

Abstract
Purpose
We investigated the effect of chemoradiotherapy with PP2 and temozolomide (TMZ) on malignant glioma cells using clonogenic assays and in vivo brain tumor model.
Materials and Methods
The effect of PP2 on radiosensitivity of U251 and T98G cells was investigated using clonogenic assays. The expression of E-cadherin, matrix metalloproteinases 2 (MMP2), Ephrin type-A receptor 2 (EphA2), and vascular endothelial growth factor (VEGF) was measured by Western blotting and an accumulation of γH2AX foci 6 hours after radiotherapy was measured after PP2 treatment. The effect of PP2 on migration, invasion, and vasculogenic mimicry formation (VMF) of U251 cells was evaluated. In an orthotopical brain tumor model with U251 cells, PP2 was injected intraperitoneally with or without oral TMZ before, during and after whole brain radiotherapy. Bioluminescence images were taken to visualize in vivo tumors and immunohistochemical staining of VEGF, CD31, EphA2, and hypoxia-inducible factor 1a was performed.
Results
PP2 increased radiosensitivity of U251 and T98G cells without decreasing survival of normal human astrocytes. Chemoradiotherapy with PP2 and TMZ resulted in increased accumulation of γH2AX foci. PP2 induced overexpression of E-cadherin and suppression of MMP2, VEGF, and EphA2. PP2 also compromised invasion, migration, and VMF of U251 cells. In brain tumors, chemoradiotherapy with PP2 and TMZ decreased tumor volume best, but not statistically significantly compared with chemoradiotherapy with TMZ. The expression of VEGF and CD31 was suppressed in PP2-treated tumors.
Conclusion
PP2 enhances radiosensitivity of malignant glioma cells and suppresses invasion and migration of U251 cells. Chemoradiotherapy with PP2 and TMZ resulted in non-significant tumor volume decrease.
Keywords: SRC tyrosine kinase inhibitor, PP2, Temozolomide, Glioblastoma, Radiotherapy
Introduction
Malignant gliomas are the most common neoplasm arising in the central neural system in adults. Among these, glioblastoma (GBM) has the worst prognosis despite aggressive multimodality treatment. The standard treatment for GBM for several decades was radiotherapy. With the advent of temozolomide (TMZ), TMZ was incorporated as an essential component into a standard treatment for GBM [1]. Several molecular markers are used in clinical diagnosis of GBM [2] and among them, methylation of the O6-methylguanine-DNA methyltransferase (MGMT) promoter gene is predictive of treatment outcomes of GBM. Nonetheless, the actuarial survival rate of patients with GBM remains disappointing because overall survival of patients with MGMT promoter methylation is only 46% at 2 years despite of chemoradiotherapy with TMZ [3]. Thus, it is essential to improve therapeutic efficacy while minimizing toxicity.
Infiltrative growth of GBM often results in incomplete surgical removal of tumors and large treatment volume owing to wider margin of radiotherapy. Inhibiting invasion or migration of GBM is therefore a promising strategy to improve treatment outcomes. Because SRC tyrosine kinase regulates actin dynamics and invasion of malignant glial cells [4], it is a strong candidate. SRC tyrosine kinase is an enzyme encoded by the SRC gene that belongs to a family of non-receptor tyrosine kinases (RTKs) [5]. It interacts with many intracellular proteins including growth factor receptors, integrins, and Eph kinase, and mediates signals from the extracellular matrix. SRC tyrosine kinase activity is elevated in GBM cells relative to normal brain cells [6,7] and SRC tyrosine kinase inhibitors (TKIs) inhibit cell invasion and migration in GBM cell lines [7-9]. Ionizing radiation enhances invasion of glioma cells through SRC/epidermal growth factor receptor–mediated p38/AKT and phosphatidylinositol 3-kinase/AKT signaling pathways [10]. In addition, SRC TKI AZD0530 blocks invasion and may act as a radiosensitizer in lung cancer cells [11] and SRC TKI Su6656 enhances antiangiogenic effect of irradiation [12].
Therefore, we investigated whether combined treatment of TMZ and SRC TKI, PP2 would inhibit migration and growth of glioma cells and improve the therapeutic effect of radiotherapy using in vitro clonogenic assays and an in vivo human brain tumor xenograft model in nude mice.
Materials and Methods
1. Cell lines and culture conditions
Human glioma cell lines U251 and T98G (American Tissue Culture Collection, Rockville, MD) were authenticated by the company’s routine Cell Biology Program and used within 6 months of receipt. Cells were maintained and cultured in Dulbecco’s modified Eagle medium (DMEM; Lonza, Allendale, NJ) media containing 10% fetal bovine serum (FBS; Gibco, Grand Island, NY) at 37°C in 5% CO2 using standard techniques.
2. Inhibitors
The SRC TKI PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo[3,4-d]pyrimidine; Calbiochem, Darmstadt, Germany) was prepared as a concentrated stock solution in dimethyl sulfoxide (Sigma-Aldrich, St. Louis, MO). TMZ (Schering-Plough, Kenilworth, NJ) was prepared by dissolving in 10 μM sodium chloride. After serial experiments to test the effect of 2.5-20 μM of PP2, 25-100 μM of TMZ, and combination of both inhibitors by optical density measurement of cell suspension, 10 μM PP2 and 25 μM TMZ were selected for further experiments.
3. Clonogenic assay
These experiments were performed as described previously using 6 megavoltage X-rays (Clinac 21EX, Varian Medical Systems, Palo Alto, CA) [13]. On the first day of treatment the medium was replaced with DMEM-lacking FBS and containing vehicle control or 10 μM PP2, with or without 25 μM TMZ. After 24 hours, the medium was replaced with DMEM containing 10% FBS. Survival data were fitted to a linear-quadratic model using Kaleidagraph ver. 3.51 (Synergy Software, Reading, PA). Sensitizer enhancement ratios (SER) of human glioma cells was calculated as radiation dose needed with radiation alone divided by the radiation dose needed with inhibitors at surviving fraction (SF).
4. γH2AX foci formation assay
After treating inhibitors for 24 hours, cells were irradiated with 6 Gy. After 6 hours of radiotherapy, γH2AX foci formation assay was performed as described previously [14].
5. Modified Boyden chamber assay
Cell migration was measured using a Transwell system (Corning, Rochester, NY) that allows cells to migrate through 8-μm pores in polycarbonate membranes. Inserts containing cancer cells were incubated in 24-well plates in serum-free medium and cells were harvested by trypsinization. An aliquot of 104 cells was added to the upper chamber and DMEM was placed in the lower chamber of Transwells, and incubated for 24 hours with or without inhibitors. The inserts were fixed in methanol, stained with 1% crystal violet, and three representative fields were photomicrographed.
6. Wound healing assay
Cells were cultured until confluent in 6-well plates (Sonic-Seal Slide, Nalge Nunc, Rochester, NY) and starved for 24 hours. Each well was divided into a 2×3 grid. A linear wound was made in each hemisphere of the well using a 1-mL pipette tip and the medium was replaced with starvation medium. After 24 hours of inhibitor treatment, images were obtained of the intersections of the linear cell wound and each grid line. Migration rate was estimated from the distance that the cells moved, determined microscopically.
7. Vasculogenic mimicry formation assay
Vasculogenic mimicry (VM) formation assay was performed using a commercial Matrigel assay kit (BD Biosciences, Le-Pont-de-Claix, France). ECM Matrigel (200 μL) was placed in 48-well tissue culture plates and incubated at 37°C for 2 hours. Cells were treated with TMZ (25 μM) with or without PP2 (10 μM), and seeded onto the coated plate. After incubation for 24 hours with or without inhibitors, VM formation was assessed using an inverted microscope.
8. Western blotting
These experiments were performed as described previously [14]. Primary antibodies against p-SRC (Thy416), phopho-epidermal growth factor receptor (p-EGFR) (Tyr 1068), p-AKT (Ser473), p-ERK (Tyr202/204), E-cadherin, matrix metalloproteinase 2 (MMP2), and Ephrin type-A receptor 2 (EphA2) were used at 1:1,000 dilutions. Primary antibodies against vascular endothelial growth factor (VEGF) and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA) were used at a dilution of 1:500 and 1:1,000, respectively.
9. In vivo imaging of a human brain tumor model in nude mice
U251 cells (3×105 cells) were injected intracranially using a 26-G needle attached to a Hamilton syringe with freehand technique over 1 minute. On day 7 of cell implantation, the pGL4 luciferase reporter vector (Promega, Madison, WI) 150 mg/kg was injected intraperitoneally and after 15 minutes, mice were anesthetized with 1%-2% isoflurane as usual. In vivo bioluminescence images were obtained using the IVIS Lumina II (Xenogen, Alameda, CA) to identify intracranial implants.
Mice were randomly assigned to the experimental or control group and were treated over three weeks as follows: (1) radiotherapy alone group: phosphate buffered saline was injected intraperitoneally on days 8, 10, 12, 15, 17, 19, 22, 24, and 26; (2) inhibitor treatment group: PP2 (10 mg/kg) was injected intraperitoneally and TMZ (50 mg/kg) was given orally per day on the same treatment day as in the control group.
Two hours after inhibitor treatment, external radiotherapy was performed with a 6 MeV electron beam (Clinac 21EX) to cover the whole brain of the mouse. A total of 9 Gy was delivered with a daily dose of 3 Gy on days 15, 17, and 19. Bioluminescence images were obtained 2 weeks after radiotherapy as described. Mice were sacrificed on day 35 according to the Institutional Animal Care and Use Committee (IACUC) protocol unless they became symptomatic from the intracranial tumor burden.
10. Cryosection and immunohistochemical stains of harvested tumors
Cryosections were performed as described by Fischer et al. [15]. Slides were incubated overnight with primary antibody against VEGF, CD31, EphA2, and HIF1α (Cell Signaling Technology, Danvers, MA), followed by incubation with secondary Alexa Fluor 488-conjugated donkey anti-goat antibody (Molecular Probes, Eugene, OR) for 1 hour. Slides were treated with 1 μg/mL 4′,6-diamidino-2-phenylindole for 5 minutes to counterstain nuclei and examined on an Axio Scope A1 Microscope (Carl Zeiss, Gottingen, Germany). Images were captured and acquired using AxioCam MRc5 and acquisition software AxioVision v.4.4 (Carl Zeiss).
11. Statistical analysis
Statistical analysis used IBM SPSS ver. 20 (IBM Corp., Armonk, NY). Student’s t test was used to analyze statistical significance as applicable. Null hypotheses of no difference were rejected if p-values were less than 0.05.
Results
1. Radiosensitizing effect of PP2 in U251 and T98G cells
We first performed clonogenic assays to evaluate the radiosensitizing effect of PP2 (Fig. 1). Combination of PP2 with TMZ resulted in an additive cytotoxic effect in U251 and T98G cells. The SER at a SF 0.5 for PP2 was 1.15, for TMZ 1.41, and PP2 plus TMZ 1.54 in U251 cells. In T98G cells, the SER at a SF 0.5 for PP2 was 1.16, for TMZ 1.26, and PP2 plus TMZ 1.38. U251 cells, which have a high proportion of methylated MGMTwith low MGMT protein expression [16], were more radiosensitive by TMZ. In contrast, T98G cells with a low proportion of methylated MGMT and intermediate MGMT protein expression were less radiosensitive by TMZ. PP2 induced additional cytotoxic effect on glioma cells irrespective of MGMT methylation status.
Fig. 1.

Clonogenic assays of U251 and T98G cells after treating PP2, temozolomide (TMZ), or both. Cell surviving curves after treating PP2 (10 μM), TMZ (25 μM), or both were generated. The points indicate the mean surviving fractions at each dose level (±standard error). The lines are fitted to linear-quadratic equations. An addition of PP2 to TMZ showed an additive cytotoxic effect both in U251 and T98G cells. Sensitizer enhancement ratios (SER) were defined as isoeffective dose at surviving fraction 0.5 or 0.05. PP2 induced radiosensitization in U251 and T98G cells irrespective of MGMT promoter methylation. EGFR, epidermal growth factor receptor; RT, radiotherapy.
2. Impairment of DNA damage repair following irradiation
Since DNA double strand breaks (DSBs) induced by radiotherapy are responsible for cell death, repair of DSBs is crucial for cellular survival. We examined whether pretreatment with SRC TKI before radiotherapy inhibits DNA DSBs repair by measuring γH2AX foci formation 6 h after radiotherapy. Pretreatment with PP2 combined with TMZ resulted in increased accumulation of γH2AX foci, indicating PP2 induced delayed repair of DNA DSBs (Fig. 2). All differences between the groups (6 Gy vs. 6 Gy+TMZ, p=0.014; 6 Gy+ TMZ vs. 6 Gy+PP2, p=0.020; 6 Gy+PP2 vs. 6 Gy+TMZ+PP2, p=0.016) were statistically significant.
Fig. 2.

Representative images of γH2AX foci formation in U251 cells. γH2AX foci formation was measured in U251 cells after treating PP2 (10 μM), temozolomide (TMZ, 25 μM), or both. The nuclei stained by DAPI are shown in blue, while the γH2AX foci stained by FITC are shown in green. Increased accumulation of γH2AX foci after treating PP2 and TMZ was confirmed by indirect immunofluorescence 6 hours after radiotherapy (6 Gy), indicating delayed DNA double strand breakage repair. All differences between the groups (6 Gy vs. 6 Gy+TMZ, p=0.014; 6 Gy+TMZ vs. 6 Gy+PP2, p=0.020; 6 Gy+PP2 vs. 6 Gy+TMZ+PP2, p=0.016) were statistically significant.
3. PP2 inhibits glioma cell invasion, migration, and VM formation
We evaluated whether PP2 can inhibit invasion and migration of U251 cells. In a modified Boyden chamber assay, cell migration was inhibited by PP2, with or without TMZ (Fig. 3A). TMZ did not affect migration of U251 cells and the number of migrating cells in the control did not differ statistically from that of TMZ-treated cells (p=0.468). However, pretreatment with PP2 markedly suppressed migration of U251 cells. The relative migration of inhibitortreated cells compared to the control was 0.993±0.131 (p=0.465) for TMZ, 0.252±0.078 (p < 0.001) for PP2, and 0.200±0.066 (p < 0.001) for TMZ plus PP2.
Fig. 3.
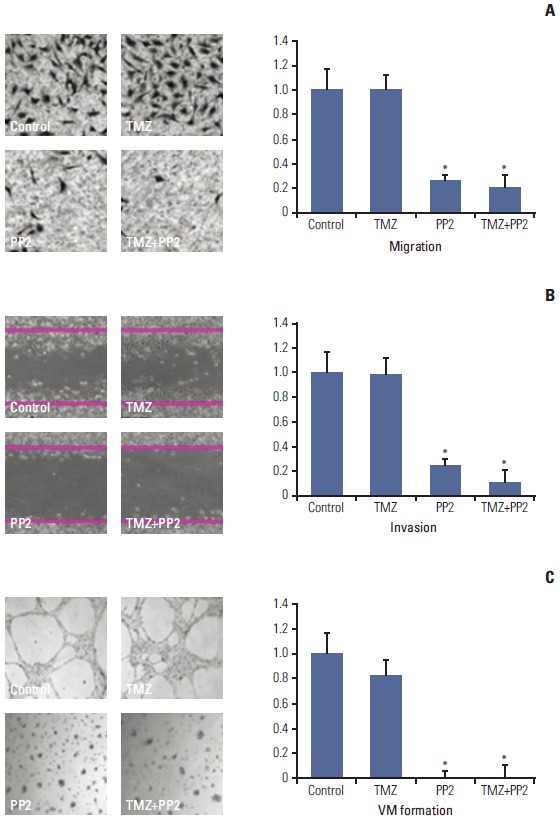
Representative images of modified Boyden chamber assay (A), wound healing assay (B), and vasculogenic mimicry (VM) formation assay (C). (A) U251 cells were treated with PP2 (10 μM), temozolomide (TMZ, 25 μM), or both for 24 hours, and cell invasion was measured using a Transwell system with 8-μm pores in polycarbonate membranes. Results are expressed as a ratio of radiotherapy alone (control). (B) U251 cells were grown to confluence in 6-well plates and then starved for 24 hours. A linear scratch was made in each hemisphere of the well with 1-mL pipette tip. Images were taken of the intersections of linear cell wound and each grid line. (C) Two hundred microliters ECM Matrigel was dropped in 48-well tissue culture plates and then incubated at 37°C for 2 hours. Column, bar, and asterisk represent the mean of three independent experiments, standard error, and statistical significance of p < 0.05 when compared with control, respectively.
In a wound healing assay, PP2 inhibited invasion of U251 cells, whereas TMZ did not (Fig. 3B). The relative invasion ability of inhibitor-treated cells compared to the control was 0.992±0.122 (p=0.461) for TMZ, 0.257±0.050 (p < 0.001) for PP2, and 0.116±0.010 (p < 0.001) for TMZ plus PP2. PP2 combined with TMZ did not differ from PP2 alone in suppressing invasion of U251 cells. As malignant gliomas can exhibit VM, which is related to aggressiveness and mesenchymal transition of cancer cells or poor survival of patients, we investigated the effect of PP2 on VM formation. As shown in Fig. 3C, pretreating PP2, with or without TMZ, inhibited VM formation.
4. Changes in protein expression following PP2 and TMZ treatment
To investigate changes in protein expression associated with invasion or migration of U251 cells, proteins related to cell adhesion or vascular proliferation, including E-cadherin, MMP2, EphA2, and VEGF, were evaluated using Western blotting. As shown in Fig. 4, pretreatment with TMZ did not alter the expression of these proteins, whereas PP2 treatment resulted in suppression of MMP2, EphA2, and VEGF and overexpression of E-cadherin in U251 cells.
Fig. 4.

Western blotting of U251 cells after treating inhibitors. After PP2 (10 μM) treatment, E-cadherin was overexpressed, while expression of matrix metalloproteinase 2 (MMP2), Ephrin type-A receptor 2 (EphA2), and vascular endothelial growth factor (VEGF) were suppressed. TMZ, temozolomide.
5. Effect of PP2 on normal human astrocytes
To assess whether SRC TKI has a cytotoxic effect on normal human astrocytes (NHAs) in the concentration of 10 μM, we examined the SF2 of NHAs after treating inhibitors (Fig. 5). PP2 did not decrease SF2 compared with control, suggesting that SRC TKI does not affect the viability of NHAs. SF2 of PP2 plus TMZ was not different from that of TMZ alone.
Fig. 5.
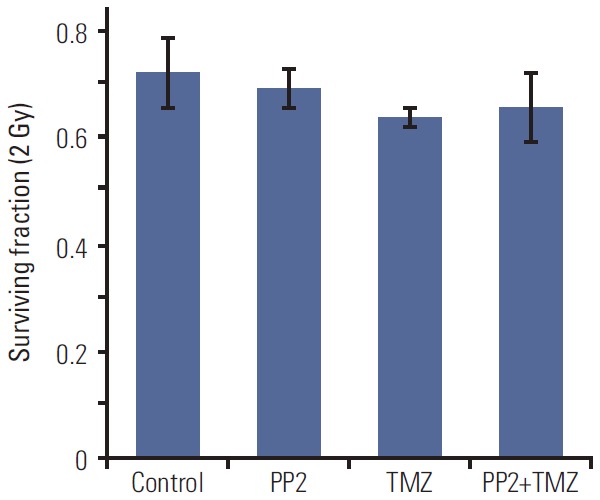
The surviving fraction at 2 Gy (SF2) of X-ray in normal human astrocytes. SF2 was measured after treating PP2 (10 μM), temozolomide (TMZ, 25 μM), or both. The points indicate the mean surviving fractions at each dose level. Column and bar represent mean of three independent experiments and standard error, respectively.
6. In vivo tumor response after PP2 treatment and radiotherapy
As shown Fig. 1, in vitro experiments revealed that PP2 induced additional cell death of U251 cells irrespective of TMZ. We investigated whether adding PP2 to TMZ would result in enhanced local control of brain gliomas using a human brain tumor xenograft model in nude mice. Radiotherapy alone induced tumor shrinkage, but failed to achieve complete remission (Fig. 6A). Chemoradiotherapy with TMZ markedly shrank tumors and chemoradiotherapy with PP2 and TMZ led to non-visualization of tumors, indicating an additional radiosensitization by PP2.
Fig. 6.

(A) Representative bioluminescence images of human brain tumor xenograft in nude mice. U251 cells were injected orthotopically into thalamic area using a 26G needle with syringe. Bioluminescence images were obtained 7 days after injection, and then randomly assigned to control or experimental groups as follows: control, radiotherapy (XRT), XRT+PP2, XRT+temozolomide (TMZ), and XRT+TMZ+PP2, respectively. Chemoradiotherapy with TMZ exhibited marked tumor shrinkage and chemoradiotherapy with PP2 and TMZ led to non-visualization of tumors, indicating an additional radiosensitization by PP2. (B) In semi-quantitative analyses of relative luminescence area, combination of PP2 and TMZ resulted in a smaller area of luminescence than TMZ or PP2 alone, but it was not statistically significant. Bar represents standard error. (C) TMZ-treated groups had smaller mean intensity than control or radiotherapy alone. However, the difference between TMZ and PP2 plus TMZ group was not significant.
As semi-quantitative analyses, the area and mean intensity of luminescence were measured and compared to each other. There was a significant difference between control and inhibitor-treated groups in the area of luminescence (p=0.025, p=0.004, and p=0.004 for PP2, TMZ, and PP2 plus TMZ, respectively). Fig. 6B shows a trend that combination of PP2 and TMZ results in a smaller area of luminescence than TMZ or PP2 alone, but it was not statistically significant. TMZ-treated groups had smaller mean intensity than control or radiotherapy alone (Fig. 6C). However, the difference between TMZ and PP2 plus TMZ group was not significant.
7. Immunohistochemical stains of brain tumors
To elucidate changes of protein expression induced by PP2 in vivomodel, we performed immunohistochemical staining with antibodies against VEGF, CD31, EphA2, and HIF1α. As shown in Fig. 7, expression of VEGF and CD31 were down-regulated in PP2-treated tumors relative to radiotherapy alone or RT plus TMZ tumors, suggesting that PP2 may suppress angiogenesis in vivo tumor as well as in vitro cells. Expression of EphA2 and HIF1α were not influenced by PP2 in vivo tumors.
Fig. 7.
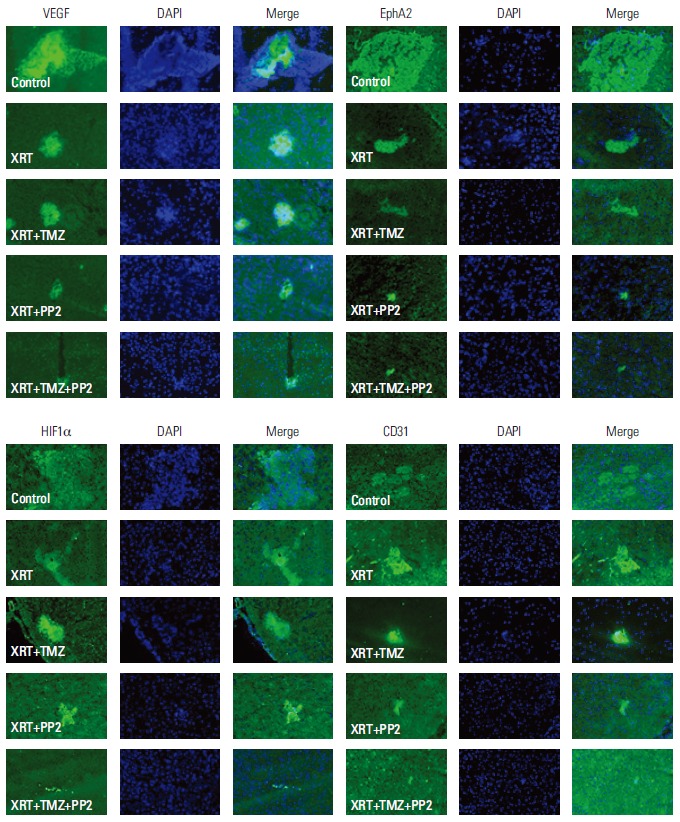
Immunohistochemical stains of in vivo human brain tumors in nude mice. Immunohistochemical staining with antibodies against vascular endothelial growth factor (VEGF), CD31, Ephrin type-A receptor 2 (EphA2), and hypoxia-inducible factor 1α (HIF1α) was performed. The expression of VEGF and CD31 were down-regulated in PP2-treated tumors relative to those with XRT alone or XRT plus temozolomide (TMZ) tumors, suggesting that PP2 may suppress angiogenesis in in vivo tumor as well as in in vitro cells. Expression of EphA2 and HIF1α were not influenced by PP2 treatment in in vivo tumors. XRT, radiotherapy.
Discussion
In this study, we evaluated the effect of combined therapy with TMZ and SRC TKI PP2 on malignant glioma cells using in vitro and in vivo experiments. Our data demonstrated that pretreating PP2 (10 μM) leads to a small, but additive cytotoxic effect in in vitro experiments and this effect is independent of MGMT promoter methylation status of glioma cells when combined with radiotherapy, suggesting that SRC tyrosine kinase can be a novel target for GBM. PP2 did not induce cytotoxicity of NHAs at the same concentration. As shown in Fig. 2, pretreating PP2 demonstrated delayed repair of DNA DSBs induced by radiotherapy. The combination of PP2 with standard treatment of malignant gliomas (radiotherapy with TMZ) was tested in an in vivo brain tumor model using nude mice, in which the PP2 combination resulted in a trend of tumor volume decrease, suggesting avenues for future research.
Infiltrative growth is a key characteristic of malignant gliomas, which hinders complete surgical removal of gliomas and often results in local recurrence despite aggressive treatment. Suppressing invasion and/or migration of glioma cells is a possible strategy to overcome the dismal prognosis of GBM.
In a modified Boyden chamber assay and wound healing assay that measures invasion and migration ability of cancer cells, PP2 significantly inhibited both invasion and migration of U251 cells (Fig. 3). It is generally accepted that loss of E-cadherin, which may affect cell-to-cell adhesion and relate to invasive property, is a fundamental event in epithelialmesenchymal transition. In this study, pretreating PP2 resulted in overexpression of E-cadherin, and suppression of MMP2 and EphA2 in U251 cells (Fig. 4). These suggest that PP2 may inhibit invasion and migration ability of U251 cells through affecting adhesion molecules such as E-cadherin. These results are consistent with other studies, in which SRC regulates actin dynamics and invasion of malignant glial cells. [4]
Neovascularization is essential for the rapid growth of tumors and is a crucial characteristic of malignant gliomas. Some targeting agents, such as bevacizumab, a monoclonal antibody to VEGF-A, aim to suppress neovascularization of tumors and as a consequence, inhibit tumor growth [17]. In clinical practice, bevacizumab is a second-line drug to salvage recurrent GBM [18]. In this study, we measured the effect of PP2 on the expression of VEGF using Western blotting and tested VM formation of U251 cells. VEGF expression was down-regulated and VM formation is strongly inhibited by PP2 in U251 cells, suggesting that SRC TKI inhibits neovascularization of malignant gliomas. In the orthotopic brain tumor model, the expression of VEGF and CD31 in implanted brain tumors was also suppressed as shown in Fig. 7. These results strongly suggest an anti-angiogenic potential of SRC TKI in malignant gliomas. Moreover, suppressing SRC family kinase (SFK) signaling can inhibit Bevacizumab-induced GBM cell invasion, suggesting a possible strategy to overcome treatment resistance of GBM [19].
Suppression of MMP2 might help restore radiation sensitivity in glioma cells. MMP2 is implicated in the invasiveness of glioma cells, which converts the pro-form of MMP2 into the active form through the urokinase-type plasminogen activator-plasmin cascade [20]. Ionizing radiation enhances MMP2 secretion and facilitates invasion of glioma cells through SRC/EGFR-mediated phosphoinositide 3-kinase (PI3K)/AKT pathways [10], providing a possible mechanism for the development of resistance to radiotherapy in malignant glioma. Our findings showed that PP2 decreased the expression of MMP2 in glioma cells, suggesting a possible mechanism of action regarding migratory inhibition of PP2 in glioma cells.
Erythropoietin-producing human hepatocellular carcinoma (Eph) receptors constitute the largest family of RTKs in the human genome [21]. In general, Eph receptors influence normal development of brain vasculature and affect brain function, including regulation of synaptic structure. Eph receptors are subdivided into EphA and EphB subgroups based on sequence homology and ligand-binding specificity. Among these, EphA2 is highly overexpressed in several carcinomas including breast, gastric, and prostate cancer and is a novel target for anti-cancer therapy. Several authors have demonstrated a relationship between EphA2 expression and proliferative activity of gliomas [22]. Up-regulation of EphA2 promotes angiogenesis in brain tumors and is associated with poor prognosis of malignant gliomas [23]. Zhuang et al. [24] reported that in breast cancer, EphA2 related to trastzumab resistance is regulated by SRC tyrosine kinase. In contrast, in T-cell lymphoma, EphA2 activation leads to phosphorylation of SFK and PI3K activation [25]. In this study, EphA2 was down-regulated in U251 cells by PP2 treatment. In sum, these findings suggest a possibility of crosstalk between SRC tyrosine kinase and EphA2 in cancer cells. However, the expression of EphA2 was not suppressed by PP2 in in vivo brain tumors; thus further study of the mechanism is warranted.
A drawback of this study lies in the failure to demonstrate a significant difference in tumor volume decrease between TMZ and combination with TMZ and PP2. This may have derived from the dramatic volume decrease of TMZ-treated tumors that make it difficult to detect a significant difference between standard treatment and PP2 combination. However, as shown in Fig. 5, brain tumors in the combination treatment group were non-visualized and an addition of PP2 to TMZ showed a trend toward volume decrease.
Conclusion
In this study, effects of chemoradiotherapy with SRC TKI, PP2 and TMZ in U251 cells are reported. These data show that adding PP2 to TMZ leads to additive cytotoxicity of U251 cells and decreased migration and invasion ability of U251 cells. Delayed DSBs repair after radiotherapy was observed when PP2 was administered. The underlying mechanism appears to involve suppression of MMP2, which might restore sensitivity to radiotherapy, and overexpression of E-cadherin, which might affect cell-to-cell adhesion. Moreover, PP2 is related to suppression of VEGF, CD31 and EphA2. These data provide evidence for combined chemoradiotherapy with TMZ and PP2 on glioma cells and present a feasible strategy to improve the therapeutic outcome in malignant gliomas.
Acknowledgments
This work was supported by grants (#2012-0004867, NRF-2013R1A1A2074531, and NRF-2013M2A2A7043683) from the National Research Foundation, Korean Ministry of Future Creative Science to In Ah Kim.
Footnotes
Conflict of interest relevant to this article was not reported.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Masui K, Cloughesy TF, Mischel PS. Review: molecular pathology in adult high-grade gliomas: from molecular diagnostics to target therapies. Neuropathol Appl Neurobiol. 2012;38:271–91. doi: 10.1111/j.1365-2990.2011.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 4.Angers-Loustau A, Hering R, Werbowetski TE, Kaplan DR, Del Maestro RF. SRC regulates actin dynamics and invasion of malignant glial cells in three dimensions. Mol Cancer Res. 2004;2:595–605. [PubMed] [Google Scholar]
- 5.Stehelin D, Fujita DJ, Padgett T, Varmus HE, Bishop JM. Detection and enumeration of transformation-defective strains of avian sarcoma virus with molecular hybridization. Virology. 1977;76:675–84. doi: 10.1016/0042-6822(77)90250-1. [DOI] [PubMed] [Google Scholar]
- 6.Stettner MR, Wang W, Nabors LB, Bharara S, Flynn DC, Grammer JR, et al. Lyn kinase activity is the predominant cellular SRC kinase activity in glioblastoma tumor cells. Cancer Res. 2005;65:5535–43. doi: 10.1158/0008-5472.CAN-04-3688. [DOI] [PubMed] [Google Scholar]
- 7.Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol. 2009;27:77–83. doi: 10.1038/nbt.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ding Q, Stewart J, Jr, Olman MA, Klobe MR, Gladson CL. The pattern of enhancement of Src kinase activity on platelet-derived growth factor stimulation of glioblastoma cells is affected by the integrin engaged. J Biol Chem. 2003;278:39882–91. doi: 10.1074/jbc.M304685200. [DOI] [PubMed] [Google Scholar]
- 9.Jallal H, Valentino ML, Chen G, Boschelli F, Ali S, Rabbani SA. A Src/Abl kinase inhibitor, SKI-606, blocks breast cancer invasion, growth, and metastasis in vitro and in vivo. Cancer Res. 2007;67:1580–8. doi: 10.1158/0008-5472.CAN-06-2027. [DOI] [PubMed] [Google Scholar]
- 10.Park CM, Park MJ, Kwak HJ, Lee HC, Kim MS, Lee SH, et al. Ionizing radiation enhances matrix metalloproteinase-2 secretion and invasion of glioma cells through Src/epidermal growth factor receptor-mediated p38/Akt and phosphatidylinositol 3-kinase/Akt signaling pathways. Cancer Res. 2006;66:8511–9. doi: 10.1158/0008-5472.CAN-05-4340. [DOI] [PubMed] [Google Scholar]
- 11.Rothschild SI, Gautschi O, Haura EB, Johnson FM. Src inhibitors in lung cancer: current status and future directions. Clin Lung Cancer. 2010;11:238–42. doi: 10.3816/CLC.2010.n.030. [DOI] [PubMed] [Google Scholar]
- 12.Cuneo KC, Geng L, Tan J, Brousal J, Shinohara ET, Osusky K, et al. SRC family kinase inhibitor SU6656 enhances antiangiogenic effect of irradiation. Int J Radiat Oncol Biol Phys. 2006;64:1197–203. doi: 10.1016/j.ijrobp.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 13.Kim IA, No M, Lee JM, Shin JH, Oh JS, Choi EJ, et al. Epigenetic modulation of radiation response in human cancer cells with activated EGFR or HER-2 signaling: potential role of histone deacetylase 6. Radiother Oncol. 2009;92:125–32. doi: 10.1016/j.radonc.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 14.Lee KM, Choi EJ, Kim IA. microRNA-7 increases radiosensitivity of human cancer cells with activated EGFR-associated signaling. Radiother Oncol. 2011;101:171–6. doi: 10.1016/j.radonc.2011.05.050. [DOI] [PubMed] [Google Scholar]
- 15.Fischer AH, Jacobson KA, Rose J, Zeller R. Cryosectioning tissues. CSH Protoc. 2008;2008: doi: 10.1101/pdb.prot4991. [DOI] [PubMed] [Google Scholar]
- 16.Chahal M, Abdulkarim B, Xu Y, Guiot MC, Easaw JC, Stifani N, et al. O6-Methylguanine-DNA methyltransferase is a novel negative effector of invasion in glioblastoma multiforme. Mol Cancer Ther. 2012;11:2440–50. doi: 10.1158/1535-7163.MCT-11-0977. [DOI] [PubMed] [Google Scholar]
- 17.Hanson JA, Hsu FP, Jacob AT, Bota DA, Alexandru D. Antivascular endothelial growth factor antibody for treatment of glioblastoma multiforme. Perm J. 2013;17:68–74. doi: 10.7812/TPP/13-081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong ET, Gautam S, Malchow C, Lun M, Pan E, Brem S. Bevacizumab for recurrent glioblastoma multiforme: a meta-analysis. J Natl Compr Canc Netw. 2011;9:403–7. doi: 10.6004/jnccn.2011.0037. [DOI] [PubMed] [Google Scholar]
- 19.Huveldt D, Lewis-Tuffin LJ, Carlson BL, Schroeder MA, Rodriguez F, Giannini C, et al. Targeting Src family kinases inhibits bevacizumab-induced glioma cell invasion. PLoS One. 2013;8: doi: 10.1371/journal.pone.0056505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le DM, Besson A, Fogg DK, Choi KS, Waisman DM, Goodyer CG, et al. Exploitation of astrocytes by glioma cells to facilitate invasiveness: a mechanism involving matrix metalloproteinase- 2 and the urokinase-type plasminogen activator-plasmin cascade. J Neurosci. 2003;23:4034–43. doi: 10.1523/JNEUROSCI.23-10-04034.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilkinson DG. Multiple roles of EPH receptors and ephrins in neural development. Nat Rev Neurosci. 2001;2:155–64. doi: 10.1038/35058515. [DOI] [PubMed] [Google Scholar]
- 22.Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–51. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- 23.Zhou N, Zhao WD, Liu DX, Liang Y, Fang WG, Li B, et al. Inactivation of EphA2 promotes tight junction formation and impairs angiogenesis in brain endothelial cells. Microvasc Res. 2011;82:113–21. doi: 10.1016/j.mvr.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010;70:299–308. doi: 10.1158/0008-5472.CAN-09-1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holen HL, Shadidi M, Narvhus K, Kjosnes O, Tierens A, Aasheim HC. Signaling through ephrin-A ligand leads to activation of Src-family kinases, Akt phosphorylation, and inhibition of antigen receptor-induced apoptosis. J Leukoc Biol. 2008;84:1183–91. doi: 10.1189/jlb.1207829. [DOI] [PubMed] [Google Scholar]