the dominant hepatic expression of OCT1 mRNA and minimal expression in other non‐hepatic tissues (Nies et al., 2009). In addition, a cognate E‐box interacting with USF1 and USF2 has been identified in the core proximal promoter region that co‐ordinately augments the HNF4α‐mediated transactivation of the OCT1 gene in an additive manner (Kajiwara et al., 2008).
In contrast to the situation with hepatocytes, contradictory data has been observed in non‐hepatic, blood cells. Austin et al. have recently published results in which OCT1 mRNA is up‐regulated in presence of the PXR ligand in both primary CML cells and in the CML cell line KCL22 (Austin et al., 2015). This discrepancy can be explained by the absence of HNF4α in peripheral blood cells. We observed a similar phenomenon in non‐hepatic Hela cells, where rifampicin activated the OCT1 promoter by PXR activation in HNF4α‐deficient HeLa cells. However, concomitant PXR and HNF4α co‐expression in the HeLa cells reversed the effect of rifampicin (our unpublished data). Silencing of HNF4α in hepatocyte cells also did not lead to OCT1 mRNA up‐regulation after rifampicin treatment (Figure 3B and D). Thus, the phenomenon of tissue‐specific OCT1 gene regulation requires further investigation.
In the mechanistic analysis of the PXR‐mediated repression of OCT1 transactivation in hepatic cells, OCT1 promoter transactivation was repressed by the ligand‐activated wild‐type PXR, GAL4‐PXR LBD fusion chimera and constitutively active PXR LBD mutant T248D with activation domain (AF2), but not by the PXR T422D AF2 mutant that fails to bind to coactivators (Figure 2C). These data indicate that PXR coactivation is critical in the phenomenon. In the next experiments, we found that the silencing of PXR and HNF4α abolished the suppressive effect of rifampicin on OCT1 mRNA expression in HepaRG cells (Figure 3B and C) and in primary human hepatocytes (Figure 3D). These results showed that both nuclear receptors were necessary for the phenomenon of OCT1 down‐regulation, a finding which suggests that competition for a common coactivator may underlie the PXR‐mediated inhibition of OCT1 expression. Both PXR and HNF4α are coactivated by SRC‐1 (NCOA1) and PGC1α coactivators (Wang et al., 1998). In the following series of experiments, increasing amounts of SRC‐1 were co‐transfected in HepG2 cells together with the OCT1 reporter construct and PXR in the presence of rifampicin. Consistent with a squelching mechanism, the gradual overexpression of SRC‐1 in HepG2 cells reversed the repressive effect of rifampicin on OCT1 transactivation (Figure 4A). In contrast, increasing the amount of PGC1α in the HepG2 cell line did not de‐repress PXR‐mediated OCT1 promoter inhibition. These data suggested that activation of PXR reduced OCT1 expression by squelching SRC‐1, but not PGC1α, the coactivator in HNF4α‐controlled OCT1 transactivation.
Because PXR binds to DNA motifs, including DR‐3, DR4, everted ER6 and the ER8 motifs (Kliewer et al., 2002), it is unlikely that PXR would inhibit HNF4α‐binding to the OCT1 promoter DR‐2 motifs. In addition, OCT1 gene reporter construct transactivation is attenuated by cotransfection with the GAL4‐PXR LBD construct without the PXR DNA‐binding domain (Figure 2 C). This evidence showed that only PXR‐LBD with a functional AF2 domain binding to coactivators is necessary for the phenomenon.
In a ChIP assay, we employed two sets of DNA probes to quantify SRC‐1 binding to DR‐2 HNF4α response elements and to the E‐box of the OCT1 gene promoter. Consistent with the other results, the decreased binding of SRC‐1 to these response elements was observed after treatment with the PXR ligand rifampicin (Figure 6), confirming the squelching of SRC‐1 in HNF4α and USFs transactivation.
This conclusion is supported by the results of gene reporter experiments employing the mutated or truncated OCT1 gene promoter construct. The PXR‐mediated OCT1 suppression was markedly diminished when DR‐2 HNF4α response elements or the E‐box were mutated. In the double‐mutant HNF4α/E‐box OCT1 gene reporter construct, we observed no suppressive effect of unliganded PXR and a slightly stimulating effect of rifampicin on transactivation (Figure 5, line 4). These data again confirm our hypothesis.
Competition for the common coactivators PGC1α, GRIP‐1 or SRC‐1 has been recently reported as a putative mechanism of crosstalk between CAR and the oestrogen receptor (for GRIP‐1) (Min et al., 2002), PXR and CAR (for SRC‐1) (Saini et al., 2005) and HNF4A and CAR (for PGC1α and GRIP‐1) (Miao et al., 2006), as well as LXR and the retinoid‐related orphan receptor α (Wada et al., 2008). Competition for a common SRC‐1 coactivator has been reported for PXR‐CAR crosstalk (Saini et al., 2005). However, for the HNF4α‐CAR crosstalk (Miao et al., 2006) or for CAR‐LXRα interaction, the effect was not statistically significant in a CHIP assay and competition for the SRC‐1 coactivator does not appear to be the crucial mechanism of the crosstalk (Zhai et al., 2010).
We can conclude that, to the best of our knowledge, this is the first report showing squelching of the SRC‐1 coactivator, between PXR and HNF4α, as an underlying mechanism of PXR‐mediated HNF4α target gene down‐regulation. Significantly, our results show for the first time the mechanism for PXR‐mediated down‐regulation of a drug transporter involved in xenobiotic handling or clearance. Thus, it is possible that other xenobiotic metabolizing enzymes or drug transporters dominantly regulated in the liver by HNF4α might be regulated via similar coactivator squelching by PXR ligands including drugs. Finally, we can hypothesize that PXR‐mediated squelching of coactivators in the regulation of OCT1 can be proposed as a new molecular mechanism of regulating the entry of cationic drugs into hepatocytes. Nevertheless, further experiments are needed to confirm this possibility.
Author contributions
All authors substantially contributed to the conception or design of the work or the acquisition, analysis or interpretation of data for the work and critically revised the manuscript and its final version. They guarantee accuracy and integrity of the work. L.H., T.S., A.C., S.M. and J.M. mainly performed the research and analysed the data. F.T., S.G.C. and P.P. designed some experiments. P.P. designed the research study. L.H. and P.P. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1 Primer sequences
Table S2 Primer sequences
Figure S1 Cotransfection of SRC‐1 significantly stimulates PXR‐mediated transactivation of CYP3A4 gene promoter p3A4‐luc construct.
Supporting info item
Acknowledgements
This work was funded by the Czech Scientific Agency (GACR 303/12/G163 to P.P.) and by SVV project 260 293.
Hyrsova, L. , Smutny, T. , Carazo, A. , Moravcik, S. , Mandikova, J. , Trejtnar, F. , Gerbal‐Chaloin, S. , and Pavek, P. (2016) The pregnane X receptor down‐regulates organic cation transporter 1 (SLC22A1) in human hepatocytes by competing for (“squelching”) SRC‐1 coactivator. British Journal of Pharmacology, 173: 1703–1715. doi: 10.1111/bph.13472.
References
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Cidlowski JA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Nuclear hormone receptors. Br J Pharmacol 172: 5956–5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin G, Holcroft A, Rinne N, Wang L, Clark RE (2015). Evidence that the pregnane X and retinoid receptors PXR, RAR and RXR may regulate transcription of the transporter hOCT1 in chronic myeloid leukaemia cells. Eur J Haematol 94: 74–78. [DOI] [PubMed] [Google Scholar]
- Badolo L, Bundgaard C, Garmer M, Jensen B (2013). The role of hepatic transport and metabolism in the interactions between pravastatin or repaglinide and two rOatp inhibitors in rats. Eur J Pharm Sci 49: 767–772. [DOI] [PubMed] [Google Scholar]
- Becker ML, Visser LE, van Schaik RH, Hofman A, Uitterlinden AG, Stricker BH (2009). Genetic variation in the organic cation transporter 1 is associated with metformin response in patients with diabetes mellitus. Pharmacogenomics J 9: 242–247. [DOI] [PubMed] [Google Scholar]
- Boxberger KH, Hagenbuch B, Lampe JN (2014). Common drugs inhibit human organic cation transporter 1 (OCT1)‐mediated neurotransmitter uptake. Drug Metab Dispos 42: 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill MA, Ernst WH, Janknecht R, Nordheim A (1994). Regulatory squelching. FEBS Lett 344: 105–108. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doricakova A, Novotna A, Vrzal R, Pavek P, Dvorak Z (2013). The role of residues T248, Y249 and T422 in the function of human pregnane X receptor. Arch Toxicol 87: 291–301. [DOI] [PubMed] [Google Scholar]
- Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I et al. (2002). Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci U S A 99: 15655–15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgendorf C, Ahlin G, Seithel A, Artursson P, Ungell AL, Karlsson J (2007). Expression of thirty‐six drug transporter genes in human intestine, liver, kidney, and organotypic cell lines. Drug Metab Dispos 35: 1333–1340. [DOI] [PubMed] [Google Scholar]
- Huang S, Li X, Yusufzai TM, Qiu Y, Felsenfeld G (2007). USF1 recruits histone modification complexes and is critical for maintenance of a chromatin barrier. Mol Cell Biol 27: 7991–8002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Tang Y, Guo C, Wang J, Boral D, Nie D (2012). Nuclear receptors in the multidrug resistance through the regulation of drug‐metabolizing enzymes and drug transporters. Biochem Pharmacol 83: 1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SK, Yoon JS, Lee MG, Lee DH, Lim LA, Park K et al. (2011). Rifampin enhances the glucose‐lowering effect of metformin and increases OCT1 mRNA levels in healthy participants. Clin Pharmacol Ther 89: 416–421. [DOI] [PubMed] [Google Scholar]
- Jigorel E, Le Vee M, Boursier‐Neyret C, Parmentier Y, Fardel O (2006). Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug‐sensing receptors in primary human hepatocytes. Drug Metab Dispos 34: 1756–1763. [DOI] [PubMed] [Google Scholar]
- Jonker JW, Schinkel AH (2004). Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1‐3). J Pharmacol Exp Ther 308: 2–9. [DOI] [PubMed] [Google Scholar]
- Jonker JW, Wagenaar E, Van Eijl S, Schinkel AH (2003). Deficiency in the organic cation transporters 1 and 2 (Oct1/Oct2 [Slc22a1/Slc22a2]) in mice abolishes renal secretion of organic cations. Mol Cell Biol 23: 7902–7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiwara M, Terada T, Asaka J, Aoki M, Katsura T, Ikai I et al. (2008). Regulation of basal core promoter activity of human organic cation transporter 1 (OCT1/SLC22A1). Am J Physiol Gastrointest Liver Physiol 295: G1211–G1216. [DOI] [PubMed] [Google Scholar]
- Kamiyama Y, Matsubara T, Yoshinari K, Nagata K, Kamimura H, Yamazoe Y (2007). Role of human hepatocyte nuclear factor 4alpha in the expression of drug‐metabolizing enzymes and transporters in human hepatocytes assessed by use of small interfering RNA. Drug Metab Pharmacokinet 22: 287–298. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Goodwin B, Willson TM (2002). The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev 23: 687–702. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C (2007). Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24: 1227–1251. [DOI] [PubMed] [Google Scholar]
- Le Vee M, Jigorel E, Glaise D, Gripon P, Guguen‐Guillouzo C, Fardel O (2006). Functional expression of sinusoidal and canalicular hepatic drug transporters in the differentiated human hepatoma HepaRG cell line. Eur J Pharm Sci 28: 109–117. [DOI] [PubMed] [Google Scholar]
- Mandikova J, Volkova M, Pavek P, Cesnek M, Janeba Z, Kubicek V et al. (2013). Interactions with selected drug renal transporters and transporter‐mediated cytotoxicity in antiviral agents from the group of acyclic nucleoside phosphonates. Toxicology 311: 135–146. [DOI] [PubMed] [Google Scholar]
- Miao J, Fang S, Bae Y, Kemper JK (2006). Functional inhibitory cross‐talk between constitutive androstane receptor and hepatic nuclear factor‐4 in hepatic lipid/glucose metabolism is mediated by competition for binding to the DR1 motif and to the common coactivators, GRIP‐1 and PGC‐1alpha. J Biol Chem 281: 14537–14546. [DOI] [PubMed] [Google Scholar]
- Min G, Kim H, Bae Y, Petz L, Kemper JK (2002). Inhibitory cross‐talk between estrogen receptor (ER) and constitutively activated androstane receptor (CAR). CAR inhibits ER‐mediated signaling pathway by squelching p160 coactivators. J Biol Chem 277: 34626–34633. [DOI] [PubMed] [Google Scholar]
- Nies AT, Koepsell H, Winter S, Burk O, Klein K, Kerb R et al. (2009). Expression of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) is affected by genetic factors and cholestasis in human liver. Hepatology 50: 1227–1240. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulcova A, Krausova L, Smutny T, Vrzal R, Dvorak Z, Jover R et al. (2013). Glucocorticoid receptor regulates organic cation transporter 1 (OCT1, SLC22A1) expression via HNF4alpha upregulation in primary human hepatocytes. Pharmacol Rep 65: 1322–1335. [DOI] [PubMed] [Google Scholar]
- Saborowski M, Kullak‐Ublick GA, Eloranta JJ (2006). The human organic cation transporter‐1 gene is transactivated by hepatocyte nuclear factor‐4alpha. J Pharmacol Exp Ther 317: 778–785. [DOI] [PubMed] [Google Scholar]
- Saini SP, Mu Y, Gong H, Toma D, Uppal H, Ren S et al. (2005). Dual role of orphan nuclear receptor pregnane X receptor in bilirubin detoxification in mice. Hepatology 41: 497–505. [DOI] [PubMed] [Google Scholar]
- Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA et al. (2007). Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest 117: 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smutny T, Bitman M, Urban M, Dubecka M, Vrzal R, Dvorak Z et al. (2014). U0126, a mitogen‐activated protein kinase kinase 1 and 2 (MEK1 and 2) inhibitor, selectively up‐regulates main isoforms of CYP3A subfamily via a pregnane X receptor (PXR) in HepG2 cells. Arch Toxicol 88: 2243–2259. [DOI] [PubMed] [Google Scholar]
- Wada T, Kang HS, Jetten AM, Xie W (2008). The emerging role of nuclear receptor RORalpha and its crosstalk with LXR in xeno‐ and endobiotic gene regulation. Exp Biol Med 233: 1191–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y (2002). Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther 302: 510–515. [DOI] [PubMed] [Google Scholar]
- Wang DS, Kusuhara H, Kato Y, Jonker JW, Schinkel AH, Sugiyama Y (2003). Involvement of organic cation transporter 1 in the lactic acidosis caused by metformin. Mol Pharmacol 63: 844–848. [DOI] [PubMed] [Google Scholar]
- Wang JC, Stafford JM, Granner DK (1998). SRC‐1 and GRIP1 coactivate transcription with hepatocyte nuclear factor 4. J Biol Chem 273: 30847–30850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AJ, Garrison WD, Duncan SA (2003). HNF4: a central regulator of hepatocyte differentiation and function. Hepatology 37: 1249–1253. [DOI] [PubMed] [Google Scholar]
- Zhai Y, Wada T, Zhang B, Khadem S, Ren S, Kuruba R et al. (2010). A functional cross‐talk between liver X receptor‐alpha and constitutive androstane receptor links lipogenesis and xenobiotic responses. Mol Pharmacol 78: 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primer sequences
Table S2 Primer sequences
Figure S1 Cotransfection of SRC‐1 significantly stimulates PXR‐mediated transactivation of CYP3A4 gene promoter p3A4‐luc construct.
Supporting info item