

Abstract
Purpose
The purpose of this study is to determine whether luminacin, a marine microbial extract from the Streptomyces species, has anti-tumor effects on head and neck squamous cell carcinoma (HNSCC) cell lines via autophagic cell death.
Materials and Methods
Inhibition of cell survival and increased cell death was measured using cell viability, colony forming, and apoptosis assays. Migration and invasion abilities of head and cancer cells were evaluated using wound healing, scattering, and invasion assays. Changes in the signal pathway related to autophagic cell death were investigated. Drug toxicity of luminacin was examined in in vitro HaCaT cells and an in vivo zebrafish model.
Results
Luminacin showed potent cytotoxicity in HNSCC cells in cell viability, colony forming, and fluorescence-activated cell sorting analysis. In vitro migration and invasion of HNSCC cells were attenuated by luminacin treatment. Combined with Beclin-1 and LC3B, Luminacin induced autophagic cell death in head and neck cancer cells. In addition, in a zebrafish model and human keratinocyte cell line used for toxicity testing, luminacin treatment with a cytotoxic concentration to HNSCC cells did not cause toxicity.
Conclusion
Taken together, these results demonstrate that luminacin induces the inhibition of growth and cancer progression via autophagic cell death in HNSCC cell lines, indicating a possible alternative chemotherapeutic approach for treatment of HNSCC.
Keywords: Luminacin, Head and neck neoplasms, Autophagy, Streptomyces, Cytotoxicity
Introduction
Cancer is a major cause of death, and head and neck squamous cell carcinoma (HNSCC) ranks seventh in annual incidence worldwide [1]. Treatment of HNSCC involves surgery, external beam radiation, or platinum-based chemotherapy. In cases of locally advanced HNSCC, these three modalities should be administered concurrently or sequentially. Despite intensive treatments, over 50% of patients with advanced HNSCC eventually experienced locoregional or distant failure within 2 years [2]. Despite a number of technical advances in chemotherapeutics, including targeted therapy for epidermal growth factor receptor, the survival rates of HNSCC have not improved significantly [2]. Therefore, efforts to develop alternative anti-cancer agents should be continued. The limited efficacy of current synthetic chemotherapeutics has necessitated the search for and evaluation of novel bioactive drugs, particularly from natural compounds.
Compounds derived from natural sources have played an important role in the development of novel anticancer therapeutics. From the 1940s to 2010, of a total of 206 approved anti-cancer drugs, 112 naturally-derived anti-cancer drugs were discovered, purified, and finally approved [3]. The ocean is one of the best sources of bioactive natural products. To date, over 22,000 marine natural compounds have been identified and reported in the literature [4]. Some of these compounds showed considerable tumor suppressing activity and are currently being investigated in clinical trials or used as lead compounds for development of new anti-cancer drugs. We hypothesized that luminacin, a metabolite from the marine Streptomyces species, is a promising alternative therapeutic agent for HNSCC. In this investigation, we determined whether luminacin has anti-proliferative and anti-progression effects on HNSCC cell lines, particularly via the autophagic cell death pathway.
Materials and Methods
1. Cytotoxic effects on zebrafish
Freshly fertilized embryos (6-hour post-fertilization) were treated with luminacin (0, 0.1, or 1 μg/mL) (Fig. 1A). The hatching rate of embryos was assessed visually by light microscopy at 24-hour intervals up to 3-day post-fertilization (dpf). Mortality was determined by the lack of a heartbeat, coagulation of the embryos, a non-detached tail, and failure to develop somites. The morphology was assessed visually using an Axiovert 200 light transmission microscope (Carl Zeiss, Göttingen, Germany) at a magnification of 60-100×. Hair cell lateral line neuromasts were labeled using 2 μM YO-PRO1 (Molecular Probes, Eugene, OR) for 30 minutes. The zebrafish were rinsed three times (5 minutes per wash) in embryo medium and anesthetized with 8 μg/mL 3-aminobenzoic acid ethyl ester methanesulfonate salt (MS-222, Sigma-Aldrich, St. Louis, MO), and then mounted with methylcellulose on a depression slide for observation under a fluorescence microscope.
Fig. 1.
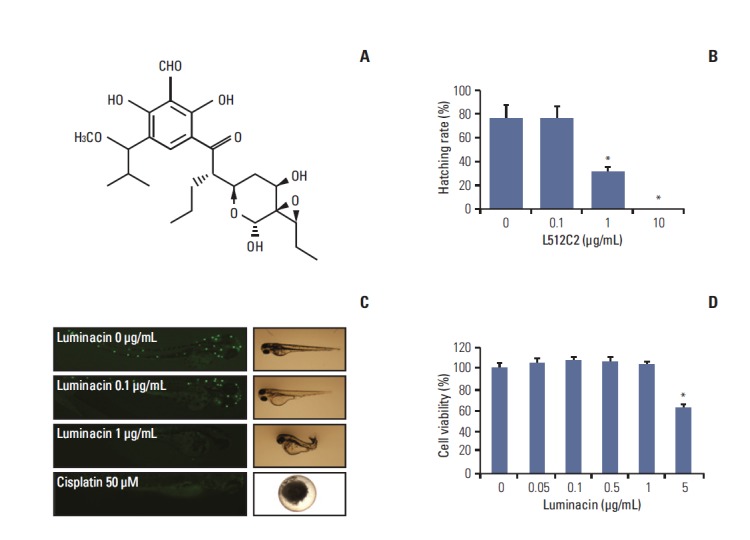
In vivo and in vitro toxicity of luminacin tested on zebrafish embryos and HaCaT cells. (A) Structure of luminacin. Embryos were exposed to luminacin at 6-hour post-fertilization. (B) Hatching rate of zebrafish embryos. (C) Staining of neuromasts in zebrafish embryos with YO-PRO1 after treatment with luminacin. Neuromasts were stained as white dots. Treatment with 0.1 μg/mL luminacin did not decrease the number of neuromasts. (D) HaCaT cells were exposed to various concentrations of luminacin (0-5 μg/mL). Cell viability was measured by MTT assay. Luminacin decreased viability of HaCaT cells only at a concentration of 5 μg/mL. Data represent the mean±standard deviation of three independent experiments. *p < 0.05.
2. Cell lines
Seven established human HNSCC cell lines—SCCQLL1, SCC15, SCC25, SCC1483, MSKQLL1, HN6 (oral cancer cell lines), HNE1 (nasopharyngeal cancer cell line), and human keratinocytes (HaCaT) cell lines were obtained from American Type Culture Collection (Manassas, VA) and Korean Cell Line Bank (Seoul, Korea). The cells were grown in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and penicillin-streptomycin at 100 U/mL (Gibco, Carlsbad, CA) at 37°C in a humidified atmosphere with 5% CO2 and 95% air. Luminacin was dissolved in autoclaved water as a stock solution for in vitro studies.
3. Cell viability assay
To determine cell viability, the HNSCC cell lines and HaCaT cells were seeded onto 96-well plates at densities of 5×103 cells/well in 1 mL complete medium with various concentrations of luminacin (0-50 μg/mL). MTT, also known as 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (Sigma-Aldrich), was added to 40 μL of the cell suspension for 4 hours. After three washes with phosphate buffered saline (PBS, pH 7.4), the insoluble formazan product was dissolved in dimethyl sulfoxide (DMSO). The optical density of each culture well was measured using a microplate reader (Bio-Tek, Winooski, VT) at 540 nm.
4. Colony forming assay
Colony forming assays were performed as previously described [5]. To determine long-term effects, HNE1 cells were untreated or treated with 30 ng/mL hepatocyte growth factor (HGF) after treatment with luminacin (0, 0.1, or 1 μg/mL) in 24-well plates. After rinsing with fresh medium, cells were allowed to grow for 3 days to form colonies, which were stained with 4% crystal violet (Sigma-Aldrich). More than 2 mm of the cells were counted.
5. Wound healing assay
Cell migration ability was measured using the wound healing assay as previously described [5]. Briefly, cells were grown to confluent monolayers, which were wounded by scratching the surface, as uniformly as possible, with a 1-mL pipette tip. MSKQLL1 cells were pre-treated with HGF (0, 1, or 10 ng/mL) followed by treatment with luminacin (0, 0.05, 0.1, or 0.5 μg/mL). Images of the wound area were captured on day 0 (day of scratching) and 12 hours after scratching using an Olympus SC 35 camera (Tokyo, Japan) connected to an inverted microscope.
6. Scattering assay
MSKQLL1 cells were seeded in 24-well tissue culture dishes at a density of 1×105 per well and incubated under serum deprivation conditions for 24 hours. The cells were then cultured with or without luminacin (0, 0.05, or 0.1 μg/mL) and HGF (10 ng/mL). After 24 hours, the cells were stained with 0.1% crystal violet solution (dissolved in 20% methanol). The number of cell colonies, their sizes, and the degree of scattering were observed, and representative images were captured using light microscopy.
7. Invasion assay
The invasion assay was performed as previously described [6] using 24-well transwell filters with an 8-μm pore size and coated collagen in filters. MSKQLL1 cells (2×104) in the upper chamber were pretreated with luminacin (0, 0.05, or 0.1 μg/mL) and then with or without 10 ng/mL HGF. Both inserts and lower wells were treated with vehicle control (DMSO), luminacin, and HGF. The chambers were incubated for 16 hours at 37°C in an atmosphere containing 5% CO2. After 16 hours, the cells in the insert were gently removed using a cotton swab. Cells on the lower surface of the filter were fixed and stained using hematoxyin and eosin staining solutions. The number of invading cells was counted in four representative fields per membrane using light microscopy at 40× magnification.
8. Western blot assay
Cells were pre-treated with HGF (30 ng) or PBS for 10 mintues, and then washed twice with cold PBS, and subsequently treated with luminacin (0, 1, 5, or 10 μg/mL). Total proteins were extracted using the ProteoExtract Subcellular Proteome Extraction Kit (Calbiochem, La Jolla, CA) following the manufacturer's instructions. Protein concentrations were measured using the BCA assay (Pierce, Rockford, IL). The proteins were separated by electrophoresis on 12% and 10% sodium dodecyl sulfate polyacrylamide gels. An equal amount of protein (10 μg) was loaded in each lane. After electrophoresis, the proteins were transferred onto polyvinylidene difluoride membranes. The membrane was blocked in Tris-buffered saline Tween-20 (TBST) containing 5% non-fat milk for 1 hour, followed by incubation overnight at 4°C with primary antibodies. All primary antibodies were purchased from Cell Signaling Technology (Danvers, MA). After washing the membrane extensively, incubation with horseradish peroxidase-conjugated secondary antibody (1:1,000, Cell Signaling Technology) was performed for 1 hour at room temperature. Protein bands on the blots were visualized using ECL Plus Western Blot detection reagents (Amersham Pharmacia Biotech, Piscataway, NJ).
9. Annexin V–fluorescein isothiocynate (FITC)/propidium iodide (PI) double staining
Quantitative analysis of apoptotic cell death caused by Luminacin was performed using the FITC Annexin V Apoptosis Detection kit II (Becton Dickinson, Franklin Lakes, NJ), following the manufacturer’s protocols. Briefly, cells were plated at 1×106 cells/well in a 6-well plate, incubated for 16 hours, and then treated with luminacin (0, 1, 2.5, 5, 10, or 20 μM) for 24 hours. The cells were harvested, washed with cold PBS, and subjected to Annexin V–FITC and PI staining in binding buffer at room temperature for 10 minutes in the dark. The stained cells were analyzed by fluorescence-activated cell sorting (FACS ARIA3, BD Biosciences, San Jose, CA) using WinMDI 2.9 software (Purdue University Cytometry Laboratories, West Lafayette, IN).
10. Immunocytochemistry
SCC15 cells (5×104) were incubated on a coverslip in a 12-well plate followed by treatment with 10 μg/mL luminacin for 24 hours. Cells were washed twice with PBS, and then incubated with 4% paraformaldehyde for 20 minutes at room temperature and treated with 0.075% Triton X-100 for 5 minutes to allow the antibody to permeabilize the cell membranes. After washing twice with PBS and blocking with 2% bovine serum albumin/PBS for 30 minutes, the slides were incubated with primary antibody, anti-LC3B I/II antibody (1:100, Cell Signaling Technology), overnight at 4°C. The slides were then washed three times with PBS and incubated with secondary antibody (Alexa Flouor 488, goat anti-rabbit IgG, Invitrogen, Grand Island, NY) for 30 minutes at room temperature. Digital images of stained cells were captured with the confocal microscope A1R-A1 (Nikon, Tokyo, Japan) and randomly selected.
11. Statistical analyses
All values were expressed as mean±standard deviation and statistical analysis was performed using the Kruskall-Wallis test and the Mann-Whitney U test (ver. 17, SPSS Inc., Chicago, IL). A p-value of < 0.05 was considered statistically significant.
Results
1. Luminacin-induced in vivo and in vitro toxicity in zebrafish embryos and HaCaT cells
To determine in vivo toxicity of luminacin, zebrafish embryos at 6 hours post-fertilization were exposed to luminacin (0, 0.1, or 1 μg/mL) and the effects of this treatment on their morphologic appearance and survival up to 3 dpf of development were measured. Luminacin decreased hatching rates of the zebrafish embryos at 1 μg/mL concentration (p < 0.05) (Fig. 1B). The distribution of neuromasts in live 3 dpf zebrafish, as detected by staining with YO-PRO1, is shown in Fig. 1C. Luminacin exposure did not result in a significant loss of YO-PRO1 staining in neuromasts and few morphological changes were observed in zebrafish embryos treated with 0.1 μg/mL luminacin; however, a 1 μg/mL concentration of luminacin caused total loss of neuromasts and definite morphological changes, including shortened body axis and reduced pigmentation (Fig. 1C). In cisplatin (50 μM) treated zebrafish, total loss of neuromasts was detected (Fig. 1C). As shown in Fig. 1D, luminacin treatment did not decrease the viability of HaCaT cells, except in high concentration (5 μg/mL).
2. Luminacin inhibited cell growth in various head and neck cancer cells
As shown in Fig. 2, treatment with luminacin resulted in decreased viability of various HNSCC cells. In examining the effects of different concentrations of luminacin on HNSCC cells, we found that luminacin significantly inhibited HNSCC cell growth in a dose-dependent manner (Fig. 2). In SCC15, HN6, and MSKQLL1 cells, 1 μg/mL luminacin showed a statistically significant cytotoxic effect (p < 0.001).
Fig. 2.
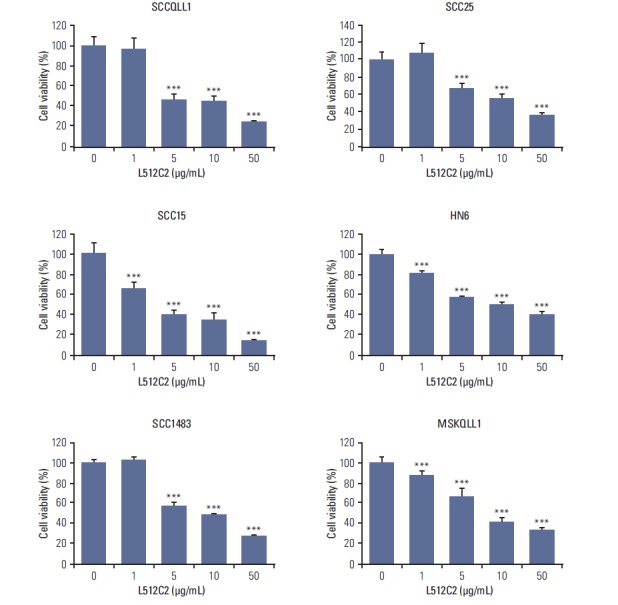
Effects of luminacin on proliferation in various head and neck carcinoma cell lines. Results of the cell proliferation assay. Head and neck cancer cells were exposed to various concentrations of luminacin (0-50 μg/mL). At 5 days after treatment, cell viability was measured by MTT assay. Luminacin significantly inhibited the proliferation of head and neck carcinoma cell lines. Data represent the mean±standard deviation of three independent experiments. ***p < 0.001 compared to control.
3. Luminacin decreased viability, migration, and invasion capability induced by HGF in head and neck cancer cells
HGF is widely accepted as a potential enhancer of invasive growth in HNSCC. A colony-forming assay was performed for analysis of the suppressive effect of luminacin on the survival of the HNSCC cell lines. As shown in Fig. 3A, pre-treatment with luminacin resulted in complete suppression of the survival of HNSCC cells not treated with HGF. In addition, the survival rate of cells co-treated with HGF and luminacin was significantly decreased (Fig. 3A). The wound healing assay was used to determine the migration capability of HNSCC cells. HGF enhanced the proliferation and migration abilities of the cells; this enhancement was successfully inhibited by luminacin treatment (Fig. 3B). The degree of scattering in HNSCC cells treated with or without HGF/luminacin was observed. HGF-treated cells showed increased scattering behavior, while HGF/luminacin co-treated cells showed a discernible reduction in HGF-induced cell scattering (Fig. 3C). The effect of luminacin on HNSCC cell invasion was further addressed using transwells, and cotreatment with luminacin and HGF significantly inhibited cell invasion compared to treatment with HGF alone (Fig. 3D), indicating that luminacin inhibited HGF-induced HNSCC cellular migration and invasion.
Fig. 3.
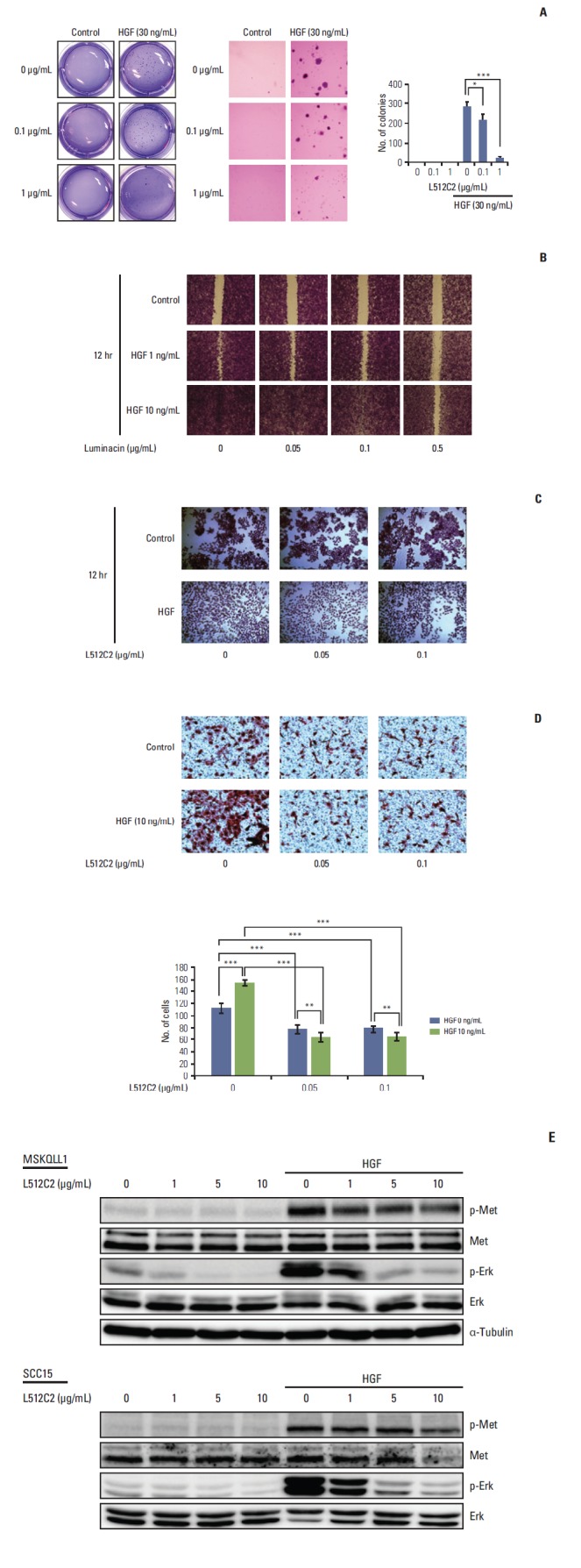
Effect of luminacin on hepatocyte growth factor (HGF)–induced viability, migration, and invasion capability in head and neck carcinoma cells. Investigation of cell viability, migration, and invasion capability after HGF/luminacin treatment using colony forming, wound healing, cell scattering, and invasion assays. (A) HNE1 cells were treated or untreated with 30 ng/mL HGF and luminacin (0, 0.1, or 1 μg/mL) and incubated for 3 days to form colonies. After 4% crystal violet staining, more than 2 mm of the colonies were counted. Luminacin treatment significantly inhibited the survival rates of cells treated with HGF. (B) Confluent monolayers of MSKQLL1 cells were wounded by scratching the surface as uniformly as possible with a 1-mL pipette tip. Cells were treated or untreated with 10 ng/mL HGF and luminacin (0, 0.05, 0.1, or 0.5 μg/mL), and then cultivated for another 12 hours. Percent closure of wound areas was measured. Luminacin treatment resulted in significant dose-dependent inhibition of HGF-induced enhancement of cell proliferation and migration. (C) MSKQLL1 cells were cultured with or without luminacin (0, 0.05, or 0.1 μg/mL) and HGF (10 ng/mL). After 24 hours, the cells were stained in a 0.1% crystal violet solution. The number of cell colonies, their sizes, and the degree of scattering were observed. HGF/luminacin co-treated cells showed a discernible reduction in HGF-induced cell scattering. (D) MSKQLL1 cells (2×104) in the upper chamber were pretreated with luminacin (0, 0.05, or 0.1 μg/mL) and then with or without 10 ng/mL HGF. After 16 hours, the number of invading cells was counted in four representative fields per membrane. Co-treatment with luminacin and HGF significantly inhibited cell invasion compared to treatment with HGF alone. (E) Immunoblot of luminacin/HGF treated cells stained with antibodies against Met, p-Met, Erk, and p-Erk. Luminacin inhibited HGF-induced enhancement of phosphorylation of p-Met and the downstream target, p-Erk. Data represent the mean±standard deviation of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we determined whether HGF-induced changes at the protein level were restored by luminacin. A representative western blot is shown in Fig. 3E; the results confirmed increased protein levels or phosphorylation of p-Met and p-Erk after HGF treatment. Luminacin inhibited the HGF-induced increase in phosphorylation of p-Met and the downstream target, p-Erk (Fig. 3E). These results suggest that the cytotoxic effect and the inhibited migration and invasion by luminacin may correlate with reduced phosphorylation of Met.
4. Luminacin induced cytotoxicity through autophagic cell death
To determine whether luminacin induced apoptosis in HNSCC cells, cells were treated with luminacin (0, 1, 2.5, 5, 10, and 20 μg/mL) before analysis of annexin/PI fluorescence-activated cell sorting apoptosis. As shown in Fig. 4, the percentages of apoptotic cells in luminacin-treated groups did not exceed 15% in SCC15 or 40% in MSKQLL1 cells, even when treated with 20 μg/mL luminacin. Our results demonstrated that luminacin-induced cell death was not caused primarily by apoptosis or necrosis, but another cell death mechanism.
Fig. 4.
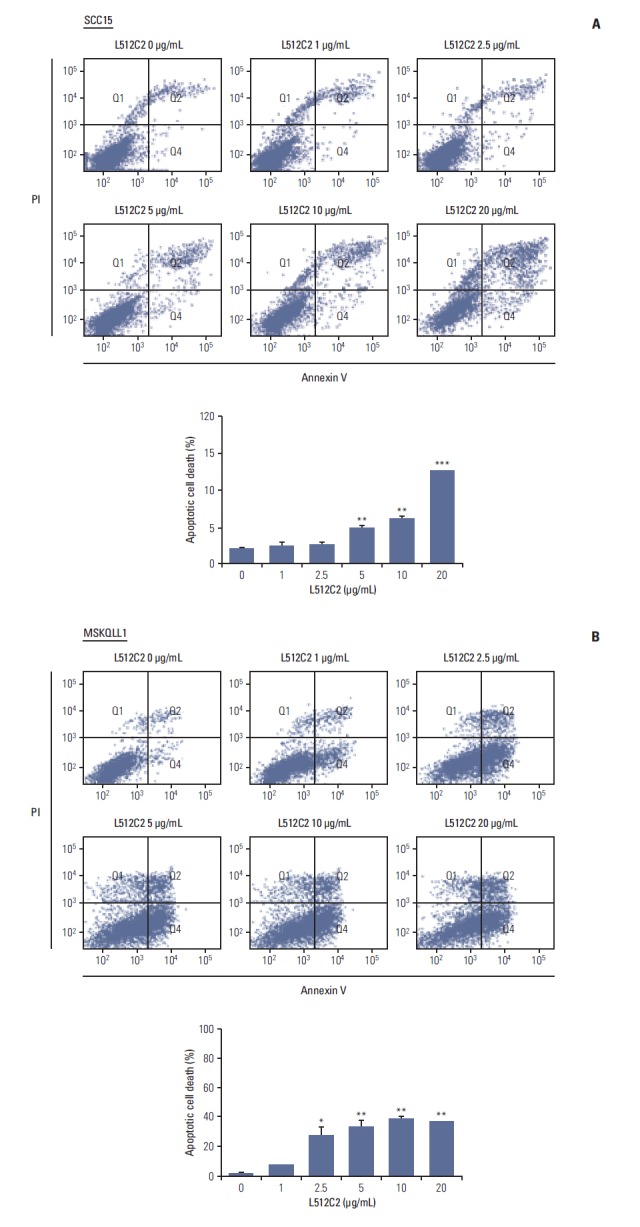
Apoptotic cell death induced by luminacin treatment in head and neck cancer cells. Cells were treated with luminacin (0-20 μg/mL). Flow cytometry was used for quantification of luminacin-induced apoptosis: annexin V–fluorescein isothiocynate (FITC) and propidium iodide (PI) staining were used for analysis of the percentage of apoptotic cells treated with luminacin. The percentages of apoptotic cells in luminacin-treated groups did not exceed 15% in SCC15 cells (A) and 40% in MSCQLL1 cells (B), even when treated with 20 μg/mL luminacin. Data represent the mean±standard deviation of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we determined whether the cell death induced by luminacin treatment was mediated mainly through autophagy. As determined by western blot analysis, luminacin increased the expression of Beclin-1 and LC3B I/II, proteins required for autophagosome formation (Fig. 5A). In addition, confocal microscopy showed that luminacin-treated cells expressed LC3B, indicating that cells underwent autophagic cell death (Fig. 5B). Next we performed western blot analysis to determine whether Akt and mitogen-activated protein kinase (MAPK) signaling, which contribute to autophagy, were blocked by luminacin. As shown in Fig. 5C, luminacin treatment resulted in increased levels of p-JNK and p-p38 protein, subsequently resulting in decreased phosphorylation of p-Akt. These results suggest that the mechanism by which luminacin induces autophagic cell death may involve the Akt and MAPK signaling pathways.
Fig. 5.
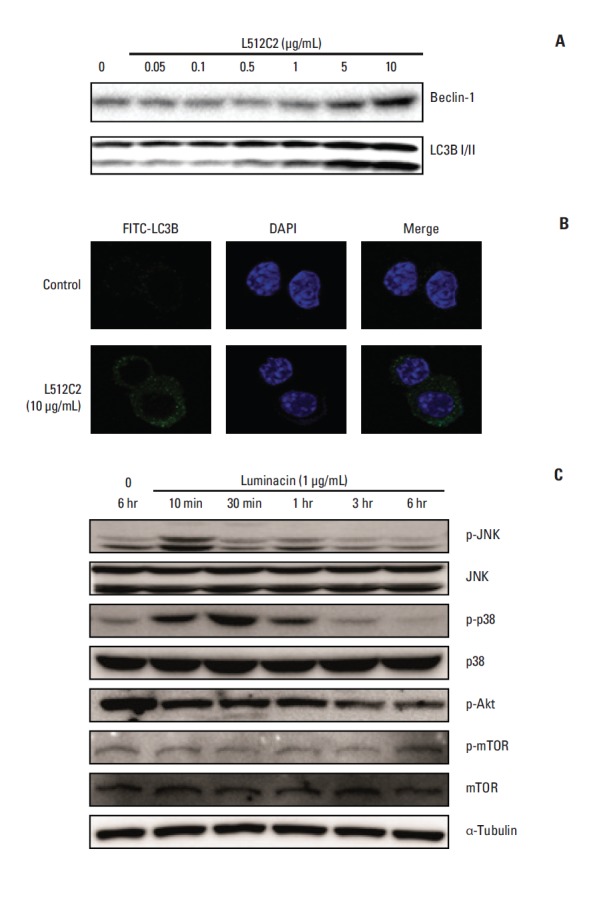
Autophagic cell death in head and neck cancer cells induced by luminacin treatment. (A) Cell lysates were collected, electrophoresed through an sodium dodecyl sulfate–polyacrylamide gel, and subjected to immunoblot analysis with antibodies against Beclin-1 and LC3B I/II. Luminacin increased the expression of Beclin-1 and LC3B I/II, which are known as autophagosome formation proteins. (B) Immunocytochemistry of LC3B was performed after treatment with luminacin (10 μg/mL) for 24 hours. Results from confocal microscopy showed that luminacin-treated cells expressed LC3B, indicating that cells underwent autophagic cell death process. (C) Representative western blot analysis of Akt and mitogen-activated protein kinase signaling indicated involvement of luminacin-induced cell death in these signaling pathways.
For measurement of autophagic flux, monitoring of LC3 turnover was performed by the observation of LC3-II degradation in autolysosomes. Treatment with a lysosomal inhibitor, NH4Cl (20 mM), resulted in increased LC3-II, and treatment with luminacin (10 μg/mL) further increased LC3-II levels (Fig. 6A and B).
Fig. 6.
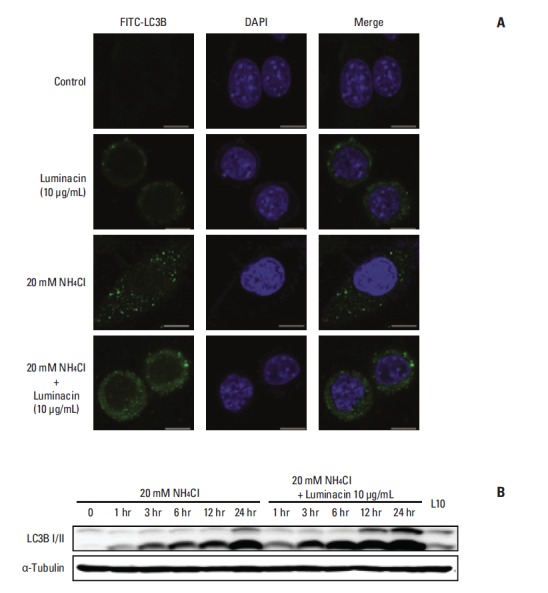
Autophagic flux assay using a lysosomal inhibitor, NH4Cl. Representative immunocytochemistry (A) and western blot analysis (B) with antibodies against LC3B I/II after treatment with luminacin with/without NH4Cl.
Discussion
Nature-derived products have infinite potential for use in biomedical and pharmaceutical applications, thus they can also be applied to diverse biotechnological indications [7-9]. The biomedical and pharmaceutical application of a natural substance is related to its anti-viral, anti-tumor, anti-microbial, and general cytotoxic properties [10]. Natural product drugs might have advantages of shorter development periods and lower research costs compared with completely synthetic compounds [4]. From 1981 to 2010, over 50% of new chemical entities were developed from natural substances, either natural products themselves or natural products that were modified in one way or another [3]. In the anti-cancer drug category, only 20.2% of anti-cancer drugs were developed using completely synthetic methods.
As sources of novel bioactive substances, several marine microbes have come into the spotlight in the field of drug development. One promising group of microbes belongs to the genus Streptomyces, members of the Actinomycetes, which are known to produce several antibiotics and bioactive compounds [11,12]. Some Streptomyces have biosynthetic gene groups that produce over 30 secondary metabolites. Thus, only a single Streptomyces strain can be a source of more than 30 bioactive substances [13]. luminacin, one of the metabolites produced by Streptomyces, was originally found in the course of screening for brand-new angiogenesis inhibitors. Luminacins are composed of 14 components with similar structures, A1, A2, B1, B2, C1, C2, D, E1, E2, E3, F, G1, G2, and H [12]. Because luminacin C2 was available in the largest quantity and had the strongest bioactive potency, investigators mainly focused on this component. Several subsequent studies have demonstrated the biological efficacy of luminacin, however little has been revealed to date, particularly regarding the anti-cancer capability of this component. Atatreh et al. [14] reported that luminacin suppresses nontyrosine kinase interactions that play a role in growth factor signal transduction and cell adhesion in human colorectal carcinoma cells. We hypothesized that luminacin might be a promising alternative anti-cancer molecule for HNSCC. The aim of the current study was to determine the safety and effectiveness of luminacin on proliferation, migration, and invasion of HNSCC cell lines in vitro and in vivo in a zebrafish model.
In this study, luminacin showed a cytotoxic effect on the survival of HNSCC cell lines. In terms of drug toxicity, luminacin showed toxicity on survival and neural development of zebrafish embryos in a toxic concentration to HNSCC cell lines, but was not more toxic compared to a conventional platinum-based chemotherapeutic, cisplatin. Luminacininduced cell death was detected by cell viability assay in a number of HNSCC cell lines. Luminacin successfully inhibited HGF-induced proliferation, migration, and invasion of HNSCC cell lines. HGF-induced activation of Met and downstream Erk were also blocked by luminacin treatment. Regarding the type of cell death, luminacin increased Beclin-1 and LC3B expression, indicating involvement of autophagic cell death. The JNK, p38 MAPK, and Akt pathways were involved in the cell death mechanism induced by luminacin treatment. These findings suggest that anti-tumor effects of luminacin on HNSCC cell lines might be achieved via autophagic cell death.
Autophagy is an important type of cell death, a cellular degradation process in response to stressed conditions. However, the role of autophagic cell death in cancer development, progression, or treatment is still unclear and controversial. Autophagy might operate as a protective mechanism that inhibits uncontrolled cell proliferation [15]. Supporting this hypothesis, some cancers respond to agents that trigger autophagy, indicating the possibility of anti-cancer therapy that targets autophagic cell death induction. Several anti-cancer drugs, including 5-fluorouracil, rapamycin, and tamoxifen are known to induce autophagic cell death [16]. Another point of view in cancer biology is that autophagy is a prosurvival mechanism. Growing evidence indicates that autophagy enables cancer cell survival under stressed conditions induced by hypoxia, starvation, or cancer treatment [17]. Inhibition of autophagy facilitates the anti-cancer efficacy of chemotherapeutic agents including oxaliplatin or doxorubicin [18,19]. In this study, luminacin consistently showed cytotoxic effects on different HNSCC cell lines, and in most cases, cell death was not caused by apoptosis or necrosis. Given the increased expression of key autophagy proteins such as Beclin-1 or LC3B, autophagy may play an important role in the cytotoxicity of luminacin.
As demonstrated in several studies, some agents exert cytotoxicity not only through autophagy, but also through apoptosis, depending on the cell type and experimental conditions. Many studies have reported an inter-relationship of apoptosis and autophagy, because activation of these two death modalities involves the same molecular pathway and occurs simultaneously [20]. In this study, apoptosis by luminacin treatment was responsible for 15% of SCC15 and 40% of MSCQLL1 cell death. Our results demonstrated an anticancer potential for luminacin by showing that autophagy works with the help of apoptosis to induce cell death. Further investigations should be conducted to determine which conditions can induce autophagy or apoptosis for cancer therapy with luminacin.
Several molecular pathways are involved in the induction of autophagy. Deregulation of Akt serine/threonine kinase is a well-known cause of anti-apoptosis and uncontrolled cell proliferation. Hennessy et al. [21] reported that autophagy is induced by regulation of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway. Baicalin, a possible anti-cancer agent derived from a natural substance, is reported to trigger autophagy in human bladder cancer cells via modulation of the Akt pathway [22]. In addition, MAPK, including JNK and p38 MAPK, has also been reported to regulate autophagy [23]. In recent studies, activation of JNK was shown to be closely involved in the induction of autophagy [24]. p38 MAPK, which regulates cell differentiation, migration, and invasion, is also strongly associated with autophagy [25]. In this investigation, incr-eased expression of p-JNK, p-p38, and decreased expression of p-Akt phosphorylation by treatment with luminacin demonstrated the possible involvement of Akt and the JNK, p38 MAPK pathways in the induction of autophagy and cell death.
Conclusion
Inhibition of growth and progression via autophagic cell death were successfully induced in HNSCC cells by treatment with luminacin, a metabolite of the marine Streptomyces. The in vitro HaCaT cells and in vivo zebrafish model confirmed acceptable toxicity of luminacin, even at a cytotoxic concentration to HNSCC. Our results demonstrate that Luminacin is a plausible chemotherapeutic agent for HNSCC, even though further characterization is necessary.
Acknowledgments
This research was supported by the Bio & Medical Technology Development Program (2012M3A9B2052870) and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and future Planning (2015R1A2A1A010029 68).
Footnotes
Conflict of interest relevant to this article was not reported.
References
- 1.Jung KW, Won YJ, Kong HJ, Oh CM, Lee DH, Lee JS. Cancer statistics in Korea: incidence, mortality, survival, and prevalence in 2011. Cancer Res Treat. 2014;46:109–23. doi: 10.4143/crt.2014.46.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takes RP, Rinaldo A, Silver CE, Haigentz M, Jr, Woolgar JA, Triantafyllou A, et al. Distant metastases from head and neck squamous cell carcinoma. Part I. Basic aspects. Oral Oncol. 2012;48:775–9. doi: 10.1016/j.oraloncology.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–35. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerwick WH, Moore BS. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem Biol. 2012;19:85–98. doi: 10.1016/j.chembiol.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shin HA, Shin YS, Kang SU, Kim JH, Oh YT, Park KH, et al. Radioprotective effect of epicatechin in cultured human fibroblasts and zebrafish. J Radiat Res. 2014;55:32–40. doi: 10.1093/jrr/rrt085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee BS, Kang S, Kim KA, Song YJ, Cheong KH, Cha HY, et al. Met degradation by SAIT301, a Met monoclonal antibody, reduces the invasion and migration of nasopharyngeal cancer cells via inhibition of EGR-1 expression. Cell Death Dis. 2014;5: doi: 10.1038/cddis.2014.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomas TR, Kavlekar DP, LokaBharathi PA. Marine drugs from sponge-microbe association: a review. Mar Drugs. 2010;8:1417–68. doi: 10.3390/md8041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Proksch P, Edrada RA, Ebel R. Drugs from the seas: current status and microbiological implications. Appl Microbiol Biotechnol. 2002;59:125–34. doi: 10.1007/s00253-002-1006-8. [DOI] [PubMed] [Google Scholar]
- 9.Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol Mol Biol Rev. 2007;71:295–347. doi: 10.1128/MMBR.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang G. Diversity and biotechnological potential of the sponge-associated microbial consortia. J Ind Microbiol Biotechnol. 2006;33:545–51. doi: 10.1007/s10295-006-0123-2. [DOI] [PubMed] [Google Scholar]
- 11.Naruse N, Kageyama-Kawase R, Funahashi Y, Wakabayashi T. Luminacins: a family of capillary tube formation inhibitors from Streptomyces sp. I. Taxomony, fermentation, isolation, physico-chemical properties and structure elucidation. J Antibiot (Tokyo) 2000;53:579–90. doi: 10.7164/antibiotics.53.579. [DOI] [PubMed] [Google Scholar]
- 12.Wakabayashi T, Kageyama-Kawase R, Naruse N, Funahashi Y, Yoshimatsu K. Luminacins: a family of capillary tube formation inhibitors from Streptomyces sp. II. Biological activities. J Antibiot (Tokyo) 2000;53:591–6. doi: 10.7164/antibiotics.53.591. [DOI] [PubMed] [Google Scholar]
- 13.Bentley SD, Chater KF, Cerdeno-Tarraga AM, Challis GL, Thomson NR, James KD, et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2) Nature. 2002;417:141–7. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 14.Atatreh N, Barraclough J, Welman A, Cawthorne C, Bryce RA, Dive C, et al. Difluoro analogue of UCS15A triggers activation of exogenously expressed c-Src in HCT 116 human colorectal carcinoma cells. J Enzyme Inhib Med Chem. 2007;22:638–46. doi: 10.1080/14756360701485760. [DOI] [PubMed] [Google Scholar]
- 15.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Wu D, Shen J, Zhou M, Lu Y. Rapamycin induces autophagy in the melanoma cell line M14 via regulation of the expression levels of Bcl-2 and Bax. Oncol Lett. 2013;5:167–72. doi: 10.3892/ol.2012.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding ZB, Hui B, Shi YH, Zhou J, Peng YF, Gu CY, et al. Autophagy activation in hepatocellular carcinoma contributes to the tolerance of oxaliplatin via reactive oxygen species modulation. Clin Cancer Res. 2011;17:6229–38. doi: 10.1158/1078-0432.CCR-11-0816. [DOI] [PubMed] [Google Scholar]
- 18.Shi Y, Tang B, Yu PW, Tang B, Hao YX, Lei X, et al. Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in Caco-2 cells. PLoS One. 2012;7: doi: 10.1371/journal.pone.0051076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sishi BJ, Loos B, van Rooyen J, Engelbrecht AM. Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity. Biochem Pharmacol. 2013;85:124–34. doi: 10.1016/j.bcp.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Wu YT, Tan HL, Huang Q, Kim YS, Pan N, Ong WY, et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy. 2008;4:457–66. doi: 10.4161/auto.5662. [DOI] [PubMed] [Google Scholar]
- 21.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 22.Lin C, Tsai SC, Tseng MT, Peng SF, Kuo SC, Lin MW, et al. AKT serine/threonine protein kinase modulates baicalin-triggered autophagy in human bladder cancer T24 cells. Int J Oncol. 2013;42:993–1000. doi: 10.3892/ijo.2013.1791. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, Shen T, Shang C, Luo Y, Liu L, Yan J, et al. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. Oncotarget. 2014;5:10140–50. doi: 10.18632/oncotarget.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358:17–26. doi: 10.1016/j.canlet.2014.11.046. [DOI] [PubMed] [Google Scholar]