

This phase I study evaluated combined panobinostat and epirubicin therapy in patients with advanced solid tumors, establishing an MTD, and demonstrating a correlation between neutropenia, PBMC histone acetylation, and clinical benefit. In a sarcoma expansion cohort, more than half of patients having previously failed anthracycline therapy benefited, suggesting that HDAC inhibition reverses resistance.
Keywords: histone deacetylase, panobinostat, sarcoma, epirubicin, topoisomerase
Abstract
Background
Treatment options for sarcoma are limited. Histone deacetylase inhibitors increase the efficacy of topoisomerase II inhibitors by promoting access to chromatin and by down-regulating DNA repair. Thus, combined panobinostat and epirubicin therapy was evaluated to treat refractory sarcoma.
Patients and methods
Patients with advanced solid tumors were enrolled in a 3 + 3 dose-escalation phase I trial of panobinostat given on days 1, 3, and 5 followed by 75 mg/m2 of epirubicin on day 5 in 21-day cycles, with a dose expansion at maximum tolerated dose (MTD) in 20 sarcoma patients. Peripheral blood mononucleocyte histone acetylation was also evaluated.
Results
Forty patients received 20–60 mg panobinostat. Dose-limiting toxicities included thrombocytopenia, febrile neutropenia, and fatigue at 60 mg, defining a panobinostat MTD at 50 mg. Four responses were seen in 37 assessable patients, all after progression on prior topoisomerase II inhibitors. For those with sarcoma, 12 of 20 derived clinical benefit (1 partial response and 11 stable disease, median overall survival 8.3 months), including 8 of 14 previously progressed on topoisomerase II therapy. Treatment benefits correlated with increased histone acetylation and decreased neutrophil count on day 5.
Conclusions
Panobinostat and epirubicin treatment is well tolerated and may reverse anthracycline resistance. Changes in histone acetylation and associated decrease in neutrophil count correlated with clinical benefit and warrant investigation as predictive biomarkers.
Clinical trial
This trial is registered at www.Clinicaltrials.gov, Identifier: NCT00878904.
introduction
More than half of patients treated for localized soft tissue sarcoma will experience relapse. Anthracycline-based chemotherapy remains the standard of care in the first-line setting [1], including topoisomerase II inhibitors doxorubicin and epirubicin that act by increasing DNA damage and promoting apoptosis [2]. Response rates are low and resistance to doxorubicin is common [3, 4]. Treatment with pazopanib modestly improves progression-free survival (PFS) from 1.6 to 4.6 months, but not overall survival (OS) 12.5 versus 10.7 months [5]. Trabectedin was recently approved with similar benefit for liposarcoma and leiomyosarcoma; eribulin showed PFS of 2.6 months and OS of 13.5 months [6].
Histone deacetylases (HDACs) modulate gene expression and protein activity by regulating protein acetylation. HDAC inhibitors, vorinostat and romidepsion, have been approved for T-cell lymphoma and panobinostat for myeloma [7]. Preclinical studies have shown that HDAC inhibitors potentiate DNA-damaging chemotherapeutics in various cancer types, including sarcoma [8–11]. Prior clinical studies evaluating HDAC inhibitors in combination with anthracyclines demonstrated efficacy [12, 13]. Supportive preclinical studies suggest a role for epigenetic regulation in sarcoma tumorigenesis [14]. In particular, HDAC2 is highly expressed in sarcomas [15]. HDAC inhibition was shown to reverse repression of gene expression by translocation products such as PAX3/FKHR in alveolar rhabdomyosarcoma, EWS/FLI in Ewing's sarcoma, and SS18/SSX in synovial sarcoma [16]. Furthermore, HDAC inhibition was shown to promote differentiation and apoptosis in sarcoma [17–19].
The primary objective of this study was to determine safety, tolerability, and the recommended phase II dose (RPTD) of panobinostat in combination with epirubicin in patients with advanced solid tumors and in an expansion cohort with sarcoma.
patients and methods
Eligible patients had metastatic solid tumor malignancies and any number of prior therapies with adequate organ function and normal cardiac output [left ventricular ejection fraction (LVEF) >55%]. Prior anthracycline exposure was limited to 300 mg/m2 of doxorubicin and 480 mg/m2 of epirubicin (refer to supplementary Data, available at Annals of Oncology online for further criteria).
study treatment
Consent was received from patients after federal and institutional reviews. In a single-institution phase I study with a 3 + 3 dose-escalation design, patients received panobinostat at escalating doses (20, 30, 40, 50, and 60 mg) orally once daily on days 1, 3, and 5, followed by epirubicin (75 mg/m2) on day 5, every 21 days. Dose escalation included patients with any solid tumor and restricted to 20 patients with sarcoma at the maximum tolerated dose (MTD). The primary end points were safety and toxicity evaluation and determination of a phase II recommended dose. Secondary end points included time to progression, objective response, and correlative pharmacokinetic and peripheral blood mononucleocyte (PBMC) pharmacodynamics studies.
Cumulative epirubicin exposure was restricted to 750 mg/m2 of epirubicin (∼1.8-fold doxorubicin-equivalent). Exceptions were allowable for patients with documented tumor response and approval by the safety review board.
treatment assessment
Toxicities were assessed by CTCAE 4.0 criteria weekly in cycle 1. Dose-limiting toxicities (DLTs) were defined as grade 3 or 4 non-hematological toxicity, and grade 4 hematological toxicity other than grade 4 neutropenia <8 days, or toxicities reducing dose delivery in cycle 1 to <75% of planned dose. Disease restaging by RECIST criteria v1.1 and LVEF assessment by ECHO or MUGA were carried out for every two cycles.
pharmacokinetic studies
Panobinostat levels were determined from plasma on day 5, 2 h post-panobinostat administration, using a validated LC–MS/MS method.
correlative studies
Whole blood was collected pretreatment on day 1 and on days 3 and 5, 2 h post-panobinostat treatment, and PBMCs were evaluated for histone acetylation as previously described [20].
statistical methods
Descriptive statistics were used to summarize patient results. Clinical benefit was defined as complete or partial response or stable disease for >3 months. A two-sided t-test and Pearson's correlation coefficient method were used to evaluate correlations between two variables (SigmaPlot, Systat, Inc.).
results
patient characteristics
Twenty patients with metastatic solid tumors were enrolled in five dose-escalation cohorts, and 20 patients with advanced sarcoma in the dose expansion cohort at the MTD. Of these, 17 (43%) had received prior topoisomerase II inhibitor-based chemotherapy (e.g. doxorubicin and etoposide) and a median of three prior systemic regimens (Table 1).
Table 1.
Patient characteristics (N = 40)
| Gender, n (%) | |
| Female | 27 (67) |
| Male | 13 (33) |
| Age, median (range), years | 49 (22–79) |
| ≤64 | 32 (80) |
| ≥65 | 8 (20) |
| Race, n (%) | |
| Caucasian | 31 (77) |
| Asian | 9 (23) |
| African-American | 0 (0) |
| Ethnicity, n (%) | |
| Non-Hispanic | 32 (80) |
| Hispanic/Latino | 8 (20) |
| ECOG performance status, median (range) | 1 (0–2) |
| Tumor histology, n (%) | |
| Melanoma | 6 (15) |
| Breast | 5 (13) |
| Ovarian | 2 (5) |
| Lung | 2 (5) |
| Other (neuroblastoma, testicular, colon, and pancreas) | 4 (10) |
| Sarcoma | 21 (52) |
| Leiomyosarcoma | 5 (12) |
| Chondrosarcoma | 4 (10) |
| Liposarcoma | 3 (8) |
| Phyllodes | 2 (5) |
| Othera | 7 (17) |
| Number of prior systemic regimens, median (range) | 3 (0–8) |
| Number of regimens, n (%) | |
| 0 | 3 (8) |
| 1 | 4 (10) |
| 2 | 12 (30) |
| 3+ | 21 (53) |
| Number of prior systemic regimens for sarcoma cohort, median (range) | 2 (0–5) |
| Number of patients receiving prior topoisomerase II inhibitors, n (%) | 17 (40) |
| Number of patient receiving prior radiation therapy, n (%) | 19 (48) |
aOne each: peripheral nerve sheath, fibrosarcoma, epitheliod hemangiosarcoma, alveolar soft part, synovial, pleiomorphic and sarcomatoid carcinoma.
patient disposition, DLT, safety, and tolerability
Panobinostat was escalated from 20–60 mg/day on days 1, 3, and 5. All patients received 75 mg/m2 of epirubicin. At 50 mg panobinostat, one patient experienced atrial fibrillation (AFIB) with a rapid ventricular response, which was considered a DLT due to its temporal relationship with study drug. At 60 mg panobinostat, two of four patients experienced a DLT (1: grade 4 thrombocytopenia and febrile neutropenia and 2: grade 3 fatigue; Table 2). Hence, the RPTD was set as 50 mg/day panobinostat on days 1, 3, and 5, and 75 mg/m2 of epirubicin on day 5. Twenty patients with metastatic sarcoma were treated at this dose.
Table 2.
DLT and grade 3/4 toxicities of dose-escalation and expansion cohorts
| Cohort | Panobinostat days 1, 3, and 5a (mg) | N | DLTs | Grade (N): toxicity | Responses (N) |
|---|---|---|---|---|---|
| 1 | 20 | 3 | 0 | 3 (2): neutropenia, WBC | SD: melanoma, neuroblastoma |
| 2 | 30 | 3 | 0 | 3 (1): neutropenia | SD: ovarian |
| 3 (1): fatigue | |||||
| 3 | 40 | 3 | 0 | 3 (1): thrombocytopenia | |
| 3 (2): neutropenia, WBC | |||||
| 4 (1): neutropenia, WBC | |||||
| 4 | 50 | 7 | Atrial fibrillation | 3 (1): neutropenia, WBC | PR: breast (2) |
| 4 (2): neutropenia, WBC | SD: NSCLC | ||||
| 5 | 60 | 4 | Febrile neutropenia | 3 (4): neutropenia, WBC | PR: SCLC |
| Fatigue | 3 (2): fatigue | ||||
| Thrombocytopenia | |||||
| MTD | 50 | 20 | Febrile neutropenia | (see supplementary Table S1, available at Annals of Oncology online) | PR: liposarcoma |
| Neutropenia | SD: LMS, aveolar soft part, nerve sheath, chondrosarcoma, hemangioepithelioma, sarcamoid, synovial, liposarcoma (2), phyllodes (2) |
aEpirubicin (75 mg/m2) administered on day 5 every 21 days.
SD, stable disease ≥12 weeks; PR, partial response; NSCLC, non-small-cell lung cancer; SCLC, small-cell lung cancer; LMS, leiomyosarcoma; WBC, leukocytopenia.
Forty patients were evaluable for treatment-emergent toxicity. A summary of grade ≥2 adverse events (AEs) is shown in supplementary Table S1, available at Annals of Oncology online. Twenty-four patients (60%) experienced at least one grade 3/4 treatment-related AE, including neutropenia (45%), leukopenia (35%), lymphopenia (22.5%), thrombocytopenia (17.5%), anemia (15%), and febrile neutropenia (7.5%). The most common grade 3 non-hematologic toxicities included fatigue (15%), vomiting (5%), hepatic dysfunction (5%), and elevated blood glucose levels (2.5%). One patient was hospitalized for AFIB with a rapid ventricular response, which was asymptomatic and resolved spontaneously within hours. Due to the timing of the event, a causal relationship to panobinostat could not be excluded.
Three patients had asymptomatic prolonged QTc-interval of grade 1 or 2. No significant declines in LVEF were observed on study. All patients with a partial response received more than the planned doses of epirubicin (up to 975 mg/m2) without observing any cardiac determinant.
response and clinical benefits
Overall, 37 of 40 patients were evaluable for response. The patient with AFIB was withdrawn from study without receiving epirubicin. A breast cancer patient with extensive chest wall disease withdrew for personal reasons after being hospitalized for chest wall bleeding following initiation of panobinostat. This serious AE was attributed to her treatment response resulting in a near complete response. Subsequently, this patient experienced grade 4 thrombocytopenia and withdrew consent. One patient withdrew from study for febrile neutropenia and infection and was not evaluable for response. In 37 assessable patients, 4 (11%) achieved a partial response and 17 (46%) had a stable disease (Figure 1). The median time to progression and median overall survival for the overall study cohort were 3.1 (95% CI: [1.8–4.6]) and 7.3 (95% CI: [5.9–10.3]) months, respectively. All four patients with objective partial response had progressed on prior topoisomerase II inhibitors. Reduced tumor burden was seen in two additional patients with prior exposure to topoisomerase II inhibitors. In total, 17 patients had received prior topoisomerase II inhibitors (e.g. 16 anthracycline and 1 etoposide), with 14 experiencing disease progression while on this regimen. Of these 14 patients, 8 (57%) benefited from panobinostat and epirubicin, including 3 with partial response.
Figure 1.
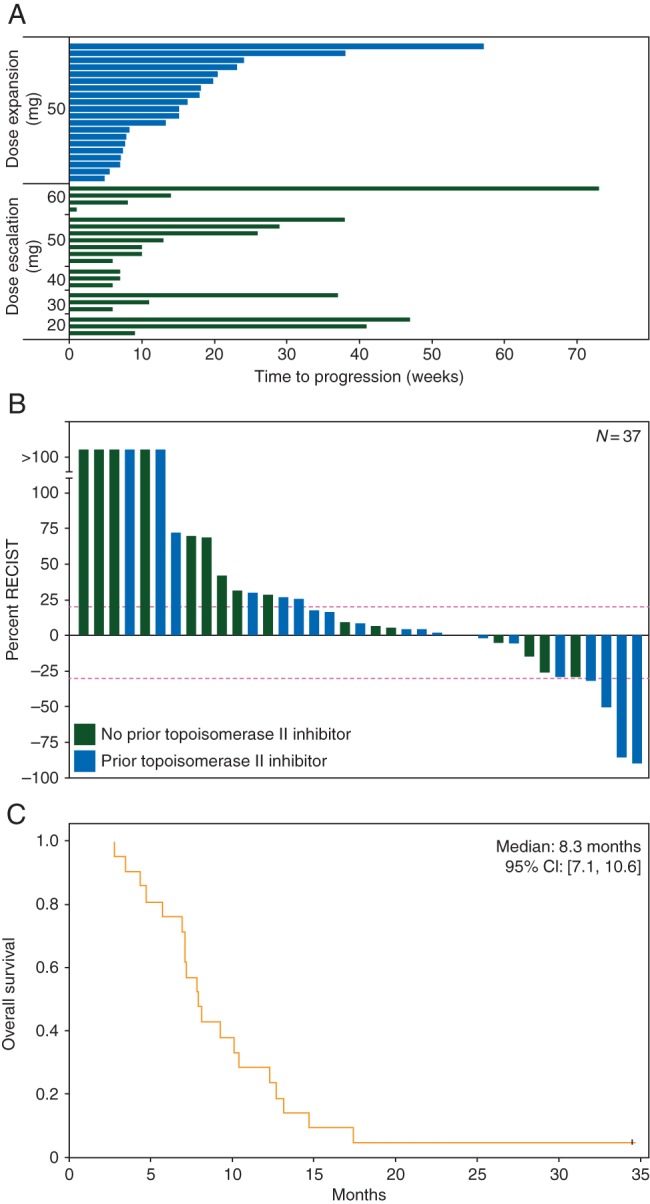
Time to progression, RECIST, and overall survival. (A) Time to progression in weeks for dose-escalation [the black (green online)] and expansion [the gray (blue online)] cohorts. (B) RECIST of assessable patients is presented. Dotted lines denote boundaries for achieving partial response (−30%) and progressive disease (20%). Prior treatment with a topoisomerase II inhibitor [the gray (blue online)] or no prior [the black (green online)] treatment is indicated. (C) Kaplan–Meier graph of overall survival in patients with sarcoma.
In the sarcoma dose expansion cohort (n = 20), 1 partial response (myxoid liposarcoma) and 11 additional patients with stable disease were observed for >3 months. The median PFS and median overall survival was 3.4 (95% CI: [2.6–5.2]) and 8.3 (95% CI: [7.1–10.6]) months, respectively.
correlative studies
There was a weak correlation for a dose-dependent increase in panobinostat plasma levels (R = 0.361, P = 0.023) with significant interpatient variability at the 50-mg dose (Figure 2A). Patients with higher plasma panobinostat levels were more likely to experience grade 3 or 4 toxicity (P = 0.034; Figure 2B). However, plasma panobinostat concentrations were not significantly associated with treatment benefit (P = 0.333; Figure 2B).
Figure 2.

Relationship between panobinostat plasma concentration and dose, response, and toxicity. (A) Panobinostat plasma concentration (ng/ml) exhibits a positive correlation with dose (mg). (B) Box plots of panobinostat plasma concentrations (ng/ml) in patients who immediately progressed (PD) versus those who experienced clinical benefit (CB) and in patients who experienced grade 1/2 toxicities versus those who experienced grade 3/4 toxicities. Correlations were conducted using Pearson's correlation coefficient method.
We have previously shown that PMBCs are a valid surrogate for tumor histone acetylation in response to HDAC inhibitors [12, 20, 21]. We found no significant correlation or association between PBMC histone acetylation and panobinostat dose (R = 0.062), panobinostat plasma concentrations (R = 0.124, P = 0.538), or increased toxicities (P = 0.915; Figure 3A and B). Patients with higher PBMC histone acetylation, however, were significantly more likely to exhibit clinical benefit [i.e. stable disease (SD) ≥12 weeks or objective response; median 14.5-fold increase in PBMC histone acetylation in those with clinical benefit versus median sixfold increase in those without treatment benefit, P = 0.041; Figure 3C].
Figure 3.

Peripheral blood mononucleocyte histone acetylation relationship to panobinostat dose and plasma concentration, response, toxicity, and white cell count. Histone acetylation neither correlates with panobinostat dose (A) nor plasma concentration (ng/ml, B). (C) Box plots of histone acetylation in patients who did (CB) versus those who did not (PD) benefit from treatment and in patients who experienced grade 1/2 versus grade 3/4 toxicity. (D) Dot plots of all leukocytes (WBC), neutrophils (ANC), and lymphocytes (Lympho) on day 5 of treatment normalized to pretreatment on day 1 of cycle 1 comparing patients who did (CB) and did not (PD) benefit from treatment. The bar indicates the mean. Correlations were conducted using Pearson's correlation coefficient method.
Furthermore, neutropenia (a relative decrease in absolute neutrophil count) on day 5 versus baseline level was significantly greater in the group of patients with clinical benefit (P = 0.0054; Figure 2D). This change in neutrophil count did not correlate with panobinostat plasma concentration (R = 0.013, P = 0.537; data not shown).
discussion
This phase 1 study was based on preclinical data, suggesting enhanced efficacy of DNA-damaging agents after pretreatment with HDAC inhibitors. Decondensation of chromatin and depletion of Ataxia Telangiectasia mutated by HDAC inhibitors seem indeed to be a prerequisite for synergy [22]. Hence, panobinostat was administered on days 1, 3, and 5 followed by epirubicin. Pulse dosing of HDAC inhibitors allowed administration of higher doses than the approved dose as a single agent for myeloma (20 mg thrice per week). At the MTD, myelotoxicity, nausea/vomiting, and fatigue were the major toxicities for panobinostat, requiring dose modification in 26% of patients [23]. No overlapping toxicities were seen with regard to cardiac toxicity. In fact, several patients received cumulative doses of epirubicin exceeding 750 mg/m2 without cardiac compromise.
Clinical benefit was observed in a substantial number of patients despite prior exposure to multiple regimens. Of patients enrolled, 17 of 40 had been exposed to prior topoisomerase II inhibitor, including all 4 patients with a partial objective tumor response and 2 patients with minor responses. In 8 of 14 patients, acquired topoisomerase resistance was reversed. Given the limited allowable prior exposure to anthracyclines, patients with prior anthracycline resistance had progressed on it after three cycles. Durable disease control was achieved in a subset of patients, with 25% (5/20) of patients in the dose-escalation cohort and 20% (4/20) of patients in the expansion cohort exhibiting stable disease or better for more than 6 months. One patient with small-cell lung cancer maintained disease control for 18 months. These efficacy results compare favorably with historical PFS with currently approved therapeutics (e.g. pazopanib) in patients with treatment-resistant sarcoma [24]. Prolonged disease stabilization (>6 months) was seen in two of the four patients with liposarcoma, one patient with nerve sheath tumor, and a fourth patient with chondrosarcoma. The potential for prolonged treatment with anthracycline in combination with an HDAC inhibitor speaks to the tolerability of this regimen. This study suggests that further investigation of HDAC inhibition in combination with DNA-damaging agents in defined advanced sarcoma subtypes to validate these preliminary findings is warranted.
A major challenge in HDAC inhibitor therapy is the absence of biomarkers, which was a focus of this study. Prior studies have shown that PBMC histone acetylation is a reliable surrogate for tumor histone acetylation [12, 20, 21]. This study showed that patients with a pronounced degree of PBMC histone acetylation were more likely to benefit from treatment. Furthermore, a decrease in neutrophil count from days 1 to 5 of cycle 1 was correlated with clinical benefit. The changes in histone acetylation and induction of neutropenia were not correlated with panobinostat plasma levels. Panobinostat-induced PBMC histone acetylation and neutropenia are host-specific biomarkers of therapeutic effect linked to the host's ability to respond to epigenetic modification, rather than a dose-dependent pharmacodynamic effect. We found that increasing doses of the HDAC inhibitor do not result in increased hyperacetylation, but are associated with increased toxicity such as fatigue, nausea, and diarrhea, without better treatment effect. Determining a patient's response to HDAC inhibition ex vivo, before treatment, may allow for the enrichment of patients most likely to benefit from HDAC inhibitor-based treatment. Thus, prospective studies evaluating an ex vivo assay of HDAC inhibitor-induced PBMC histone acetylation could present a new means for identifying patient's likely to benefit.
funding
This work was supported in part by Novartis International AG and the National Cancer Institute at the National Institutes of Health (grant numbers RO1 1RC1CA145425-01 and R01 CA122657).
disclosure
PNM receives research support from Novartis for this and other clinical trials. All remaining authors have declared no conflicts of interest.
Supplementary Material
references
- 1.Bramwell VH. Management of advanced adult soft tissue sarcoma. Sarcoma 2003; 7: 43–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zunino F, Capranico G. DNA topoisomerase II as the primary target of anti-tumor anthracyclines. Anticancer Drug Des 1990; 5: 307–317. [PubMed] [Google Scholar]
- 3.Antman K, Crowley J, Balcerzak SP et al. . A Southwest Oncology Group and Cancer and Leukemia Group B phase II study of doxorubicin, dacarbazine, ifosfamide, and mesna in adults with advanced osteosarcoma, Ewing's sarcoma, and rhabdomyosarcoma. Cancer 1998; 82: 1288–1295. [PubMed] [Google Scholar]
- 4.Edmonson JH, Ryan LM, Blum RH et al. . Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J Clin Oncol 1993; 11: 1269–1275. [DOI] [PubMed] [Google Scholar]
- 5.van der Graaf WT, Blay JY, Chawla SP et al. . Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012; 379: 1879–1886. [DOI] [PubMed] [Google Scholar]
- 6.Schöffski P, Maki RG, Italiano A et al. . Randomized, open-label, multicenter, phase III study of eribulin versus dacarbazine in patients (pts) with leiomyosarcoma (LMS) and adipocytic sarcoma (ADI). J Clin Oncol 2015; 33: Supplemental Abstr LBA 10502. [Google Scholar]
- 7.Thurn KT, Thomas S, Moore A, Munster PN. Rational therapeutic combinations with histone deacetylase inhibitors for the treatment of cancer. Future Oncol 2011; 7: 263–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchion DC, Bicaku E, Daud AI et al. . In vivo synergy between topoisomerase II and histone deacetylase inhibitors: predictive correlates. Mol Cancer Ther 2005; 4: 1993–2000. [DOI] [PubMed] [Google Scholar]
- 9.Lopez G, Liu J, Ren W et al. . Combining PCI-24781, a novel histone deacetylase inhibitor, with chemotherapy for the treatment of soft tissue sarcoma. Clin Cancer Res 2009; 15: 3472–3483. [DOI] [PubMed] [Google Scholar]
- 10.Sampson ER, Amin V, Schwarz EM et al. . The histone deacetylase inhibitor vorinostat selectively sensitizes fibrosarcoma cells to chemotherapy. J Orthop Res 2011; 29: 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thurn KT, Thomas S, Raha P et al. . Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol Cancer Ther 2013; 12: 2078–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munster P, Marchion D, Bicaku E et al. . Clinical and biological effects of valproic acid as a histone deacetylase inhibitor on tumor and surrogate tissues: phase I/II trial of valproic acid and epirubicin/FEC. Clin Cancer Res 2009; 15: 2488–2496. [DOI] [PubMed] [Google Scholar]
- 13.Munster P, Marchion D, Bicaku E et al. . Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol 2007; 25: 1979–1985. [DOI] [PubMed] [Google Scholar]
- 14.Bennani-Baiti IM. Epigenetic and epigenomic mechanisms shape sarcoma and other mesenchymal tumor pathogenesis. Epigenomics 2011; 3: 715–732. [DOI] [PubMed] [Google Scholar]
- 15.Pacheco M, Nielsen TO. Histone deacetylase 1 and 2 in mesenchymal tumors. Mod Pathol 2012; 25: 222–230. [DOI] [PubMed] [Google Scholar]
- 16.Su L, Sampaio AV, Jones KB et al. . Deconstruction of the SS18-SSX fusion oncoprotein complex: insights into disease etiology and therapeutics. Cancer Cell 2012; 21: 333–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hrzenjak A, Moinfar F, Kremser ML et al. . Valproate inhibition of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cells. Mol Cancer Ther 2006; 5: 2203–2210. [DOI] [PubMed] [Google Scholar]
- 18.Sakimura R, Tanaka K, Yamamoto S et al. . The effects of histone deacetylase inhibitors on the induction of differentiation in chondrosarcoma cells. Clin Cancer Res 2007; 13: 275–282. [DOI] [PubMed] [Google Scholar]
- 19.Hrzenjak A, Moinfar F, Kremser ML et al. . Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol Cancer 2010; 9: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munster PN, Thurn KT, Thomas S et al. . A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer 2011; 104: 1828–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Munster PN, Marchion D, Thomas S et al. . Phase I trial of vorinostat and doxorubicin in solid tumours: histone deacetylase 2 expression as a predictive marker. Br J Cancer 2009; 101: 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchion DC, Bicaku E, Daud AI et al. . Sequence-specific potentiation of topoisomerase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem 2004; 92: 223–237. [DOI] [PubMed] [Google Scholar]
- 23.Wolf JL, Siegel D, Goldschmidt H et al. . Phase II trial of the pan-deacetylase inhibitor panobinostat as a single agent in advanced relapsed/refractory multiple myeloma. Leuk Lymphoma 2012; 53: 1820–1823. [DOI] [PubMed] [Google Scholar]
- 24.Van Glabbeke M, Verweij J, Judson I et al. . Progression-free rate as the principal end-point for phase II trials in soft-tissue sarcomas. Eur J Cancer 2002; 38: 543–549. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.