

Abstract
Background
Congenital heart disease is the most common type of birth defect, affecting ≈2% of the population. Malformations involving the cardiac outflow tract and semilunar valves account for >50% of these cases predominantly because of a bicuspid aortic valve, which has an estimated prevalence of 1% in the population. We previously reported that mutations in NOTCH1 were a cause of bicuspid aortic valve in nonsyndromic autosomal‐dominant human pedigrees. Subsequently, we described a highly penetrant mouse model of aortic valve disease, consisting of a bicuspid aortic valve with thickened cusps and associated stenosis and regurgitation, in Notch1‐haploinsufficient adult mice backcrossed into a Nos3‐null background.
Methods and Results
Here, we described the congenital cardiac abnormalities in Notch1 +/− ;Nos3 −/− embryos that led to ≈65% lethality by postnatal day 10. Although expected Mendelian ratios of Notch1 +/− ;Nos3 −/− embryos were found at embryonic day 18.5, histological examination revealed thickened, malformed semilunar valve leaflets accompanied by additional anomalies of the cardiac outflow tract including ventricular septal defects and overriding aorta. The aortic valve leaflets of Notch1 +/− ;Nos3 −/− embryos at embryonic day 15.5 were significantly thicker than controls, consistent with a defect in remodeling of the semilunar valve cushions. In addition, we generated mice haploinsufficient for Notch1 specifically in endothelial and endothelial‐derived cells in a Nos3‐null background and found that Notch1 fl/+;Tie2‐Cre +/− ;Nos3 −/− mice recapitulate the congenital cardiac phenotype of Notch1 +/− ;Nos3 −/− embryos.
Conclusions
Our data demonstrate the role of endothelial Notch1 in the proper development of the semilunar valves and cardiac outflow tract.
Keywords: bicuspid aortic valve, cardiovascular genetics, congenital heart defect, conotruncal heart defects, Notch1 signaling
Subject Categories: Developmental biology, Animal Models of Human Disease, Genetically Altered and Transgenic Models, Valvular Heart Disease
Introduction
Bicuspid aortic valve (BAV) is the most common congenital anomaly, with an estimated incidence of 1.3% in the population.1 Along with BAV, developmental anomalies of the semilunar valves and cardiac outflow tract (OFT) account for a significant proportion of congenital heart defects and result in a substantial clinical burden for affected patients.2, 3, 4 Multiple cell lineages are responsible for the development of the cardiac OFT and semilunar valves.5 During early heart development, cells from the anterior second heart field (SHF) contribute to formation of the cardiac OFT, also known as the conotruncus.6, 7, 8 In addition, a subpopulation of endothelial cells that line the OFT undergo endothelial‐to‐mesenchymal transition to form the OFT cushions. These mesenchymal cells are joined by the migrating cardiac neural crest cells, which are required for septation of the aorta and pulmonary artery.9, 10, 11 Proper migration and signaling of all 3 cell lineages, the SHF, cardiac neural crest and endothelial‐derived mesenchymal cells are required for normal OFT cushion development and semilunar valve remodeling.
The Notch signaling pathway has been shown to be important for multiple aspects of heart development, from endocardial cushion formation to myocardial development, and mutations in this pathway have been linked to a spectrum of congenital heart defects in humans.12 The Notch family is composed of 4 receptors responsible for various cell‐fate decisions and for vascular development and disease.12, 13 These receptors are transmembrane signaling proteins that contain a ligand‐binding extracellular domain and an intracellular domain. On binding with its ligand, a series of cleavage events release the Notch intracellular domain, which translocates to the nucleus and binds with its cofactors mastermind‐like protein and RBPJκ to regulate expression of target genes.14 Specifically, the Notch signaling family has been shown to play a critical role in cardiac OFT development because loss of the Notch signaling pathway, specifically in the SHF via a dominant‐negative truncated form of mastermind‐like protein or loss of Jagged1, results in a spectrum of OFT defects including aortic valve abnormalities.15 Furthermore, mutations in NOTCH2 and the Notch ligand JAGGED1 are responsible for Alagille syndrome, which is characterized by pulmonary stenosis, ventricular septal defects, coarctation of the aorta, and tetralogy of Fallot, among other developmental defects.16, 17 Notch1 is expressed in the endothelial cells lining the cardiac OFT during development, and mutations in NOTCH1 have been linked primarily to human BAV and other left‐sided cardiac malformations.18, 19 Although these studies indicate the important role for Notch signaling in the development of the cardiac OFT and aortic valve, the underlying mechanisms and the cell lineages in which Notch1 is required have not yet been elucidated.
We previously described reduced survival to adulthood in Notch1 +/− ;Nos3 −/− mice, suggesting a potential embryonic phenotype.20 To further investigate the cause of this lethality, we bred Notch1 +/− ;Nos3 +/− female mice with Nos3 −/− male mice and examined the resultant litters. We observed 65% neonatal lethality in Notch1 +/− ;Nos3 −/− mice and found that compound mutant embryos displayed a spectrum of congenital cardiac malformations, including thickened semilunar valves, ventricular septal defects, and overriding aorta. Using a conditional gene deletion approach (Cre/LoxP), we found that loss of endothelial Notch1 was responsible for the cardiac phenotypes observed in the Notch1 +/− ;Nos3 −/− mice. Our results indicate a novel role for endothelial Notch1 in the development of the semilunar valves and cardiac OFT.
Methods
Mice
Animal use was approved and monitored by the institutional animal care and use committee at the Research Institute at Nationwide Children's Hospital. Nos3 −/− and Notch1 +/− ;Nos3 +/− mice were bred to obtain Notch1 +/− ;Nos3 −/− mice (n=49) and littermate controls (n=216) and were genotyped, as described previously.20 For lineage‐specific deletions of Notch1 (using Tie2‐Cre,21 Mef2C‐Cre,22 Wnt1‐Cre23), Nos3 −/− ;Cre +/− male mice were bred with Notch1 flox/wt ;Nos3 +/− female mice to obtain Notch1 flox/wt ;Tie2‐Cre +/− ;Nos3 −/− (n=6) mice, Notch1 flox/wt ;Mef2C‐Cre +/− ;Nos3 −/− (n=7), Notch1 flox/wt ;Wnt1‐Cre +/− ;Nos3 −/− (n=4), and control littermates (n=4, n=4, n=4, respectively). SHF lineage tracing was completed by breeding Mef2C‐Cre +/− male mice with ROSA26 mT/mG female mice.24
Tissue Fixation and Histology
Embryos were harvested at the indicated time points and fixed in 10% formalin at 4°C overnight. Sections (6 μm) were stained with hematoxylin and eosin and imaged at ×50. Valve area was determined by the average area across 3 sections of each leaflet using AxioVision software (Zeiss). Cell density was determined by dividing the number of nuclei in each valve leaflet by the measured area. Valve excavation, as described by Dupuis et al,25 was determined by the ratio of space across the valve by sections en face and calculated by ImageJ (National Institutes of Health). A minimum of 3 sections of each valve were performed. Immunofluorescence was performed using anti–green fluorescent protein (ab290, 1:1000; Abcam) and anti‐PECAM1 (sc‐1506, 1:50; Santa Cruz Biotechnology) and was counterstained with Vector Laboratories Hardset Mounting Medium with DAPI (H‐1500).
Statistics
Statistical analysis was performed on quantitative graphs using the Mann–Whitney test because of the small number of mice used and the lack of normality, with median and 25th and 75th percentiles reported. For categorical data, the Fisher exact test was used. P<0.05 was considered significant.
Results
Neonatal Lethality in Notch1 +/− ;Nos3 −/− Mice
To determine the embryonic phenotype of Notch1 +/− ;Nos3 −/− mice, we bred Notch1 +/− ;Nos3 +/− and Nos3 −/− mice and examined the resultant litters at postnatal day 10. We found ≈65% lethality in Notch1 +/− ;Nos3 −/− pups at postnatal day 10, whereas no lethality was observed in littermate controls (Figure 1A). Interestingly, this was contrasted by expected Mendelian ratios for all genotypes between embryonic day (E) 11.5 and E18.5 (Figure 1A). Examination of Notch1 +/− ;Nos3 −/− embryos at E18.5 revealed no gross abnormalities or growth retardation compared with littermates (Figure 1B through 1E). Gross examination of embryonic hearts at E18.5 revealed abnormal cardiac morphology with an enlarged right ventricle in the Notch1 +/− ;Nos3 −/− embryos compared with control littermates, suggesting that a congenital cardiac malformation was contributing to their neonatal lethality (Figure 1F through 1I).
Figure 1.
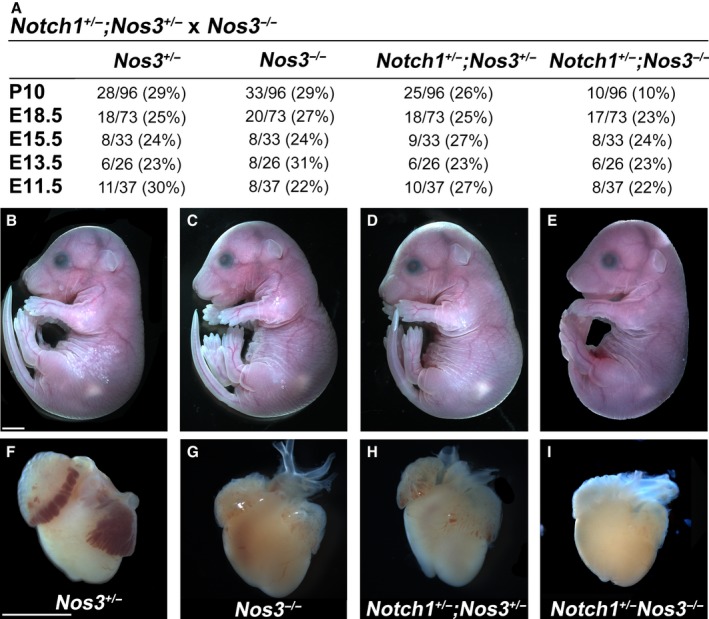
Notch1 +/− ;Nos3 −/− mice display perinatal lethality. Compound mutant mice suffered ≈65% lethality by P10, as shown in (A). Nevertheless, compared with Nos3 +/− (n=18) (B), Nos3 −/− (n=20) (C), and Nos3 +/− ;Notch1 +/− (n=18) (D) littermates, Notch1 +/− ;Nos3 −/− mice did not display any embryonic lethality (A) or growth retardation at E18.5 (n=17) (E). Compared with controls (F through H), examination of E18.5 hearts revealed right ventricle enlargement in compound mutants (I). Scale bars, 2 mm. E indicates embryonic day; P, postnatal day.
Notch1 +/− ;Nos3 −/− Mice Display Congenital Heart Malformations and Thickened Semilunar Valves
To determine whether congenital cardiac malformations were present in the compound mutant animals, histological examination of hearts at E18.5 was performed. Notch1 +/− ;Nos3 −/− hearts were found to have a spectrum of cardiac malformations, including thickened aortic and pulmonary valves, ventricular septal defects, and overriding aorta (Figure 2B through 2E), compared with control littermate hearts (Figure 2A and 2E). Although a small subset of Notch1 +/− ;Nos3 +/− animals demonstrated mildly thickened semilunar valve leaflets at E18.5, we did not observe any other malformations in the Nos3 +/−, Nos3 −/− or Notch1 +/− ;Nos3 +/− embryos (Figure 2E).
Figure 2.

Notch1 +/− ;Nos3 −/− embryos display cardiac outflow tract and semilunar valve malformations. Compared with Nos3 +/− control mice (A), compound mutant mice (B through D) displayed AoV thickening (B, arrowhead), VSD with overriding Ao (C, asterisk), and PV thickening (D, arrow), as summarized in the table (E). Nos3 +/−, n=7; Nos3 −/−, n=7; Notch1 +/− ;Nos3 +/−, n=7; Notch1 +/− ;Nos3 −/−, n=9. Scale bars=200 μm. *P<0.05. Ao indicates aorta; AoV, aortic valve; LV, left ventricle; PA, pulmonary artery; PV, pulmonary valve; RV, right ventricle; VSD, ventricular septal defect.
Further examination of the aortic valves at E18.5 demonstrated that the Notch1 +/− ;Nos3 −/− mutant valves (Figure 3D) were significantly larger than Nos3 +/−, Nos3 −/− or Notch1 +/− ;Nos3 +/− valves (Figure 3A through 3C and 3K). Although the valve leaflets of compound mutant animals were larger, they contained fewer nuclei per unit area (Figure 3I, 3J, and 3L). Remodeling of the aortic valve cushions occurs during the later stages of gestation; therefore, examination of Notch1 +/− ;Nos3 −/− embryos at E15.5 was performed. Compound mutant hearts demonstrated thickened valve leaflets (Figure 3H) compared with control animals (Figure 3E through 3G) and were accompanied by a reduction in aortic valve excavation (Figure 3M).25 These results suggest that Notch1 signaling is required for normal development of the cardiac OFT and for proper remodeling of the semilunar valves.
Figure 3.
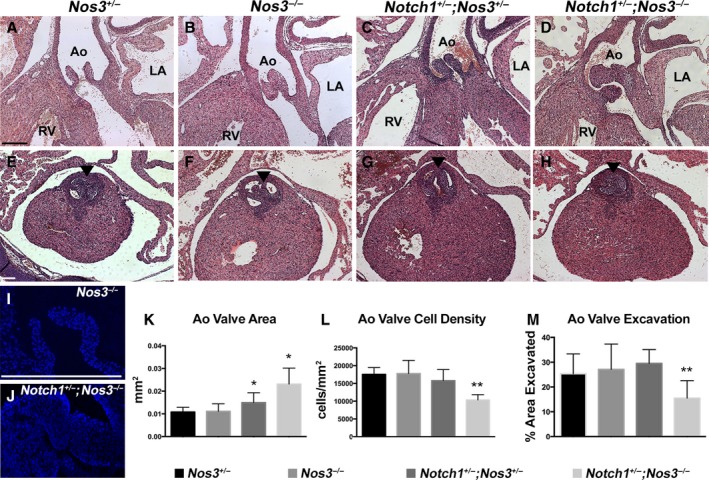
Notch1 +/− ;Nos3 −/− embryos display abnormal aortic valve remodeling. Compared with Nos3 +/− (A), Nos3 −/− (B), and Nos3 +/− ;Notch1 +/− (C) animals at embryonic day 18.5, compound mutant mice demonstrated thickened aortic valves (D), as summarized in (K) (Nos3 +/−: n=5, median 0.01 [IQR 0.01–0.01125]; Nos3 −/−: n=5, median 0.01 [IQR 0.01–0.01]; Notch1 +/− ;Nos3 +/−: n=5, median 0.015 [IQR 0.01–0.02]; Notch1 +/− ;Nos3 −/−: n=5, median 0.0225 [IQR 0.02–0.03]). Notch1 +/− ;Nos3 −/− animals also display a reduced number of nuclei per unit area (J and L) compared with Nos3 −/− animals (I) and controls (L) (Nos3 +/−: n=5, median 17 200 [IQR 15 675–19 750]; Nos3 −/−: n=5, median 18 000 [IQR 14 975–20 375]; Notch1 +/− ;Nos3 +/−: n=5, median 14 600 [IQR 14 350–16 950]; Notch1 +/− ;Nos3 −/−: n=5, median 10 400 [IQR 9000–11 767]). Notch1 +/− ;Nos3 −/− mice were found to have thickened aortic valve leaflets (arrowhead) at embryonic day 15.5 (H) compared with Nos3 +/− (E), Nos3 −/− (F) and Notch1 +/− ;Nos3 +/− (G) animals, accompanied by a decrease in aortic valve excavation (M) (Nos3 +/−: n=4, median 23.90 [IQR 18.80–33.60]; Nos3 −/−: n=4, median 25.10 [IQR 17.30–37.80]; Notch1 +/− ;Nos3 +/−: n=4, median 29.80 [IQR 24.00–32.80]; Notch1 +/− ;Nos3 −/−: n=4, median 16.80 [IQR 10.40,– 20.05]). Scale bars=200 μm. *P<0.05; **P<0.01. IQR shows the 25th and 75th percentiles. Ao indicates aorta; IQR, interquartile range; LA, left atria; RV, right ventricle.
Notch1 Is Required Within the Endothelial Cell Lineage for Proper Morphogenesis of the Aortic Valve
The mesenchymal cells of the OFT cushions are derived from multiple sources including the cardiac neural crest, the SHF, and endothelial‐derived cells. To determine the specific cell lineage in which loss of Notch1 contributes to the aortic valve phenotype in Notch1 +/− ;Nos3 −/− mice, we used mice containing a Notch1‐floxed allele.26 Because Notch1 is expressed throughout the developing valve mesenchyme in all of the aforementioned lineages, we used Cre‐specific drivers for each cell lineage. To test whether the endothelium may be critical in the role of Notch1 in valve development, we bred Notch1 fl/wt ;Nos3 +/− animals to Nos3 −/− mice harboring the endothelial‐specific driver Tie2‐Cre.21 We discovered that Notch1 fl/wt ;Tie2‐Cre +/− ;Nos3 −/− mice recapitulated the phenotypes seen in our Notch1 +/− ;Nos3 −/− animals, as summarized in Figure 4E. Notch1 fl/wt ;Tie2‐Cre +/− ;Nos3 −/− mice displayed thickened aortic (Figure 4C) and pulmonary valves (Figure 4D) compared with littermate controls (Figure 4A and 4B), and the aortic valves were also found to have reduced cell density (Figure 4F) similar to that of Notch1 +/− ;Nos3 −/− mice (Figure 3L). To determine whether Notch1 was required in other cell lineages, we used the SHF‐specific driver Mef2C‐Cre.22 Notch1 fl/wt ;Mef2C‐Cre +/− ;Nos3 −/− animals also displayed thickened aortic and pulmonary leaflets compared with littermate controls, although to a lesser extent than the Notch1 fl/wt ;Tie2‐Cre +/− ;Nos3 −/− animals (Figure 5A through 5F). SHF lineage tracing with ROSA26 mT/mG 24 mice showed that a subset of endothelial cells within the aorta and aortic valves were derived from the SHF (Figure 5G and 5H). SHF‐derived endothelial cells are also known to populate the pulmonary trunk and pulmonary valve, suggesting that the effect of Notch1 may still be limited to the endothelial cell layer.22 Because other publications have implicated the role of neural crest cells in OFT formation, we obtained neural crest–specific Wnt1‐Cre mice to ascertain the role of Notch1 specifically in the neural crest.23 We did not observe any cardiac phenotypes in Notch1 fl/wt ;Wnt1‐Cre +/− ;Nos3 −/− mice. This finding suggests that Notch1 in the neural crest is not responsible for the cardiovascular and semilunar valve phenotypes observed in the Notch1 +/− ;Nos3 −/− mice (Figure 6). Although our results do not preclude the possibility of SHF cells requiring Notch1, they demonstrate a critical role of Notch1 within the endothelial cells for the proper development of the semilunar valves and cardiac OFT.
Figure 4.
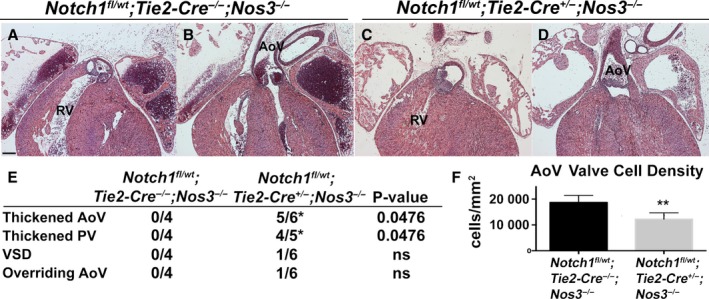
Endothelial‐specific Notch1 haploinsufficiency in a Nos3‐null background recapitulates the Notch1 +/− ;Nos3 −/− cardiac phenotype. Notch1 fl/wt;Tie2‐Cre +/−;Nos3 −/− embryos (n=6) were found to have thickened aortic and pulmonary valve leaflets (C and D) at embryonic day 18.5 compared with Notch1 fl/wt ;Tie2‐Cre −/− ;Nos3 −/− animals (n=4) (A and B). E, Table showing cardiac phenotypes in Notch1 fl/wt;Tie2‐Cre +/−;Nos3 −/− embryos, which also displayed VSD and overriding Ao. F, Reduced AoV cell density in Notch1 fl/wt;Tie2‐Cre +/−;Nos3 −/− embryos (n=5, median 12 196 [IQR 9769–14 446]) compared with Notch1 fl/wt ;Tie2‐Cre −/− ;Nos3 −/− embryos (n=4, median 17 100 [IQR 16 745–21 473]). Scale bar=200 μm. *P<0.05; **P<0.01. IQR shows the 25th and 75th percentiles. Ao indicates aorta; AoV, aortic valve; IQR, interquartile range; ns, not significant (P=1.00); PV, pulmonary valve; RV, right ventricle; VSD, ventricular septal defect.
Figure 5.
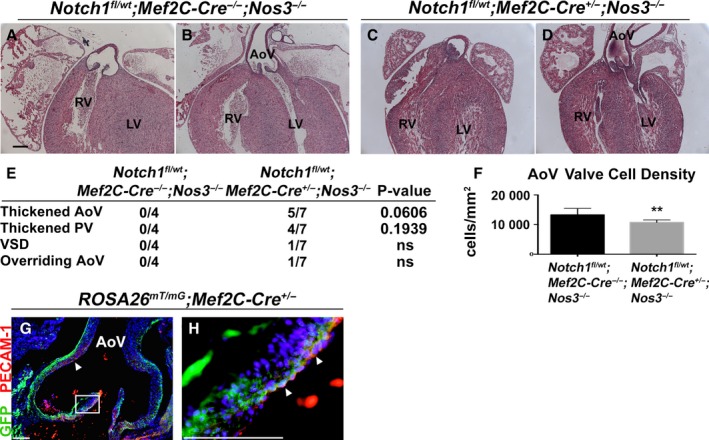
SHF‐specific Notch1 haploinsufficiency in a Nos3‐null background partially recapitulates the Notch1 +/− ;Nos3 −/− cardiac phenotype. Notch1 fl/wt ;Mef2C‐Cre +/− ;Nos3 −/− embryos (n=7) were found to have thickened AoV and PV leaflets (C and D) at embryonic day 18.5 compared with Notch1 fl/wt ;Mef2C‐Cre −/− ;Nos3 −/− animals (n=4) (A and B). Notch1 fl/wt; Mef2C‐Cre +/−;Nos3 −/− mice also displayed VSDs and overriding Ao (E), although to a lesser extent than what is seen in the Notch1 +/− ;Nos3 −/− mice, which is also observed by a reduction in AoV cell density in Notch1 fl/wt;Mef2C‐Cre +/−;Nos3 −/− embryos (n=3, median 10 455 [IQR 10 144–11 442]) compared with Notch1 fl/wt ;Mef2C‐Cre −/− ;Nos3 −/− animals (n=3, median 13 258 [IQR 11 271–14 512]) (F). Lineage tracing with ROSA26 mT/mG;Mef2C‐Cre +/− mice (n=4) revealed SHF‐derived endothelial cells (arrowheads) lining the AoV and aortic root (G and H). GFP indicates SHF‐derived cells; PECAM1 indicates endothelial cells. Scale bars=100 μm. **P<0.01. IQR shows the 25th and 75th percentiles. Ao indicates aorta; AoV, aortic valve; GFP, green fluorescent protein; IQR, interquartile range; ns, not significant (P=1.00); LV, left ventricle; PV, pulmonary valve; RV, right ventricle; SHF, second heart field; VSD, ventricular septal defect.
Figure 6.
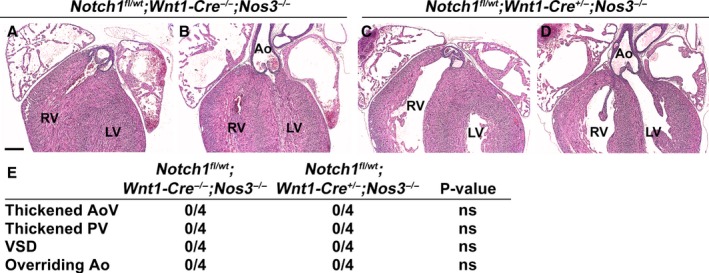
Neural crest–specific Notch1 haploinsufficiency in a Nos3‐null background does not recapitulate the cardiac phenotype of the Notch1 +/− ;Nos3 −/− mice. Notch1 fl/wt ;Nos3 +/− mice were bred to Notch1 wt/wt ;Wnt1‐Cre +/− ;Nos3 −/− male mice, creating neural crest–specific Notch1 haploinsufficiency in a Nos3 −/− background. Notch1 fl/wt;Wnt1‐Cre +/− ;Nos3 −/− embryos (n=4) were found to have normal AoV and PV leaflets (A through D) at embryonic day 18.5 compared with Notch1 fl/wt ;Wnt1‐Cre −/− ;Nos3 −/− animals (n=4) (E). There was no evidence of VSDs and overriding Ao in Notch1 fl/wt; Wnt1‐Cre +/−;Nos3 −/− mice (E). Scale bar=200 μm. Ao indicates aorta; AoV, aortic valve; ns, not significant (P=1.00); LV, left ventricle; PV, pulmonary valve; RV, right ventricle; VSD, ventricular septal defect.
Discussion
We have described the cardiac phenotype in Notch1‐haploinsufficient embryos backcrossed into a Nos3‐null background. Our studies demonstrate that although adult survivors display isolated aortic valve anomalies, mutant embryos have a spectrum of cardiac phenotypes including thickened semilunar valve leaflets, overriding aorta, and ventricular septal defects. In addition, we found that loss of Notch1 in endothelial and endothelial‐derived cells resulted in a spectrum of cardiac OFT defects and semilunar valve anomalies in our model. In summary, these findings highlight the importance of endothelial Notch1 in multiple aspects of cardiac OFT development including semilunar valve remodeling.
Notch1 is a transmembrane signaling receptor responsible for many developmental processes.14 The Notch signaling family proteins are expressed in multiple cardiac lineages and during different stages of development and have the ability to act in a noncell autonomous fashion, resulting in signal transduction to multiple cell types.12, 15 Our results indicate that Notch1 is required within the endothelial cell lineage for proper OFT development and semilunar valve remodeling; however, the cell lineages with which Notch1 communicates have not been well defined. Similar to Notch1, deletion of Gata5 or Alk2 in the OFT endothelial (endocardial) and endothelial‐derived mesenchymal cells is sufficient to cause BAV.27, 28, 29 In addition, the cardiac neural crest is critical for the development of the cardiac OFT because loss of BMP signaling via the receptor ALK2 has been shown to cause persistent truncus arteriosus, improper cardiac neural crest migration causes OFT defects, and the loss of Rho kinase signaling within neural crest cells gives rise to BAV.30, 31, 32 We did not observe any cardiac malformations using a neural crest–specific Cre driver to delete Notch1; however, this does not exclude the possibility that Notch1 in endothelial‐derived cells may be signaling to neural crest cells in the cardiac OFT. Neural crest cells have been shown to direct mesenchymal cell fate decisions within the developing cardiac OFT, and it is possible that Notch1 in endothelial and endothelial‐derived cells has the capacity to act in a similar role via the neural crest.33 Our data also demonstrate a role for Notch1 in SHF‐derived cells because use of Mef2C‐Cre mice resulted in a partial recapitulation of our observed cardiac phenotype. We reason that this is likely because of deletion of Notch1 in SHF‐derived endothelial cells but cannot exclude a potential role for Notch1 in other SHF‐derived cells.
The human aortic valve is composed of 3 leaflets called the right coronary (R), left coronary (L), and noncoronary (NC) leaflets, so named for their spatial arrangement to the coronary arteries. BAVs may result from the fusion of any leaflet, although in humans, right–left fusion is most common.34 The process of leaflet fusion is currently not well understood because leaflet fusion may occur early in cushion development or later during valve remodeling. Gata5 knockout mice display BAV, which is caused by an early fusion without leaflet thickening, unlike that of ALK2 mutant mice, which demonstrate thickened valve leaflets at early stages.27, 30 Some studies have concluded that BAV subtypes are a result of specific genetic etiologies because Gata5 −/− mice and Nos3 −/− mice display BAVs with right–noncoronary fusion.27, 35 Further research using genetic sequencing in humans has also suggested this because human BAV patients harboring rare GATA5 mutations have a higher incidence of right–left and right–noncoronary BAVs.36 Nevertheless, a study of 1849 inbred Syrian hamsters with a high probability of homozygosity found variable aortic valve morphology and suggested that environmental factors, rather than genetics, account for BAV morphology.37, 38 Gene–environment interactions may also play a role in the development of BAV because mutations in the Notch signaling pathway combined with environmental effectors have been demonstrated to increase the penetrance of disease states.39 Accordingly, we examined the BAV morphology in the Notch1 +/− ;Nos31 −/− adult mice and identified a variable morphology, with left–right, right–noncoronary, and left–noncoronary fusion all observed (n=4, data not shown).
In summary, our work demonstrates the role for Notch1 in OFT and semilunar valve development. Interestingly, examination of the families reported in the original publication linking NOTCH1 mutations and congenital heart defects demonstrates the presence of an individual with tetralogy of Fallot, a phenotype consistent with the phenotype observed in the Notch1 +/− ;Nos3 −/− embryos.18 This potential link was further supported by the identification of a microdeletion encompassing NOTCH1 in a patient with tetralogy of Fallot by chromosomal microarray.18, 40 Further recent evidence supporting a link between NOTCH1 mutations and right‐sided congenital heart defect is the identification of mutations in NOTCH1 in 2 additional families with malformations of right‐sided cardiac structures.41, 42 Further investigation is required to determine whether mutations in NOTCH1 are found in children with malformations affecting both the right (pulmonary) and left (aortic) sides of the developing OFT.
Sources of Funding
This work was supported by grants from NIH/NLHBI (R01‐HL121797) and the Saving Tiny Hearts Society to Garg. Boss was supported by an NIH T32 Training Grant. Koenig was supported by Award Number Grant TL1TR001069 from the National Center For Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center For Advancing Translational Sciences or the National Institutes of Health.
Disclosures
None.
Acknowledgments
The authors would like to acknowledge Nianyuan Huang and Darian Bauer for technical assistance in management of the mouse colony for this project and thank Dr. B.L. Black for providing us with the Mef2c‐Cre mice and Dr. M. Yanagisawa for the Tie2‐Cre mice.
(J Am Heart Assoc. 2016;5:e003075 doi: 10.1161/JAHA.115.003075)
References
- 1. Verma S, Siu SC. Aortic dilatation in patients with bicuspid aortic valve. N Engl J Med. 2014;370:1920–1929. [DOI] [PubMed] [Google Scholar]
- 2. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. 2011;108:1392–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez‐Sarano M. Burden of valvular heart diseases: a population‐based study. Lancet. 2006;368:1005–1011. [DOI] [PubMed] [Google Scholar]
- 4. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. [DOI] [PubMed] [Google Scholar]
- 5. Martin PS, Kloesel B, Norris RA, Lindsay M, Milan D, Body SC. Embryonic development of the bicuspid aortic valve. J Cardiovasc Dev Dis. 2015;2:248–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mjaatvedt CH, Nakaoka T, Moreno‐Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR. The outflow tract of the heart is recruited from a novel heart‐forming field. Dev Biol. 2001;238:97–109. [DOI] [PubMed] [Google Scholar]
- 7. Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–3188. [DOI] [PubMed] [Google Scholar]
- 8. Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10‐expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. [DOI] [PubMed] [Google Scholar]
- 9. Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. [DOI] [PubMed] [Google Scholar]
- 10. Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. [DOI] [PubMed] [Google Scholar]
- 11. Nakamura T, Colbert MC, Robbins J. Neural crest cells retain multipotential characteristics in the developing valves and label the cardiac conduction system. Circ Res. 2006;98:1547–1554. [DOI] [PubMed] [Google Scholar]
- 12. de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell. 2012;22:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gridley T. Notch signaling in the vasculature. Curr Top Dev Biol. 2010;92:277–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. High FA, Jain R, Stoller JZ, Antonucci NB, Lu MM, Loomes KM, Kaestner KH, Pear WS, Epstein JA. Murine Jagged1/Notch signaling in the second heart field orchestrates Fgf8 expression and tissue‐tissue interactions during outflow tract development. J Clin Invest. 2009;119:1986–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McDaniell R, Warthen DM, Sanchez‐Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, Costa T, Pierpont ME, Rand EB, Piccoli DA, Hood L, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nat Genet. 1997;16:243–251. [DOI] [PubMed] [Google Scholar]
- 18. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. [DOI] [PubMed] [Google Scholar]
- 19. McBride KL, Riley MF, Zender GA, Fitzgerald‐Butt SM, Towbin JA, Belmont JW, Cole SE. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand‐induced signaling. Hum Mol Genet. 2008;17:2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, LaHaye S, Tao G, Lucchesi PA, Lincoln J, Lilly B, Garg V. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. J Mol Cell Cardiol. 2013;60:27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2‐Cre transgenic mice: a new model for endothelial cell‐lineage analysis in vivo. Dev Biol. 2001;230:230–242. [DOI] [PubMed] [Google Scholar]
- 22. Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005;287:134–145. [DOI] [PubMed] [Google Scholar]
- 23. Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen‐inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. [DOI] [PubMed] [Google Scholar]
- 24. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double‐fluorescent Cre reporter mouse. Genesis. 2007;45:593–605. [DOI] [PubMed] [Google Scholar]
- 25. Dupuis LE, Osinska H, Weinstein MB, Hinton RB, Kern CB. Insufficient versican cleavage and Smad2 phosphorylation results in bicuspid aortic and pulmonary valves. J Mol Cell Cardiol. 2013;60:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999;10:547–558. [DOI] [PubMed] [Google Scholar]
- 27. Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest. 2011;121:2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laforest B, Nemer M. GATA5 interacts with GATA4 and GATA6 in outflow tract development. Dev Biol. 2011;358:368–378. [DOI] [PubMed] [Google Scholar]
- 29. Thomas PS, Sridurongrit S, Ruiz‐Lozano P, Kaartinen V. Deficient signaling via Alk2 (Acvr1) leads to bicuspid aortic valve development. PLoS One. 2012;7:e35539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. [DOI] [PubMed] [Google Scholar]
- 31. Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997;124:505–514. [DOI] [PubMed] [Google Scholar]
- 32. Phillips HM, Mahendran P, Singh E, Anderson RH, Chaudhry B, Henderson DJ. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovasc Res. 2013;99:452–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, Epstein JA. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. 2011;121:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Friedman T, Mani A, Elefteriades JA. Bicuspid aortic valve: clinical approach and scientific review of a common clinical entity. Expert Rev Cardiovasc Ther. 2008;6:235–248. [DOI] [PubMed] [Google Scholar]
- 35. Fernandez B, Duran AC, Fernandez‐Gallego T, Fernandez MC, Such M, Arque JM, Sans‐Coma V. Bicuspid aortic valves with different spatial orientations of the leaflets are distinct etiological entities. J Am Coll Cardiol. 2009;54:2312–2318. [DOI] [PubMed] [Google Scholar]
- 36. Bonachea EM, Chang SW, Zender G, LaHaye S, Fitzgerald‐Butt S, McBride KL, Garg V. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr Res. 2014;76:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fernandez B, Duran AC, Fernandez MC, Arque JM, Such M, Sans‐Coma V. Genetic contribution of bicuspid aortic valve morphology. Am J Med Genet A. 2011;155A:2897–2898; author reply 2899‐900. [DOI] [PubMed] [Google Scholar]
- 38. Sans‐Coma V, Carmen Fernandez M, Fernandez B, Duran AC, Anderson RH, Arque JM. Genetically alike Syrian hamsters display both bifoliate and trifoliate aortic valves. J Anat. 2012;220:92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sparrow DB, Chapman G, Smith AJ, Mattar MZ, Major JA, O'Reilly VC, Saga Y, Zackai EH, Dormans JP, Alman BA, McGregor L, Kageyama R, Kusumi K, Dunwoodie SL. A mechanism for gene‐environment interaction in the etiology of congenital scoliosis. Cell. 2012;149:295–306. [DOI] [PubMed] [Google Scholar]
- 40. Greenway SC, Pereira AC, Lin JC, DePalma SR, Israel SJ, Mesquita SM, Ergul E, Conta JH, Korn JM, McCarroll SA, Gorham JM, Gabriel S, Altshuler DM, Quintanilla‐Dieck Mde L, Artunduaga MA, Eavey RD, Plenge RM, Shadick NA, Weinblatt ME, De Jager PL, Hafler DA, Breitbart RE, Seidman JG, Seidman CE. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat Genet. 2009;41:931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blue GM, Kirk EP, Giannoulatou E, Dunwoodie SL, Ho JW, Hilton DC, White SM, Sholler GF, Harvey RP, Winlaw DS. Targeted next‐generation sequencing identifies pathogenic variants in familial congenital heart disease. J Am Coll Cardiol. 2014;64:2498–2506. [DOI] [PubMed] [Google Scholar]
- 42. Theis JL, Hrstka SC, Evans JM, O'Byrne MM, de Andrade M, O'Leary PW, Nelson TJ, Olson TM. Compound heterozygous NOTCH1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum Genet. 2015;134:1003–1011. [DOI] [PubMed] [Google Scholar]