

Abstract
Background
Although clinical trials have proved that statin can be used prophylactically against cardiovascular events, the direct effects of statin on plaque development are not well understood. We generated low‐density lipoprotein receptor knockout (LDLR −/−) pigs to study the effects of early statin administration on development of atherosclerotic plaques, especially advanced plaques.
Methods and Results
LDLR −/− pigs were generated by targeted deletion of exon 4 of the LDLR gene. Given a standard chow diet, LDLR −/− pigs showed atherosclerotic lesions starting at 6 months of age. When 3‐month‐old LDLR −/− pigs were fed a high‐cholesterol, high‐fat (HCHF) diet for 4 months (HCHF group), human‐like advanced coronary plaques developed. We also fed 3‐month‐old LDLR −/− pigs an HCHF diet with pitavastatin for 4 months (Statin Prophylaxis Group). Although serum cholesterol concentrations did not differ significantly between the 2 groups, intravascular ultrasound revealed 52% reduced plaque volume in statin‐treated pigs. Pathological examination revealed most lesions (87%) in the statin prophylaxis group were early‐stage lesions, versus 45% in the HCHF diet group (P<0.01). Thin‐cap fibroatheroma characterized 40% of the plaques in the HCHF diet group versus 8% in the statin prophylaxis group (P<0.01), intraplaque hemorrhage characterized 11% versus 1% (P<0.01), and calcification characterized 22% versus 1% (P<0.01).
Conclusions
Results of our large animal experiment support statin prophylaxis before the occurrence of atherosclerosis. Early statin treatment appears to retard development of coronary artery atherosclerosis and ensure lesion stability. In addition, the LDLR −/− pigs we developed represent a large animal model of human‐like advanced coronary plaque suitable for translational research.
Keywords: atherosclerosis, cholesterol, coronary disease, plaque, statins
Subject Categories: Animal Models of Human Disease, Translational Studies
Introduction
Although the general consensus is that plaque rupture and erosion are the most common causes of acute coronary syndrome, the detailed process of plaque destabilization and definitive proof of unstable plaque remain elusive.1 By applying intracoronary imaging modalities, such as intravascular ultrasound (IVUS) and optical coherence tomography, we can visualize developing plaques in vivo. However, IVUS and optical coherence tomography are usually performed only in patients who have had a heart attack requiring percutaneous coronary intervention. Even if we understand characteristics of the plaque on rupture or occlusion, it remains difficult to trace the plaque progression. To understand the natural history of and the histopathologic mechanisms responsible for atherosclerotic plaque development, an animal model in which vulnerable plaques develop and data supporting a cause‐and‐effect relation are needed.1
Animal models of atherosclerosis have been reported for several decades, but there are few that reproducibly exhibit the advanced plaques seen in humans, especially in the coronary arteries. Selective breeding of Watanabe heritable hyperlipidemic (WHHL) rabbits, however, has allowed development of advanced lesions with multiple necrotic zones in the main aortic and coronary branches.2 Advanced lesions with intraplaque hemorrhage and rupture have also been observed in the innominate arteries of ApoE−/− mice.3 However, ApoE−/− mice are resistant to coronary atherosclerosis,2 and the coronary artery in the WHHL rabbit is too small for serial in vivo same‐site intracoronary imaging of the process of plaque development. The small size also limits use of WHHL rabbit arteries in the development of human cardiovascular devices such as stents. Moreover, plaque in a main aortic branch such as the carotid artery is not similar to and only weakly correlated with plaque in the coronary artery.4 For investigation into the development of coronary artery plaques, especially advanced plaques, an animal model with human‐like coronary arteries is needed. The pig, by virtue of its similarity to humans with respect to size of the coronary artery, pathoanatomy, and physiology, is an ideal animal for the study of human coronary artery disease.5 Porcine models of atherosclerosis have gained attention, and several pig/minipig models, such as the Rapacz familial hypercholesterolemic (FH) pig and the low‐density lipoprotein receptor (LDLR) knockout Yucatan minipig, were developed recently.6, 7, 8, 9, 10, 11 These models, however, are not ideal. FH pigs show considerable variability in plasma cholesterol concentrations and atherosclerotic plaque development,8 and LDLR knockout Yucatan minipigs need at least 6 months on a 40% fat, 1% cholesterol diet for development of coronary plaques. Furthermore, although the minipig is advantageous because of its size, it is in short supply and thus around 5 times more expensive than the domestic pig, making this model cost‐prohibitive for many studies. In response to these obstacles, we knocked out LDLR from the domestic pig and generated a new LDLR knockout (LDLR−/−) pig. Under a high‐fat diet, advanced human‐like plaques developed in the LDLR−/− pigs at a very young age, allowing for investigation after a relatively short developmental period.
Several compelling clinical trials have supported statin treatment as prophylaxis against cardiovascular events.12, 13, 14, 15, 16 With development of our LDLR−/− pigs, we sought to take the next step in the research process to examine the direct effects of statin on the development of plaques, especially advanced plaques.
Methods
Ethics Statement
The study was carried out in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals.” The experimental protocol was approved by the Gene Recombination Experiment Safety Committees (No. 2010M5 for Nihon University School and No. 500035 for the National Institute of Agrobiological Sciences) and Animal Care and Use Committees (No. AP10M108 for Nihon University and No. H18‐038 for the National Institute of Agrobiological Sciences). All animals were housed at the Nihon University Medical Research Support Center, and standard animal husbandry procedures were followed.
Production of Cloned LDLR‐Targeted Pigs
A conventional targeting vector for the porcine LDLR gene was constructed so that a major part of exon 4 was replaced by the neomycin resistance gene. The vector was introduced into fetal fibroblasts from Landrace × Large White crossbred pigs with use of the Gene Pulser II electroporation system (Bio‐Rad Laboratories). The targeted cell clones were screened by polymerase chain reaction (PCR) and then used as donor cells for nuclear transfer. The nuclear transfer and subsequent embryo transfer were performed as previously described.17 The cloned fetuses were collected at 39 or 72 days of gestation to confirm the targeting events by Southern blotting and so that large populations of cells could be obtained for further nuclear transfer. The cell populations that were confirmed to be targeted were used for the secondary nuclear transfer.
F1 progeny were produced by artificial insemination or in vitro fertilization (IVF) with epididymal sperm collected from the clones. IVF was performed as described previously.18 F2 and later progeny including homozygously LDLR‐targeted (LDLR−/−) pigs were produced by using conventional breeding methods involving heterozygously LDLR‐targeted (LDLR+/−) F1 pigs.
The extracted DNA was cut with SphI or NheI and EcoRV and then subjected to electrophoresis and transferred to nylon membranes. The membranes were hybridized with digoxigenin (DIG)‐labeled 5′‐ or 3′‐end probes, and the hybridization signals were detected with CSPD (Roche Applied Science, Indianapolis, IN).
Development of Advanced Coronary Artery Plaques
To accelerate the development of advanced coronary artery plaques, 3‐month‐old male LDLR−/− pigs (n=5) were fed a high‐cholesterol, high‐fat (HCHF) diet (1 kg/d) for 4 months (HCHF diet group). The HCHF diet was made from standard pig chow with 1.5% cholesterol and 15% fat (beef tallow). The fat was heated in a microwave oven before being mixed into the chow.
Statin Prophylaxis
Pitavastatin (40 mg/d) was administered along with the HCHF chow to 3‐month‐old male LDLR−/− pigs (n=6; statin prophylaxis group) for 4 months. Pitavastatin powder was obtained from Kowa Pharmaceutical Co Ltd. The powder was freshly mixed with a small amount of feed in the laboratory and was then added to the daily ration.
Laboratory Tests
Blood samples were obtained from both the HCHF diet group and statin prophylaxis group pigs before the treatment period and then once a month during the 4‐month treatment period. The samples were collected via an ear vein after an overnight fast. Whole blood was immediately separated and sent out to commercial clinical laboratories for blood counts (MONOLIS) and biochemical analysis (SRL). Serum lipoproteins were analyzed by means of gel permeation high‐performance liquid chromatography (HPLC).19 The serum cholesterol and triglyceride concentrations of all 20 lipoprotein subclasses were measured by the LipoSEARCH testing service (Skylight Biotech, Inc). Particle sizes used to define the 20 lipoprotein subclasses are shown in Table 1. The serum C‐reactive protein (CRP) concentration, as a measure of inflammation, was determined with a commercial ELISA assay kit (Pig CRP ELISA, Life Diagnostics, Inc) according to the manufacturer's instructions. Before analysis, serum samples were diluted 1:500 or 1:1000.
Table 1.
Lipoprotein Subclasses in the LDLR+/+, LDLR+/−, and LDLR−/− Pigs, Shown By Particle Size
| Cholesterol (mg/dL) | Triglyceride (mg/dL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LDLR+/+ (n=3) | LDLR+/− (n=8) | LDLR−/− (n=6) | P1 (+/− vs +/+) | P2 (−/− vs +/+) | LDLR+/+ (n=3) | LDLR+/− (n=8) | LDLR−/− (n=6) | P1 (+/− vs +/+) | P2 (−/− vs +/+) | |
| Total | 169.8±12.8 | 158.5±6.5 | 620.3±41.1 | 0.99 | <0.01a | 35.7±5.7 | 26.2±2.1 | 41.0±4.9 | 0.23 | 0.60 |
| CM (>70 nm) | ||||||||||
| G01 (>90 nm) | 0.5±0.1 | 0.4±0.02 | 0.8±0.1 | 0.64 | 0.14 | 4.9±0.7 | 3.8±0.4 | 5.0±1.2 | 0.62 | 0.99 |
| G02 (75 nm) | 0.2±0.04 | 0.1±0.01 | 0.4±0.1 | 0.82 | 0.56 | 1.4±0.2 | 1.2±0.1 | 1.6±0.4 | 0.82 | 0.88 |
| VLDL (30–70 nm) | ||||||||||
| G03 (64 nm) | 0.3±0.1 | 0.2±0.02 | 0.7±0.1 | 0.97 | <0.01a | 1.8±0.1 | 1.5±0.2 | 2.1±0.6 | 0.82 | 0.84 |
| G04 (53.6 nm) | 0.6±0.1 | 0.3±0.1 | 2.9±0.5 | 0.78 | <0.01a | 3.1±0.4 | 2.3±0.3 | 3.4±0.6 | 0.41 | 0.88 |
| G05 (44.5 nm) | 1.2±0.3 | 1.2±0.1 | 13.6±1.2 | 0.99 | <0.01a | 6.2±1.6 | 3.9±0.6 | 5.3±0.6 | 0.14 | 0.68 |
| G06 (36.8 nm) | 2.1±1.1 | 0.8±0.1 | 35.5±4.5 | 0.93 | <0.01a | 4.1±1.7 | 2.1±0.2 | 3.2±0.3 | 0.06 | 0.52 |
| G07 (31.3 nm) | 16.3±3.5 | 13.2±0.8 | 105.0±9.7 | 0.92 | <0.01a | 1.5±0.6 | 0.7±0.1 | 1.5±0.2 | 0.04a | 0.99 |
| LDL (16–30 nm) | ||||||||||
| G08 (28.6 nm) | 32.6±4.5 | 33.1±1.7 | 186.4±12.8 | 0.99 | <0.01a | 3.4±0.9 | 2.1±0.3 | 3.5±0.4 | 0.11 | 0.99 |
| G09 (25.5 nm) | 27.5±2.6 | 33.7±2.7 | 148.4±8.4 | 0.70 | <0.01a | 3.9±0.7 | 3.6±0.3 | 6.6±0.6 | 0.89 | 0.01a |
| G10 (23 nm) | 9.7±0.3 | 13.9±1.5 | 52.1±2.9 | 0.37 | <0.01a | 1.6±0.2 | 2.0±0.2 | 4.2±0.4 | 0.53 | <0.01a |
| G11 (20.7 nm) | 3.3±0.1 | 4.8±0.6 | 18.7±1.1 | 0.37 | <0.01a | 0.5±0.1 | 0.7±0.1 | 1.5±0.1 | 0.55 | <0.01a |
| G12 (18.6 nm) | 1.5±0.2 | 1.4±0.1 | 2.7±0.2 | 0.92 | <0.01a | 0.3±0.01 | 0.3±0.02 | 0.4±0.1 | 0.99 | 0.11 |
| G13 (16.7 nm) | 0.9±0.1 | 1.0±0.1 | 3.8±0.2 | 0.85 | <0.01a | 0.2±0.03 | 0.2±0.01 | 0.3±0.03 | 0.99 | <0.01a |
| HDL (8–16 nm) | ||||||||||
| G14 (15 nm) | 1.3±0.2 | 0.7±0.1 | 1.6±0.1 | 0.03a | 0.21 | 0.2±0.01 | 0.1±0.01 | 0.2±0.03 | 0.95 | 0.88 |
| G15 (13.5 nm) | 0.3±0.2 | 0.5±0.2 | 1.8±0.1 | 0.57 | <0.01a | 0.1±0.01 | 0.1±0.02 | 0.2±0.03 | 0.99 | 0.02a |
| G16 (12.1 nm) | 6.8±2.0 | 2.2±0.6 | 2.7±0.5 | <0.01a | 0.02a | 0.3±0.1 | 0.2±0.01 | 0.2±0.04 | 0.06 | 0.31 |
| G17 (10.9 nm) | 34.6±3.4 | 29.1±1.3 | 22.5±2.0 | 0.14 | <0.01a | 0.7±0.1 | 0.6±0.04 | 0.6±0.1 | 0.69 | 0.70 |
| G18 (9.8 nm) | 14.8±2.2 | 15.8±0.8 | 14.6±0.8 | 0.71 | 0.98 | 0.7±0.3 | 0.3±0.02 | 0.4±0.1 | 0.04a | 0.14 |
| G19 (8.8 nm) | 3.7±0.4 | 4.1±0.2 | 4.2±0.2 | 0.53 | 0.35 | 0.3±0.1 | 0.3±0.02 | 0.3±0.03 | 0.46 | 0.95 |
| G20 (7.6 nm) | 1.8±0.2 | 1.7±0.1 | 2.0±0.1 | 0.93 | 0.23 | 0.6±0.1 | 0.4±0.03 | 0.5±0.04 | 0.02a | 0.14 |
CM indicates chylomicron; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; VLDL, very low‐density lipoprotein.
Significant difference.
iMAP–IVUS Analysis
At the end of the 4‐month treatment period, all pigs, under general anesthesia, underwent coronary angiography and IVUS (Atlantis SR Pro 40‐MHz Catheter; Boston Scientific Co) examinations for in vivo observation of coronary atherosclerotic plaques. IVUS images with radiofrequency signals were recorded continuously during an automatic pull‐back maneuver at a constant speed of 0.5 mm/s. All of these iMAP‐IVUS images were stored on DVD and were analyzed offline with the use of commercial software (echoPlaque 3.0; INDEC Systems). The external elastic membrane (EEM) area and lumen area were traced. The plaque area was calculated as the EEM area minus the lumen area, and plaque burden was calculated as the plaque area divided by the EEM area. In iMAP‐IVUS analysis of plaque composition, various aspects of the radiofrequency signal are processed by autoregressive modeling and matched to a database of known radiofrequency signal profiles containing the characteristics of 4 basic tissue types.20 The tissue components of the plaques were classified into the 4 basic types as fibrotic (green), lipidic (yellow), necrotic (red), and calcified (blue).
Histologic Analysis
The pigs were killed immediately after IVUS. The hearts with connected aorta roots were removed for fixation via coronary perfusion. The coronary arteries were flushed with normal saline for 30 minutes and then infused with 10% buffered formalin for 30 minutes at 150 cm H2O. The coronary arteries with surrounding tissues were removed from the heart. Sections of each coronary artery were taken perpendicular to the direction of blood flow at 1‐cm intervals and embedded in paraffin (Figure 1). These sections were stained with hematoxylin and eosin, elastic tissue fiber–Verhoeff‐van Gieson, and Masson's trichrome. The lumen, internal elastic membrane, and EEM were traced. The EEM area was defined as the vessel area. The plaque area was calculated as the internal elastic membrane area minus the lumen area. When a plaque had a fibrous cap covering a necrotic core, the thickness of the fibrous cap was measured at its thinnest point. Thin‐cap fibroatheroma (TCFA) was identified by a fibrous cap with a thickness of <65 μm.21 All measurements were made with the use of ImageJ 1.46r (National Institutes of Health).
Figure 1.
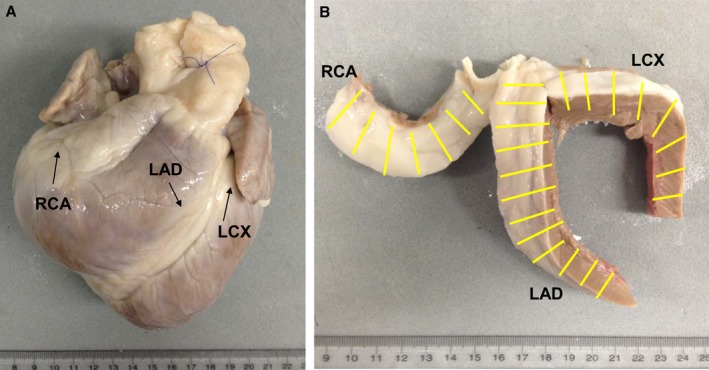
Preparation of coronary artery sections. A, Heart from an LDLR −/− pig after formalin fixation. B, Coronary arteries with surrounding tissues removed from the heart; yellow lines indicate the 1‐cm intervals at which the coronary arteries were sectioned. LAD indicates left anterior descending coronary artery; LCX, left circumflex coronary artery; RCA, right coronary artery.
To evaluate the extent of atherosclerosis, we classified all lesions into 6 types according to the Stary criteria but with some modification.22 In short, type I to III lesions were considered early‐stage atherosclerotic lesions (type I: seen as intimal thickening; type II: consisting primarily of layers of macrophage foam cells and lipid‐laden smooth muscle cells; type III: seen as scattered collections of extracellular lipid droplets), and type IV to VI lesions were considered advanced lesions with a lipid core (type IV: atheroma; type V: fibroatheroma; type VI: a type IV or type V lesion with fissure, hemorrhage, thrombus, or calcification). Lesions without a lipid core but with hemorrhage or calcification were considered type V lesions. In each coronary artery, the section with largest plaque area was selected for immunohistochemical detection of macrophages with the use of antimacrophage antibody MAC387 (Abcam). MAC387‐labeled macrophages were identified under a microscope (BZ‐X700; KEYENCE) and counted with the use of BZ‐X Analyzer (BZ‐analysis application, KEYENCE).
Statistical Analysis
Discrete variables are presented as numbers and percentages. Continuous variables are presented as mean±SE. Between‐group differences in discrete variables were assessed by χ2 test or Fisher's exact test. Between‐group differences in continuous variables were assessed by using unpaired Student t test or Wilcoxon rank sum test. Between‐group differences in the results of laboratory tests performed over time were assessed by repeated‐measures ANOVA, which was followed by Tukey's HSD or Dunnett's test for comparisons between 3 groups. A value of P<0.05 was accepted as statistically significant. JMP 10.02 (SAS Institute Inc) was used for all statistical analyses.
Results
Generation of LDLR−/− Pigs
We constructed a targeting vector for exon 4 of the porcine LDLR gene (Figure 2A) and introduced it into porcine fetal fibroblasts. After selection with G418, 413 colonies were picked up to be screened by polymerase chain reaction. One (0.24%) of these colonies was shown to be targeted and used as a donor cell population. Six nuclear transfer and subsequent embryo transfer series yielded 9 fetuses from 2 pregnant female pigs (Table 2). Seven of these were confirmed by Southern blotting to be correctly targeted. Fibroblasts from these targeted fetuses were used as donor cells for secondary nuclear transfer. Eleven nuclear transfers and subsequent embryo transfer series yielded 31 live‐born piglets, but 29 of these died within 7 months, presumably because of epigenetic abnormalities caused by somatic cloning procedures (Table 2). Because the 2 surviving pigs could not be subjected to the conventional sperm collection technique, owing to weakness of their hindlimbs, we retrieved epididymal sperm, which we used for artificial insemination or IVF. A total of 21 LDLR+/− F1 progenies were thus obtained. From this F1 population, F2 and later progenies including LDLR−/− pigs were generated (Figure 2B and 2C). Body weights, blood pressures, and appetites of the LDLR−/− progeny, LDLR+/− progeny, and their wild‐type (LDLR+/+) littermates were comparable until at least 7 months of age (data not shown).
Figure 2.
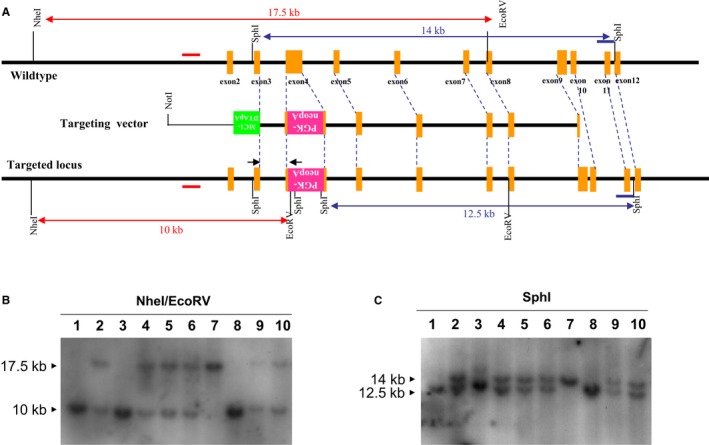
Generation of LDLR −/− pigs. A, Schematic diagram of the wild‐type LDLR locus (top line), targeting vector (middle line), and predicted targeted allele (bottom line). Positions of the 5′ and 3′ probes for Southern blotting are indicated by red and blue bars, respectively, and DNA fragments expected to be detected are indicated by the lines with arrows as both ends. B, Southern blot analysis of representative F2 siblings with 5′ probe. The wildtype allele yielded a 17.5‐kb fragment hybridized to the 5′ probe, whereas the targeted allele yielded a 10‐kb fragment. C, Southern blot analysis of representative F2 siblings with 3′ probe. The wildtype allele yielded a 14‐kb fragment hybridizing to 3′ probe, whereas the targeted allele yielded a 12.5‐kb fragment. These siblings include homozygous LDLR‐targeted pigs (lanes 1, 3, and 8), heterozygous LDLR‐targeted pigs (lanes 2, 4, 5, 6, 9, and 10), and wild‐type pigs (lane 7).
Table 2.
Results of LDLR+/− Pig Generation (by Somatic Cell Nuclear Transfer)
| Donor Cell Type | No. Reconstructed Oocytes | No. (%) Embryos Transferreda | No. Recipients | No. (%) Pregnancies by Day 30b | No. (%) Fetusesc | No. (%) Term Pregnanciesb | No. (%) Bornc | No. (%) Survivedd |
|---|---|---|---|---|---|---|---|---|
| G418‐resistant fetal fibroblaste | 2967 | 1290 (43.5)f | 6 | 2 (33.3)g | 9 (0.7) | — | — | — |
| Fetal fibroblast from targeted cloneh | 6351 | 2509 (39.5)f | 11 | — | — | 6 (50) | 31 (1.6)i | 2 (6.5) |
Expressed as percentage of surviving reconstructed oocytes.
Expressed as percentage of recipients.
Expressed as percentage of transferred embryos.
Pigs that survived for longer than 7 months; expressed as percentage of live‐born piglets.
PCR‐positive colonies obtained after TV electroporation and subsequent G418 selection.
Embryos that had developed to the 2‐ to 8‐cell stage after 2 days of culture in vitro were transferred to recipients.
Pregnancies were terminated on days 39 and 72 for isolation of embryonic fibroblasts.
Cells isolated from a cloned fetus in which targeting was confirmed.
Eight stillborn piglets not included.
Blood Counts, Blood Chemistry Values, and Lipid Concentrations
Blood counts and blood sugar, protein, electrolyte, urea nitrogen, creatinine, aspartate transaminase, alanine transaminase, lactate dehydrogenase, alkaline phoshphastase, and γ‐guanosine triphosphatase levels of the LDLR−/−, LDLR+/−, and LDLR+/+ pigs were comparable until at least 7 months of age (data not shown). The serum concentrations of lipoprotein and its 20 subclasses (G1–G20) in pigs at age 1 month are shown in Figure 3A and 3B and Table 1. Very low‐density lipoprotein cholesterol and low‐density lipoprotein cholesterol (LDL‐C) concentrations were much higher in LDLR−/− pigs (n=6) than in LDLR+/− (n=8) and LDLR+/+ pigs (n=3). The serum lipoprotein profiles of LDLR+/− and LDLR+/+ pigs were similar.
Figure 3.
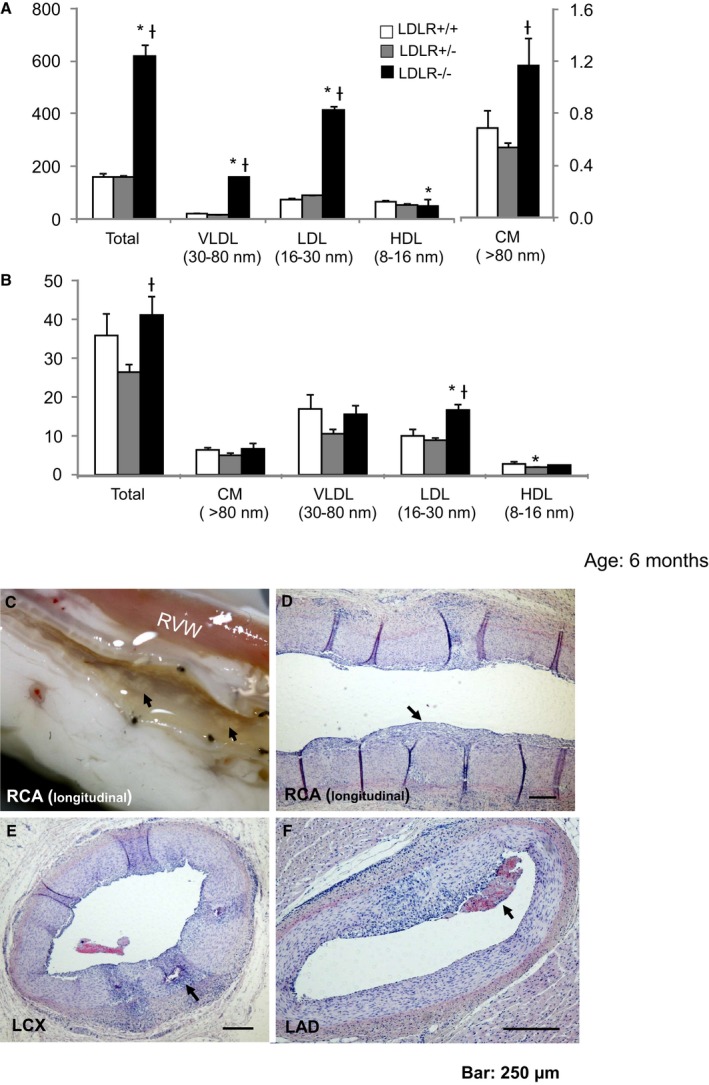
Serum lipoprotein concentrations and coronary atherosclerosis in LDLR −/− pigs fed a standard chow diet. A, Cholesterol concentrations (mg/dL) of VLDL, LDL, HDL, and CM. B, Triglyceride concentrations (mg/dL) of VLDL, LDL, HDL, and CM. C, Longitudinal macroscopic view of the RCA; arrows show atherosclerotic lesions (fatty streak). D, Longitudinal H&E‐stained section of RCA; arrow shows intimal thickening. E, H&E‐stained cross section of LCX; arrow shows calcification. F, H&E stained cross section of LAD; arrow indicates mural thrombosis. *P<0.05 vs LDLR +/+; † P<0.05 vs LDLR +/−. CM indicates chylomicron; H&E, hematoxylin and eosin; HDL, high‐density lipoprotein; LAD, left anterior descending coronary artery; LCX, left circumflex coronary artery; LDL, low‐density lipoprotein; RCA, right coronary artery; RVW, right ventricular wall; VLDL, very low‐density lipoprotein.
Coronary Atherosclerosis in LDLR−/− Pigs Fed a Standard Chow Diet
We investigated whether LDLR−/− pigs fed a standard chow diet would show coronary artery atherosclerosis by 3 or 6 months of age (n=2 each). Atherosclerosis was not apparent in the LDLR−/− pigs at age 3 months (data not shown), but raised atherosclerotic lesions with intimal thickening, mural thrombosis, foam cells, and plaque calcification were seen in the coronary artery at age 6 months (Figure 3C–3F).
Accelerated Development of Human‐Like Advanced Coronary Plaques in LDLR−/− Pigs
Although atherosclerotic lesions were apparent at age 6 months in the LDLR−/− pigs fed the standard chow diet, human‐like complicated advanced plaques were not found in these pigs. To accelerate advanced plaque development in the short term, the HCHF diet was started in 5 male LDLR−/− pigs at age 3 months and continued for an additional 4 months (HCHF diet group), as noted earlier. For controls, 2 LDLR+/+ littermates were fed the HCHF diet starting at age 3 months.
The serum cholesterol concentrations before the HCHF diet was started and at the end of the 4‐month HCHF diet period are shown in Figure 4A. Serum cholesterol concentrations were significantly increased after 4 months in both LDLR−/−, and LDLR+/+ pigs fed the HCHF diet. Very low‐density lipoprotein cholesterol concentrations, in particular, were ≈4‐fold greater in the LDLR−/− pigs than in the LDLR+/+ pigs after 4 months of being fed the HCHF diet.
Figure 4.
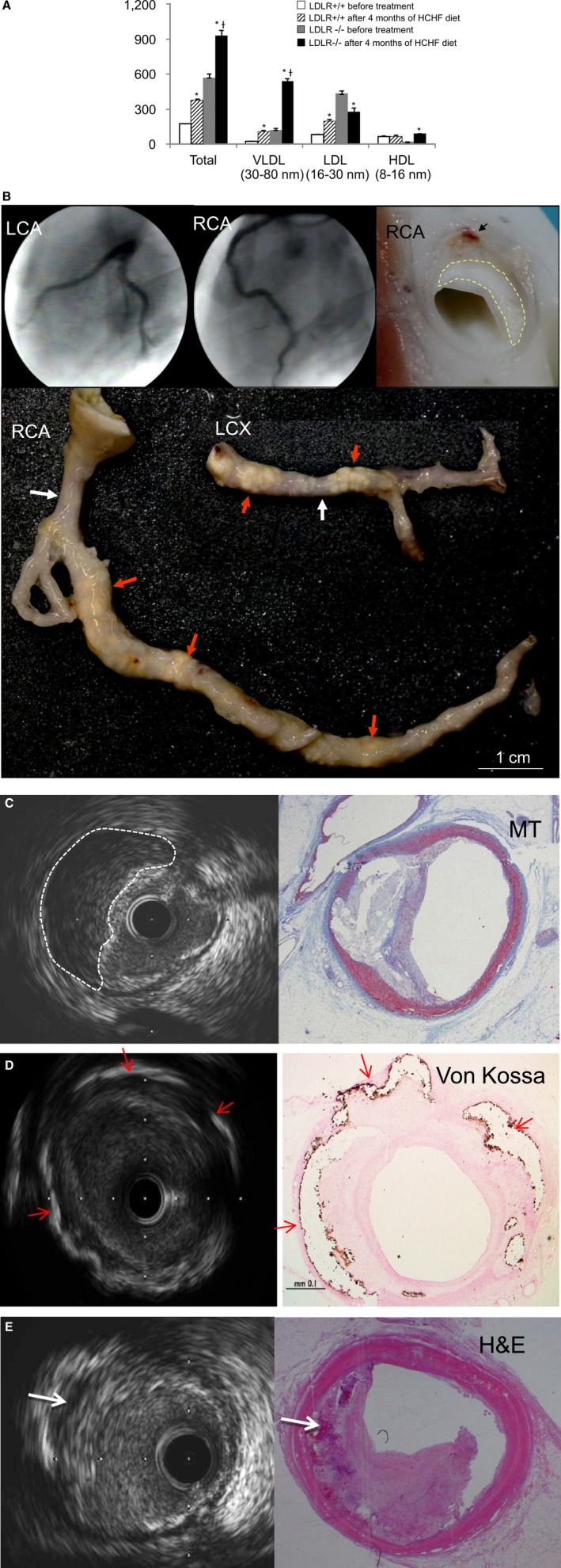
Cholesterol concentrations (mg/dL) and human‐like advanced coronary plaque in LDLR −/− pigs fed an HCHF diet for 4 months. A, Cholesterol concentrations (mg/dL) before and 4 months after HCHF diet. HDL indicates high‐density lipoprotein; LDL, low‐density lipoprotein; VLDL, very low‐density lipoprotein. *P<0.01, after vs before treatment; † P<0.01, LDLR −/− after treatment vs LDLR +/+ after treatment. B, Coronary angiography (upper left and middle images); macroscopic view of an RCA cross section (upper right); and dissected LCX and RCA (lower); yellow dashed line traces necrotic area, black arrow indicates intraplaque hemorrhage, red arrows indicate massive and rough atherosclerotic lesions with positive vessel remodeling; white arrows indicate relative noninvasive regions. C, IVUS image and MT‐stained section at same location; white dashed line traces necrotic core. D, IVUS image and Von Kossa–stained section at same location; red arrows indicate calcium deposits. E, IVUS image and H&E‐stained section at same location; white arrows indicate intraplaque hemorrhage. H&E indicates hematoxylin and eosin; HCHF, high‐cholesterol, high‐fat; LCA, left coronary artery; LCX, left circumflex coronary artery; MT, Masson's trichrome; RCA, right coronary artery.
Despite the 4‐month HCHF diet, there were no signs of coronary artery atherosclerosis in the 2 LDLR+/+ pigs (data not shown). In addition, no luminal stenosis was seen on coronary angiography performed in the LDLR−/− pigs fed the 4‐month HCHF diet (Figure 4B, upper left and middle). However, when we dissected the coronary arteries from the hearts of these LDLR−/− pigs, massive, irregular atherosclerotic lesions were seen even from the vessel surface (Figure 4B, lower images). When coronary arteries from LDLR−/− pigs were cut in cross ‐section, human‐like advanced plaques with large necrotic cores and intraplaque hemorrhage were observed (Figure 4B, upper right). In vivo IVUS and histological staining confirmed these human‐like advanced plaques with necrotic cores and fibrous caps (Figure 4C), calcium deposits (Figure 4D), and intraplaque hemorrhage (Figure 4E).
Effect of Statin Prophylaxis on Advanced Plaque Development
To test whether early statin treatment prevents the rapid development of advanced plaques found in LDLR−/− pigs fed an HCHF diet, we treated these pigs' male LDLR−/− littermates with pitavastatin (40 mg/d) starting at age 3 months (n=6), as noted earlier. The serum cholesterol concentrations in LDLR−/− pigs fed the standard chow diet, LDLR−/− pigs fed the HCHF diet, and LDLR−/− pigs fed the HCHF diet with statin are shown in Figure 5A. The serum total, very low‐density lipoprotein, and high‐density lipoprotein cholesterol values differed significantly between groups (P<0.01) and between time points (P<0.01). Specifically, in comparison to the concentrations in LDLR−/− pigs fed the standard chow diet, the concentrations were increased in LDLR−/− pigs fed the HCHF diet. However, the cholesterol concentrations did not decrease in the LDLR−/− pigs fed the HCHF diet plus statin. There were no between‐group differences in serum LDL‐C.
Figure 5.
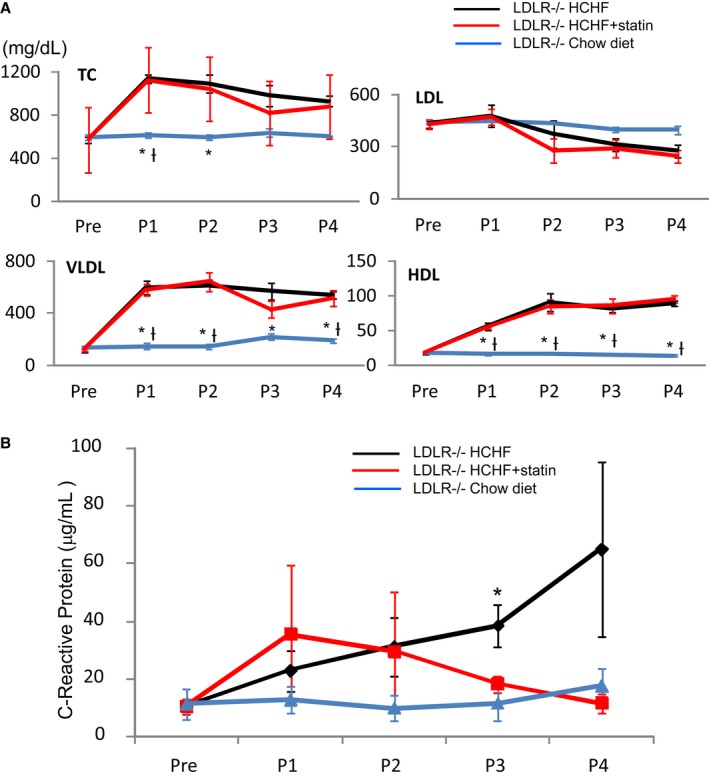
Cholesterol concentrations (mg/dL) and C‐reactive protein concentrations (μg/mL) before and after statin treatment (P1–P4). A, Cholesterol concentrations (mg/dL) before (Pre) and after statin treatment (P1–P4). HDL indicates high‐density lipoprotein; LDL, low‐density lipoprotein; TC, total cholesterol; VLDL, very low‐density lipoprotein. *P<0.05 vs HCHF; † P<0.05 vs HCHF+statin (B) C‐reactive protein concentrations (μg/mL) before (Pre) and after statin treatment (P1–P4). *P<0.05, HCHF vs chow diet. P1 indicates after 1 month; P2, after 2 months; P3, after 3 months; P4, after 4 months.
The CRP concentrations before and every month after statin treatment are shown in Figure 5B. Although there were no significant differences (P=0.09) between the 3 groups and no differences between time points, in comparison to the serum CRP concentrations in LDLR−/− pigs fed the standard chow diet, the serum CRP concentration was significantly increased in LDLR−/− pigs after 3 months of being fed the HCHF diet (HCHF versus standard chow diet: 38±7 versus 12±6 μg/mL, P<0.05). The CRP concentrations were comparable between pigs fed the standard chow diet and those fed the HCHF diet plus statin.
Results of the in vivo iMAP‐IVUS analysis of the coronary arteries after 4 months of statin treatment are shown in Figure 6. Although the lumen volumes were comparable between pigs fed the HCHF diet plus statin and those fed the HCHF diet alone, the plaque volume and vessel volume were significantly reduced in the statin prophylaxis group (Figure 6A and 6B). In terms of plaque composition, the fibrotic, lipidic, and necrotic plaque volumes were also significantly decreased (Figure 6A and 6C).
Figure 6.
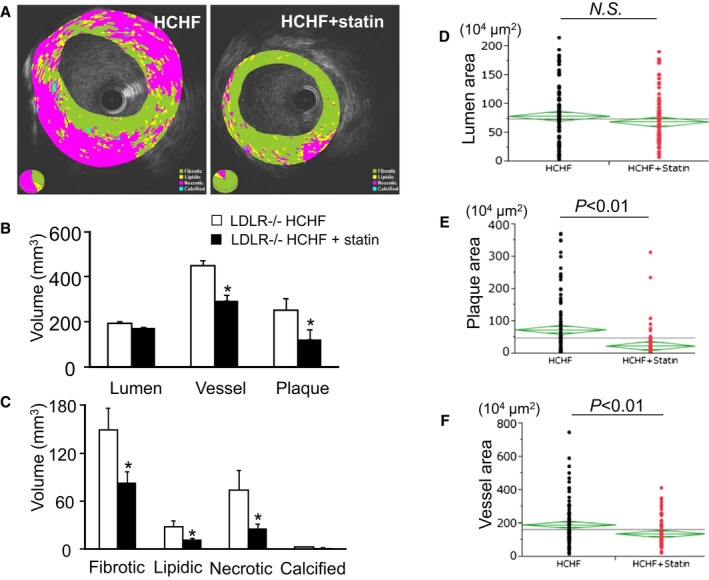
Quantitative iMAP‐IVUS and pathological analysis. A, Representative iMAP‐IVUS images; components include fibrotic (green), lipidic (yellow), necrotic (red), and calcified (blue) tissues. B, Length‐modified lumen volume, vessel volume, and plaque volume; *P<0.05. C, Length‐modified volume of fibrotic, lipidic, necrotic, and calcified components; *P<0.05. Pathological measurements of lumen area (D), plaque area (E), and vessel area (F).
Pathological examination, which was performed for all 1‐cm sections obtained from each coronary artery (HCHF: n=132 sections, HCHF plus statin: n=157 sections), confirmed the reduced vessel and plaque areas seen on IVUS (Figure 6D–6F). Atherosclerotic lesions were confirmed in 80% of the coronary artery sections obtained from LDLR−/− pigs fed the HCHF diet. However, only 67% of sections from pigs fed the HCHF diet plus statin contained atherosclerotic lesions (P<0.01) (Figure 7A).
Figure 7.
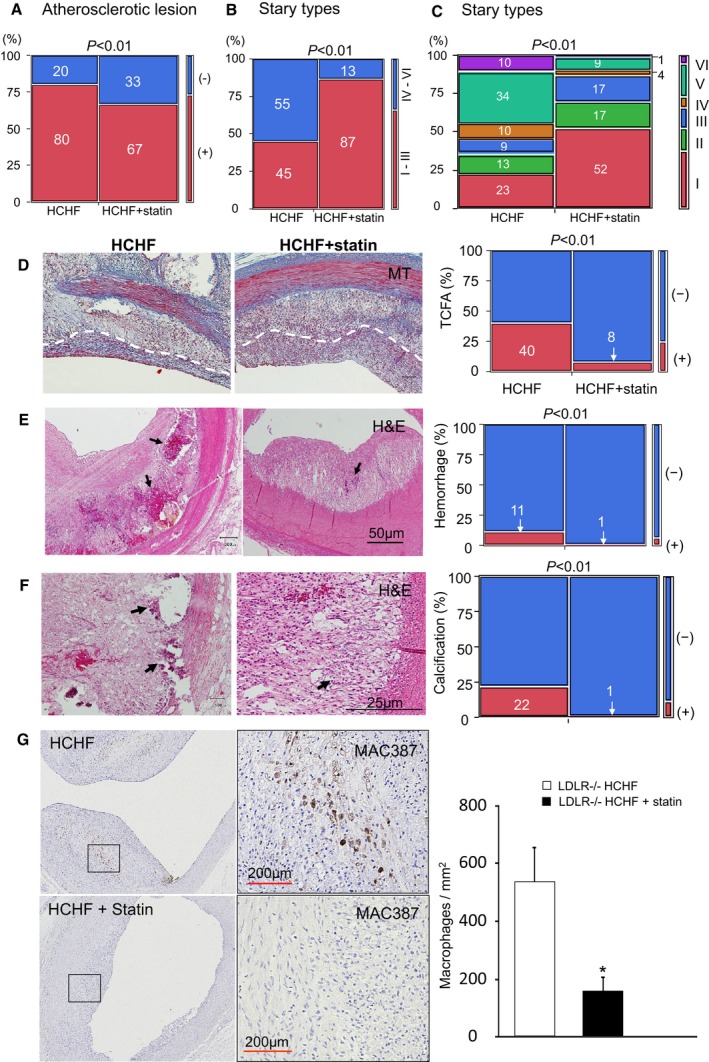
Atherosclerotic lesions, thin‐cap fibroatheroma, intraplaque hemorrhage, calcification, and macrophage invasion. A, Percentage of sections with atherosclerotic lesions. (−) atherosclerotic lesions absent; (+) atherosclerotic lesions present. B, Extent of (%) Stary type I–III lesions and Stary type IV–VI lesions. C, All lesions classified by Stary type and shown as percentages. D, MT‐stained sections; white dashed lines indicate borders of a fibrous cap. E, H&E‐stained sections; black arrows indicate intraplaque hemorrhage. F, H&E‐stained sections, black arrows indicate calcium deposits. G, MAC387 (macrophage) staining of coronary plaque; areas indicated by rectangle in high‐magnification view in right sides; *P<0.05. H&E indicates hematoxylin and eosin; HCHF, high‐cholesterol, high‐fat; MT, Masson's trichrome; IVUS, intravascular ultrasound.
The extent of atherosclerosis also differed significantly between the LDLR−/− pigs fed the HCHF diet and the LDLR−/− pigs fed the HCHF diet plus statin. As shown in Figure 7B, lesions in the statin prophylaxis group were predominantly of types I to III (HCHF versus HCHF plus statin, 45% versus 87%, respectively, P<0.01). Further, most advanced type VI plaques represented only 1% of plaques in this group, versus 10% in the HCHF diet group (P<0.01) (Figure 7C). We also investigated the proportions of sections showing TCFA, intraplaque hemorrhage and calcification, and the density of macrophage invasion, which are considered important factors in plaque instability. In LDLR−/− pigs fed the HCHF diet, 40% of the plaques with a fibrous cap were judged to be TCFAs. However, in LDLR−/− pigs fed the HCHF diet plus statin, only 8% of plaques with a fibrous cap were judged to be TCFAs (P<0.01) (Figure 7D). Similarly, the proportions of intraplaque hemorrhage differed significantly between these 2 groups of pigs (11% versus 1%, respectively; P<0.01) (Figure 7E), as did the proportions of calcification (22% versus 1%, respectively, P<0.01) (Figure 7F). The numbers of MAC387‐labeled macrophages were significantly reduced in the statin prophylaxis group (528±120/mm2 versus 150±49/mm2, respectively; P<0.01) (Figure 7G).
Discussion
We produced hypercholesterolemia‐induced human‐like advanced coronary plaques in a porcine model of atherosclerosis by knocking out the endogenous LDLR gene. Using this LDLR−/− pig, we verified that statin prophylaxis can play a role in preventing advanced plaque development. Although early statin treatment did not reduce the high serum cholesterol concentrations, it appeared to dramatically retard the development of advanced plaques, keeping most atherosclerotic lesions within the early, so‐called stable, stages.
With human‐like coronary arteries and physiology, pigs have been considered promising for development of a large animal model of coronary atherosclerosis. Before gene mutation or genetic recombination in pigs, coronary atherosclerosis was induced in pigs by feeding them an atherogenic diet and sometimes combining the diet with coronary artery balloon injury or by intramural coronary lipid injection.23, 24, 25, 26 However, most of these previous models showed early‐stage atherosclerotic lesions but no human‐like advanced plaques. Recently, techniques such as gene mutation and recombination have been applied to pigs, Rapacz FH pigs and LDLR knockout Yucatan minipigs, for example, and the resulting coronary artery atherosclerotic lesions have been more “human‐like.” In comparison to the Rapacz FH pigs and LDLR knockout Yucatan minipigs, our LDLR−/− pigs offer several advantages. Our LDLR−/− pigs show more advanced atherosclerotic lesions from an earlier age. In being fed standard chow, Rapacz FH pigs and LDLR knockout Yucatan minipigs showed development of atherosclerotic lesions with foam cells by 12 months and not until after age 6 months, respectively,11, 27 whereas our LDLR−/− pigs showed more advanced atherosclerotic lesions with foam cells, mural thrombosis, and calcification as early as 6 months of age. Eleven‐month‐old LDLR knockout Yucatan minipigs, after 6 months on an HCHF diet of as much as 40% fat, 1% cholesterol (amount fed unknown), showed development of coronary artery atherosclerotic lesions with a fibrous cap, hemorrhage, calcification, and foam cells, but without a necrotic core.11 Our 7‐month‐old LDLR−/− pigs, after 4 months on a 15% fat, 1.5% cholesterol HCHF diet (1 kg/d) showed development of more‐complicated human‐like advanced plaques including TCFA, necrotic core, hemorrhage, and calcification. With a body weight of 80 to 95 kg at age 7 months, LDLR−/− pigs are much smaller than Rapacz FH pigs (who weigh ≈200 kg at 1 year) and thus are easier to use in experiments. LDLR knockout Yucatan minipigs are even smaller at 60 to 75 kg, but these pigs are more expensive, and a prolonged experiment with an expensive HCHF diet makes this model cost prohibitive for broad application. Our LDLR−/− pigs can be used for development of human‐like advanced coronary atherosclerotic plaques and, in turn, for translational research, especially for the development of drugs aimed at plaque stabilization and development of percutaneous diagnostic and interventional devices.
Although statin use has proved effective for primary prevention of cardiovascular events,12, 13, 14, 15, 16 the underlying mechanisms are still not well understood because there has been no appropriate analysis. By producing human‐like advanced coronary plaques in our LDLR knockout pigs, we justified, on the basis of histopathologic examination, early statin treatment before the formation of atherosclerotic plaques. Early statin treatment not only reduced the proportion of coronary atherosclerotic lesions that developed in our pigs but also retarded the extent of atherosclerosis development, holding most lesions to the early stage. Specifically, development of advanced plaques with TCFA, intraplaque hemorrhage, calcification, and cellular inflammation was dramatically decreased, suggesting a protective role for early statin treatment against advanced plaque instability. Because thrombotic coronary occlusion and myocardial infarction most frequently evolve from mild to moderate stenoses,28, 29 it is difficult to identify vulnerable plaque before a heart attack, even by angiographic examination. Our animal study verified the effects of early statin administration, before development of atherosclerosis, and provided histological evidence for early statin administration for primary prevention of advanced plaque development.
Absence of the LDL receptor and differences between porcine apolipoprotein B and human apolipoprotein B may explain why the statin treatment did not decrease the LDL‐C concentration in our LDLR−/− pigs. The protective role of statin in pig coronary atherosclerosis may come about via some pleiotropic mechanism rather than via its lipid‐lowering effect. Inflammation plays a crucial role in atherogenesis,13, 30 and the inflammatory biomarker CRP independently predicts vascular events and has been shown to be a stronger predictor of coronary heart disease than is LDL‐C.31, 32, 33 The serum CRP concentration was significantly increased in our LDLR−/− pigs after 3 months of being fed the HCHF diet. However, statin treatment prevented this increase. These results are consistent with previously reported clinical data,15, 34, 35 indicating that statin treatment reduces CRP in a manner that is independent of the LDL‐C level. These results also suggest that the benefit of statin treatment is due in part to the control of serum CRP that is achieved with statin administration. Additionally, our identification of plaque macrophages leads us to believe that the statin treatment has a direct effect on inflammation at the cellular level. The statin treatment significantly reduced macrophage invasion, which is a key contributor to plaque vulnerability.36, 37
Our findings should be considered in light of our study limitations. Our LDLR−/− pigs fed an HCHF diet more closely model familial hypercholesterolemia, which often manifests in early childhood as rapidly progressive cardiovascular disease, than they model the coronary artery disease seen in older patients, in whom most atherosclerotic plaques develop slowly over many years. In addition, we observed development of coronary atherosclerosis in the pigs fed the standard chow diet only until 6 months of age, that is, until they reached puberty. For a model that more closely resembles coronary artery disease in older patients, the pigs should be maintained taking the standard chow diet and observed for 1 year or longer. However, long‐term observation of pigs is difficult because of their rapid increase in size and body weight. Thus, although LDLR−/− miniature pigs are several‐fold more expensive than LDLR−/− domestic pigs, they are more suitable for long‐term observation. For the same reason, we did not observe plaque rupture in this experiment, even though we found several plaques with a papery fibrous cap covering a large necrotic core and thus prone to rupture. Future long‐term serial in vivo observations involving IVUS and optical coherence tomography are needed for us to understand the full spectrum of atherosclerotic plaque development from appearance to rupture. We did not investigate the detailed pleiotropic mechanisms of the effects of statin on coronary artery atherosclerotic lesions. A small animal model, the ApoE−/− mouse, for example, is more suitable for investigating the signaling pathways in detail. Large animal models, such as our LDLR−/− pig model, are suitable for translational research, which bridges the gap between basic research and clinical application.
In summary, results of our large animal experiment provide support for statin treatment before the occurrence of atherosclerosis. Early statin treatment appears to retard development of coronary artery atherosclerosis and ensure lesion stability. In addition, the LDLR−/− pigs we developed represent a large animal model of human‐like advanced coronary plaque suitable for translational research.
Sources of Funding
This work was supported by a medical research grant from Takeda Science Foundation (http://www.takeda-sci.or.jp), a Grant‐in‐Aid from the Ministry of Agriculture, Forestry and Fisheries of Japan, and a research grant from Nihon University School of Medicine.
Disclosures
None.
Acknowledgments
We thank the staff at the Nihon University Medical Research Support Center and at Livestock Research Support Center of NARO Institute of Livestock and Grassland Science for their assistance with animal management. We extend special thanks to Emi Tashiro, Rie Takahashi, Yoshiki Taniguchi, and Misae Suzuki for their excellent technical assistance with the experiments.
(J Am Heart Assoc. 2016;5:e002779 doi: 10.1161/JAHA.115.002779)
References
- 1. Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30:1282–1292. [DOI] [PubMed] [Google Scholar]
- 2. Shiomi M, Ito T. The Watanabe heritable hyperlipidemic (WHHL) rabbit, its characteristics and history of development: a tribute to the late Dr. Yoshio Watanabe. Atherosclerosis. 2009;207:1–7. [DOI] [PubMed] [Google Scholar]
- 3. Rosenfeld ME, Polinsky P, Virmani R, Kauser K, Rubanyi G, Schwartz SM. Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler Thromb Vasc Biol. 2000;20:2587–2592. [DOI] [PubMed] [Google Scholar]
- 4. Adams MR, Nakagomi A, Keech A, Robinson J, McCredie R, Bailey BP, Freedman SB, Celermajer DS. Carotid intima‐media thickness is only weakly correlated with the extent and severity of coronary artery disease. Circulation. 1995;92:2127–2134. [DOI] [PubMed] [Google Scholar]
- 5. Sahni D, Kaur GD, Jit H, Jit I. Anatomy & distribution of coronary arteries in pig in comparison with man. Indian J Med Res. 2008;127:564–570. [PubMed] [Google Scholar]
- 6. Hasler‐Rapacz J, Ellegren H, Fridolfsson AK, Kirkpatrick B, Kirk S, Andersson L, Rapacz J. Identification of a mutation in the low density lipoprotein receptor gene associated with recessive familial hypercholesterolemia in swine. Am J Med Genet. 1998;76:379–386. [PubMed] [Google Scholar]
- 7. Rapacz J, Hasler‐Rapacz J, Taylor KM, Checovich WJ, Attie AD. Lipoprotein mutations in pigs are associated with elevated plasma cholesterol and atherosclerosis. Science. 1986;234:1573–1577. [DOI] [PubMed] [Google Scholar]
- 8. Grunwald KA, Schueler K, Uelmen PJ, Lipton BA, Kaiser M, Buhman K, Attie AD. Identification of a novel Arg–>Cys mutation in the LDL receptor that contributes to spontaneous hypercholesterolemia in pigs. J Lipid Res. 1999;40:475–485. [PubMed] [Google Scholar]
- 9. Prescott MF, McBride CH, Hasler‐Rapacz J, Von Linden J, Rapacz J. Development of complex atherosclerotic lesions in pigs with inherited hyper‐LDL cholesterolemia bearing mutant alleles for apolipoprotein B. Am J Pathol. 1991;139:139–147. [PMC free article] [PubMed] [Google Scholar]
- 10. Thim T, Hagensen MK, Drouet L, Bal Dit Sollier C, Bonneau M, Granada JF, Nielsen LB, Paaske WP, Botker HE, Falk E. Familial hypercholesterolaemic downsized pig with human‐like coronary atherosclerosis: a model for preclinical studies. EuroIntervention. 2010;6:261–268. [DOI] [PubMed] [Google Scholar]
- 11. Davis BT, Wang XJ, Rohret JA, Struzynski JT, Merricks EP, Bellinger DA, Rohret FA, Nichols TC, Rogers CS. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS One. 2014;9:e93457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, Heeringa J, Witteman JC, Lansberg PJ, Kastelein JJ, Sijbrands EJ. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ridker PM. The JUPITER trial: results, controversies, and implications for prevention. Circ Cardiovasc Qual Outcomes. 2009;2:279–285. [DOI] [PubMed] [Google Scholar]
- 14. Mihaylova B, Emberson J, Blackwell L, Keech A, Simes J, Barnes EH, Voysey M, Gray A, Collins R, Baigent C. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta‐analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, Nordestgaard BG, Shepherd J, Willerson JT, Glynn RJ. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 16. Raal FJ, Pilcher GJ, Panz VR, van Deventer HE, Brice BC, Blom DJ, Marais AD. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid‐lowering therapy. Circulation. 2011;124:2202–2207. [DOI] [PubMed] [Google Scholar]
- 17. Onishi A, Iwamoto M, Akita T, Mikawa S, Takeda K, Awata T, Hanada H, Perry AC. Pig cloning by microinjection of fetal fibroblast nuclei. Science. 2000;289:1188–1190. [DOI] [PubMed] [Google Scholar]
- 18. Kikuchi K, Onishi A, Kashiwazaki N, Iwamoto M, Noguchi J, Kaneko H, Akita T, Nagai T. Successful piglet production after transfer of blastocysts produced by a modified in vitro system. Biol Reprod. 2002;66:1033–1041. [DOI] [PubMed] [Google Scholar]
- 19. Usui S, Hara Y, Hosaki S, Okazaki M. A new on‐line dual enzymatic method for simultaneous quantification of cholesterol and triglycerides in lipoproteins by HPLC. J Lipid Res. 2002;43:805–814. [PubMed] [Google Scholar]
- 20. Sathyanarayana S, Carlier S, Li W, Thomas L. Characterisation of atherosclerotic plaque by spectral similarity of radiofrequency intravascular ultrasound signals. EuroIntervention. 2009;5:133–139. [DOI] [PubMed] [Google Scholar]
- 21. Narula J, Finn AV, Demaria AN. Picking plaques that pop. J Am Coll Cardiol. 2005;45:1970–1973. [DOI] [PubMed] [Google Scholar]
- 22. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr, Rosenfeld ME, Schwartz CJ, Wagner WD, Wissler RW. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–1374. [DOI] [PubMed] [Google Scholar]
- 23. Griggs TR, Bauman RW, Reddick RL, Read MS, Koch GG, Lamb MA. Development of coronary atherosclerosis in swine with severe hypercholesterolemia. Lack of influence of von Willebrand factor or acute intimal injury. Arteriosclerosis. 1986;6:155–165. [DOI] [PubMed] [Google Scholar]
- 24. Holvoet P, Theilmeier G, Shivalkar B, Flameng W, Collen D. LDL hypercholesterolemia is associated with accumulation of oxidized LDL, atherosclerotic plaque growth, and compensatory vessel enlargement in coronary arteries of miniature pigs. Arterioscler Thromb Vasc Biol. 1998;18:415–422. [DOI] [PubMed] [Google Scholar]
- 25. Turk JR, Henderson KK, Vanvickle GD, Watkins J, Laughlin MH. Arterial endothelial function in a porcine model of early stage atherosclerotic vascular disease. Int J Exp Pathol. 2005;86:335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tellez A, Schuster DS, Alviar C, Lopez‐Berenstein G, Sanguino A, Ballantyne C, Perrard XY, Schulz DG, Rousselle S, Kaluza GL, Granada JF. Intramural coronary lipid injection induces atheromatous lesions expressing proinflammatory chemokines: implications for the development of a porcine model of atherosclerosis. Cardiovasc Revasc Med. 2011;12:304–311. [DOI] [PubMed] [Google Scholar]
- 27. Granada JF, Kaluza GL, Wilensky RL, Biedermann BC, Schwartz RS, Falk E. Porcine models of coronary atherosclerosis and vulnerable plaque for imaging and interventional research. EuroIntervention. 2009;5:140–148. [DOI] [PubMed] [Google Scholar]
- 28. Ambrose JA, Tannenbaum MA, Alexopoulos D, Hjemdahl‐Monsen CE, Leavy J, Weiss M, Borrico S, Gorlin R, Fuster V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol. 1988;12:56–62. [DOI] [PubMed] [Google Scholar]
- 29. Falk E, Shah PK, Fuster V. Coronary plaque disruption. Circulation. 1995;92:657–671. [DOI] [PubMed] [Google Scholar]
- 30. Hansson GK, Libby P. The immune response in atherosclerosis: a double‐edged sword. Nat Rev Immunol. 2006;6:508–519. [DOI] [PubMed] [Google Scholar]
- 31. Ridker PM, Hennekens CH, Buring JE, Rifai N. C‐reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–843. [DOI] [PubMed] [Google Scholar]
- 32. Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GD, Pepys MB, Gudnason V. C‐reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–1397. [DOI] [PubMed] [Google Scholar]
- 33. Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C‐reactive protein and low‐density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;347:1557–1565. [DOI] [PubMed] [Google Scholar]
- 34. Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E. Long‐term effects of pravastatin on plasma concentration of C‐reactive protein. The Cholesterol and Recurrent Events (CARE) Investigators. Circulation. 1999;100:230–235. [DOI] [PubMed] [Google Scholar]
- 35. Albert MA, Danielson E, Rifai N, Ridker PM. Effect of statin therapy on C‐reactive protein levels: the pravastatin inflammation/CRP evaluation (PRINCE): a randomized trial and cohort study. JAMA. 2001;286:64–70. [DOI] [PubMed] [Google Scholar]
- 36. Silvestre‐Roig C, de Winther MP, Weber C, Daemen MJ, Lutgens E, Soehnlein O. Atherosclerotic plaque destabilization: mechanisms, models, and therapeutic strategies. Circ Res. 2014;114:214–226. [DOI] [PubMed] [Google Scholar]
- 37. Seneviratne A, Hulsmans M, Holvoet P, Monaco C. Biomechanical factors and macrophages in plaque stability. Cardiovasc Res. 2013;99:284–293. [DOI] [PubMed] [Google Scholar]