

Abstract
We present a versatile and facile method to enhance user-control of small molecule drug release from a poly(ethylene glycol)-based hydrogel using the host/guest complex formed between an azobenzene derivative guest and a β-cyclodextrin host. A model drug is formed from a short peptide containing a fluorophore and an azobenzene functional group on one terminus. Upon irradiation with UV light, azobenzene isomerization leads to decreased complex formation and an on-demand acceleratation of the release rate of an entrapped model drug.
Graphical Abstract
Photoresponsive azobenzene-cyclodextrin guest-host chemistry can be used to control the release rate of a small peptide from a PEG hydrogel with light.
Enhancing user-defined control over the rate of drug delivery remains an important consideration for biomedical researchers, as it has the potential to improve the efficacy of a large range of protein and small molecule-based therapeutics1. Poly(ethylene glycol) (PEG) hydrogels have long been effectively employed to achieve controlled release of therapeutics on account of their tunable mesh size and ease of chemical functionalization2. This control of network structure has led to a well-developed body of PEG-based diffusion mediated drug release reservoirs, as well as substrates that release a tethered drug in response to a stimulus3–5. Chemical modifications (e.g., affinity binders) have also been introduced to reduce the effective diffusion rate of drugs within PEG hydrogels and to achieve more sustained release over longer time courses6–10.
Here, we focus on one such modification that consists of encapsulating a therapeutic inside a cyclodextrin-containing hydrogel11,12. Cyclodextrin has a hydrophobic pocket that allows for favorable interactions with small hydrophobic molecules, such as adamantane, thereby reducing the effective diffusion rate of these molecules through the gel13,14. In particular, cyclodextrin and azobenzene form a host/guest system of interest based on hydrophobic interactions between azobenzene and the interior pocket of cyclodextrin in aqueous solution15–17. This complex is attractive because the binding affinity between azobenzene and cyclodextrin can be switched on demand via photoinduced isomerization of azobenzene. In the trans configuration, azobenzene is planar, and the complex formation is favorable. In contrast, in the cis configuration, the introduction of a dipole moment and change of geometry decreases the binding affinity and leads to complex disruption18. Furthermore, the strength of association can be modified by varying the functionalization of the azobenzene derivative guest or by changing the size and orientation of the cyclodextrin host19. This strategy has previously been used in the design of gating systems or as a means to change the mesh size of a drug reservoir20–23.
We hypothesized that the cyclodextrin/azobenzene system could be used to tune the release rate of a model, small drug out of a hydrogel. To achieve this, β-cyclodextrin was tethered to a PEG hydrogel as a pendant moiety, and a model drug functionalized with azobenzene was encapsulated in the hydrogel (Fig. 1). The proposed system offers two primary benefits over previously developed systems. First, the release rate is photoresponsive and can be temporally, and even spatially, controlled by irradiating only when and where release is desired. Second, the PEG network is formed using a step-growth polymerization, which leads to a more homogeneous network structure and mesh size, as well as tunable release kinetics24–26.
Fig. 1.
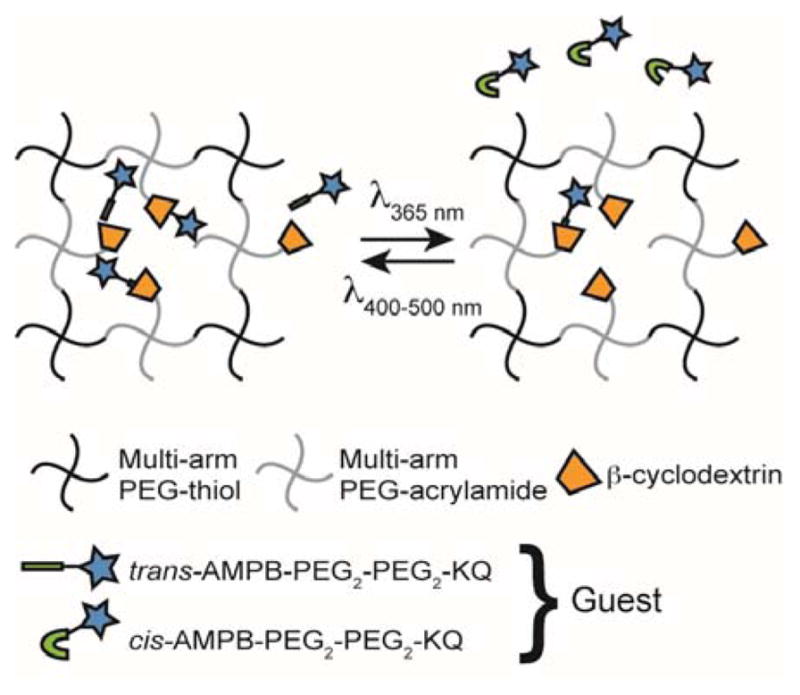
Schematic of photoinitiated release of an azobenzene-functionalized drug from a β-cyclodextrin-containing PEG-based reservoir.
A guest peptide (Fig. S3a) was synthesized by solid phase peptide synthesis (Tribute) and contained N-terminal acetylated 3-[[4-[amino]methyl)phen-yl]diazenyl]benzoic acid (3,4′-AMPB)27,28. This particular azobenzene derivative was selected on account of its previously reported long half-life of thermal relaxation, which may be related, in part, to intermolecular interactions between the meta-substituent and the peptide backbone29. The half-life was determined by UV-Vis tracking of the absorbance corresponding to the trans configuration as follows: a solution of guest peptide was irradiated with 365 nm light (5 mW/cm2) for 5 min, and the absorbance at 325 nm was measured every 10 minutes for the first half hour, every half hour for the next two hours, and every hour thereafter for 6 hours. The relaxation followed first order kinetics, and the estimated half-life was approximately 9 h for the thermal relaxation of AMPB on this particular peptide. Thus, these results indicate that a relatively short pulse of UV irradiation (5 min) could lead to a change in molecular structure over a long period (hours), thereby opening the possibility of altering release rate with short doses of light.
Next, the strength of association (Ka) of 3,4-AMPB with 3A-amino-3A-deoxy-β-cyclodextrin (β-CD, TCI), the selected host, was determined by an NMR adaptation of the Benesi-Hildebrand equation30 (further details in the ESI). β-CD was selected as the host, because prior reports suggested that the association constant between many functionalized azobenzenes is higher with β-CD than α-CD or γ-CD31. In addition, β-CD has been reported to lack the ability to form rotaxanes with PEG chains32. To obtain Ka, samples of 0.5 mM guest peptide were titrated with CD (0–20 eq.) in deuterium oxide and analyzed by 1H NMR on a 500 Hz magnet. The change in the NMR peak shift between the sample with only guest and samples containing the CD host is indicative of the degree of complex formation (Fig. 2a, distance from vertical line). The results were plotted according to the Benesi-Hildebrand equation (Fig. 2b), and Ka was calculated to be 242 M−1. Although this binding affinity is relatively weak, it is similar to that reported for β-CD and some clinically-relevant drugs, such as doxorubicin (297 M−1)33.
Fig. 2.

(a) 1H NMR spectra (bonds shown in red) of 0.5 mM guest peptide with increasing concentration of β-CD (labels are of the form [CD]:[guest]), (b) and corresponding Benesi-Hildebrand plot with linear fit giving Ka of 242 M−1.
Next, the reversibility of the guest peptide isomerization was probed by UV-Vis spectroscopy (Fig. 3a). A sample of 25 mM guest was prepared in PBS and analyzed on a Synergy H1 microplate reader (BioTek). An ambient spectrum was taken prior to any irradiation of the sample (black line, Fig. 3a). Immediately before a second spectrum was taken, the sample was irradiated for five minutes with UV light (365 nm, 5 mW/cm2, Omnicure) to convert the azobenzene units in the guest peptide to the cis conformation. As shown in Fig. 3 (green line, dotted), the peak at 325 nm significantly decreases and is blue-shifted, accompanied by the appearance of a more prominent peak at 450 nm that is characteristic of the cis configuration. Finally, the initial isomeric state of the guest peptide was recovered by irradiation for five minutes with 400–500 nm visible light (5 mW/cm2, Omnicure), as seen by the reversion of the spectrum (red line, dashed) to the shape of the pre-irradiated sample.
Fig. 3.

(a) Absorbance spectra of guest in ambient lab conditions (black), after irradiation with 365 nm UV light (green, dotted), and after subsequent irradiation with 400–500 nm light (red, dashed). (b) 1H NMR spectra of 0.5 mM guest peptide with no β-CD (bottom of each pair) and 20 equivalents of β-CD (top of each pair) in ambient lab conditions (black), after irradiation with 365 nm UV light (green), and after subsequent irradiation with 400–500 nm light (red).
While this confirmed the reversibility of the guest peptide isomerization, reversibility of the host/guest complex formation was probed using NMR (Fig. 3b). Two samples were prepared with 0.5 mM guest peptide, a control with the guest alone and one with 20 equivalents of β-CD. The same irradiation conditions were used as in Fig. 3a. As seen in Fig. 3b, the NMR signal representing the azobenzene protons shifts from 7.43 ppm to 7.46 ppm upon complexation with cyclodextrin in ambient lab conditions (two black lines). Upon irradiation with UV light, the complex is disrupted, indicated by the equivalent NMR shifts of 6.84 ppm for the cis azobenzene protons in the absence and presence of β-cyclodextrin (two green lines). Subsequent irradiation with visible light restores the difference in peak shifts (two red lines), suggesting a restoration of the complex.
After confirming the isomerization effects on complex formation in solution, we looked towards the synthesis of a drug release substrate. To synthesize a hydrogel that contains β-cyclodextrin, 3A-amino-3A-deoxy-β-cyclodextrin was thiolated using 2-iminothiolane (Sigma) for 1 h at r.t. in PBS (pH 8). HPLC purification (Waters) was used to stop the reaction, and the thiolated CD was lyophilized in preparation for the next step. A slight molar excess of tris(2-carboxyethyl)phosphine was added 15 min before HPLC purification to ensure that the thiol groups remained in their reduced form. The thiolated β-CD was then tethered to 8-arm, 10 kDa PEG-acrylamide (Creative PEGworks) via Michael addition in PBS (pH 10) with 300 mM triethanolamine (TEOA, Sigma). The degree of functionalization was approximately 1.9 CD moieties for each 8 arm PEG, as calculated by NMR (ESI). Gels with a final concentration of 10 wt% polymer were formed between this CD functionalized PEG-acrylamide and 8-arm, 10 kDa PEG-thiol via Michael addition in PBS (pH 10) with 300 mM TEOA such that there was a 1:1 ratio between acrylamide and thiol groups. The equimolar pre-polymer solutions were mixed in a microcentrifuge tube, placed in a 0.5 mm thick, 8 mm diameter rubber gasket, and allowed to gel for 3 h at 37 °C.
The guest peptide was conjugated to N-hydroxysuccinimide functionalized Alexa Fluor 647 (AF647) via the lysine residue on the guest to simulate the release of a small molecule drug and aid in quantification of release. The release was then simulated by encapsulating the guest functionalized model drug in a 10 wt% β-cyclodextrin-containing gel and monitoring supernatant fluorescence at 647 nm, beginning immediately after the gels were placed in a PBS bath (Fig. 4). Though the exact ratio is difficult to determine due to the proprietary nature of AF647, it is estimated that there is a tenfold molar excess of β-cyclodextrin relative to guest peptide in the gel prior to release. One condition consisted of a gel left in the ambient lab environment, shown as the solid black release profile in both Fig. 4a and Fig. 4b (corresponding to azobenzene trans state). Another condition consisted of a gel that was pulsed with 365 nm irradiation (5 mW/cm2) for 5 minutes every ten minutes from time 0 (corresponding to azobenzene cis state). This condition is depicted as the dotted profile in Fig. 4a. A comparison of the two profiles in Fig. 4a demonstrates that isomerization of the azobenzene-containing tether increases the effective rate of diffusion and ultimate release rate from the hydrogel, starting around 80 minutes. A final condition consisted of a gel left in the ambient lab environment for 2 h and pulsed with irradiation starting thereafter (dashed profile in Fig. 4b). Though less pronounced, a comparison of the profiles in Fig. 4b suggests that the effect of the isomerization can be initiated by the user on demand to increase the rate of drug release from the system.
Fig. 4.

Release profile of AF647 functionalized guest peptide from a hydrogel left in ambient lab conditions (solid), (a) pulsed with UV irradiation (dotted), or (b) kept at ambient conditions for 2 h (indicated by vertical blue line) and pulsed thereafter (dashed). Asterisks denote statistical significance for p = 0.1 (single asterisk) or p = 0.05 (double asterisk) as determined by a one tailed t-test.
Collectively, these data show that the release rate can be tuned with the isomeric state of the azobenzene functionality. For this system, the release is very fast due to the relatively weak binding affinity discussed above, as well as a low excess of CD in the gel. It is expected that increasing the amount of cyclodextrin would slow the release from the gel and likely magnify the observed difference in release between the cis and trans states.
To summarize, the system presented here allows for tunable release of low molecular weight drugs in highly swollen hydrogels and subsequent on-demand control of the release rate from a hydrogel reservoir by controlled exposure to irradiation. The initial rate of release can be further tailored via selection of the azobenzene guest derivative, design of the hydrogel mesh size relative to the drug molecular weight, selection and orientation of the cyclodextrin host molecule, and the ratio of the host to the guest. Such a material system adds to the versatility of stimuli-sensitive hydrogels for the localized and sustained release of small molecules, and should prove particularly useful for future directions where individual methods of modification might be layered upon one another to achieve increasingly complex and versatile delivery vehicles and substrates, especially for multiple drugs.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the Howard Hughes Medical Institute and the NSF (DMR 1408955). A portion of this work was funded by a Burroughs Wellcome Fund Postdoctoral Enrichment Program award (to AMR) and an NIH postdoctoral fellowship (5 F32 HL121986-02, to AMR). In addition, we would also like to acknowledge Professor Jason Burdick for helpful discussions and facility support, as well as Dr. Joseph Grim for his assistance with NMR analysis.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Notes and references
- 1.Nguyen MK, Alsberg E. Prog Polym Sci. 2014;39:1235–1265. doi: 10.1016/j.progpolymsci.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peppas NA, Keys KB, Torres-Lugo M, Lowman AM. J Controlled Release. 1999;62:81–87. doi: 10.1016/s0168-3659(99)00027-9. [DOI] [PubMed] [Google Scholar]
- 3.Betancourt T, Pardo J, Soo K, Peppas NA. J Biomed Mater Res A. 2010;93A:175–188. doi: 10.1002/jbm.a.32510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azagarsamy MA, Anseth KS. Angew Chem Int Ed. 2013;52:13803–13807. doi: 10.1002/anie.201308174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zustiak SP, Leach JB. Biotechnol Bioeng. 2011;108:197–206. doi: 10.1002/bit.22911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin CC, Anseth KS. Pharm Res. 2008;26:631–643. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salmaso S, Semenzato A, Bersani S, Matricardi P, Rossi F, Caliceti P. Int J Pharm. 2007;345:42–50. doi: 10.1016/j.ijpharm.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 8.Wang NX, von Recum HA. Macromol Biosci. 2011;11:321–332. doi: 10.1002/mabi.201000206. [DOI] [PubMed] [Google Scholar]
- 9.Oss-Ronen L, Seliktar D. Acta Biomater. 2011;7:163–170. doi: 10.1016/j.actbio.2010.07.017. [DOI] [PubMed] [Google Scholar]
- 10.Nie T, Baldwin A, Yamaguchi N, Kiick KL. J Controlled Release. 2007;122:287–296. doi: 10.1016/j.jconrel.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shih H, Lin CC. Biomacromolecules. 2015;16:1915–1923. doi: 10.1021/acs.biomac.5b00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mealy J, Rodell C, Burdick JA. J Mater Chem B. 2015;3:8010–8019. doi: 10.1039/C5TB00981B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uekama K, Hirayama F, Irie T. Chem Rev. 1998;98:2045–2076. doi: 10.1021/cr970025p. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Ma PX. Adv Drug Deliv Rev. 2013;65:1215–1233. doi: 10.1016/j.addr.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murakami H, Kawabuchi A, Kotoo K, Kunitake M, Nakashima N. J Am Chem Soc. 1997;119:7605–7606. [Google Scholar]
- 16.Anderson S, Claridge TDW, Anderson HL. Angew Chem Int Ed Engl. 1997;36:1310–1313. [Google Scholar]
- 17.Takashima Y, Nakayama T, Miyauchi M, Kawaguchi Y, Yamaguchi H, Harada A. Chem Lett. 2004;33:890–891. [Google Scholar]
- 18.Hartley GS, Fèvre RJWL. J Chem Soc. 1939:531–535. [Google Scholar]
- 19.Tomatsu I, Hashidzume A, Harada A. J Am Chem Soc. 2006;128:2226–2227. doi: 10.1021/ja058345a. [DOI] [PubMed] [Google Scholar]
- 20.Chiang CY, Chu CC. Carbohydr Polym. 2015;119:18–25. doi: 10.1016/j.carbpol.2014.11.043. [DOI] [PubMed] [Google Scholar]
- 21.Mei X, Yang S, Chen D, Li N, Li H, Xu Q, Ge J, Lu J. Chem Commun. 2012;48:10010. doi: 10.1039/c2cc33995a. [DOI] [PubMed] [Google Scholar]
- 22.Li J, He L, Wang J, Zhang ZT, Shi J, Zhang XZ, Cao YP, Chen Y. Express Polym Lett. 2014;8:143–153. [Google Scholar]
- 23.Lee IET, Hashidzume A, Harada A. Macromol Rapid Commun. 2015;36:2055–2059. doi: 10.1002/marc.201500389. [DOI] [PubMed] [Google Scholar]
- 24.Tibbitt MW, Kloxin AM, Sawicki LA, Anseth KS. Macromolecules. 2013;46:2785–2792. doi: 10.1021/ma302522x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin CC, Metters AT. Adv Drug Deliv Rev. 2006;58:1379–1408. doi: 10.1016/j.addr.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 26.van de Wetering P, Metters AT, Schoenmakers RG, Hubbell JA. J Controlled Release. 2005;102:619–627. doi: 10.1016/j.jconrel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 27.Rosales AM, Mabry KM, Nehls EM, Anseth KS. Biomacromolecules. 2015;16:798–806. doi: 10.1021/bm501710e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rück-Braun K, Kempa S, Priewisch B, Richter A, Seedorff S, Wallach L. Synthesis. 2009;24:4256–4267. [Google Scholar]
- 29.Schweighauser L, Strauss MA, Bellotto S, Wegner HA. Angew Chem Int Ed. 2015;54:13436–13439. doi: 10.1002/anie.201506126. [DOI] [PubMed] [Google Scholar]
- 30.Fielding L. Tetrahedron. 2000;31:6151–6170. [Google Scholar]
- 31.Yamaguchi H, Kobayashi Y, Kobayashi R, Takashima Y, Hashidzume A, Harada A. Nat Commun. 2012;3:603. doi: 10.1038/ncomms1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nepogodiev SA, Stoddart JF. Chem Rev. 1998;98:1959–1976. doi: 10.1021/cr970049w. [DOI] [PubMed] [Google Scholar]
- 33.Husain N, Ndou TT, Peña AMDL, Warner IM. Appl Spectrosc. 1992;46:652–658. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.