

Abstract
Genetic mouse models for Alzheimer’s disease (AD) have been widely used to understand aspects of the biology of the disease, but have had limited success in translating these findings to the clinic. In this review, we discuss the benefits and limitations of existing genetic models and recent advances in technologies (including high through put sequencing and genome editing) that promise more predictive models. We summarize widely used biomarkers and behavioral tests for mouse models of AD and highlight best practices that will maximize translatability of preclinical findings.
Keywords: Alzheimer’s disease, mouse model, genetic model, preclinical model, translational assays, biomarkers, genomics, neuroinflammation
1. Introduction
With an aging population, Alzheimer’s disease (AD) is on the increase with no current cures or effective treatments. Despite the obvious advantages of using mice to study complex, age-related diseases such as AD, it is a challenging time for mouse models. In some quarters, enthusiasm for using mice to model AD is waning, in part, because of the lack of success in translating findings in mouse models to the clinic. Current models utilize knowledge from early onset Alzheimer’s disease (EOAD, or familial AD), incorporating mutant forms of amyloid precursor protein (APP), presenilins, Tau (Mapt) and other genes. These models have been essential in understanding the biology of key aspects of AD, most prominently the formation of amyloid plaques and neurofibrillary tangles, but have not proven particularly effective as preclinical models. Some of this is down to the lack of the critical hallmarks of AD, notably significant neuronal cell loss, in the current models. However, the lack of success is also due to a lack of standardization of models (such as inconsistent genetic background), underpowered experiments, and less than ideal end points. Additionally, there may be significant differences between early and late onset AD (LOAD, or sporadic AD) such that treatments tested in existing models may be useful for EOAD but not for the sporadic form of AD that is much more common in the patient population.
Encouragingly, times are changing. Advances in studies of patient populations and animal models should enable the creation of more predictive mouse models; these are summarized in Table 1 and described in detail below. Genome-wide association studies (GWAS), and more so high-throughput genome sequencing projects are identifying novel variants for late onset Alzheimer’s disease that increase our knowledge of genetic susceptibility of LOAD (Figure 1). Combine these advances with the revolution in genetic and genome engineering and, although there is much work still to do, the future looks bright for developing the next generation of AD models. In this review, we aim to provide recent updates regarding current mouse models relevant to AD as well as consider emerging strategies for the generation of improved models. We also discuss how the field is moving towards improved standards for experimental design and phenotyping to maximize the benefit of mouse models as researchers seek novel therapeutic targets for AD.
Table 1.
Summary of recent advances that will aid generation of more predictive models
| Advances | Examples | |
|---|---|---|
| A. Advances in human AD characterization | 1. Identification of specific AD-relevant variations in genes using high throughput sequencing |
|
| 2. Improvements in ‘staging’ of AD |
|
|
| 3. Emerging clinical endophenotypes |
|
|
| B. Advances in mouse modeling | 1. Genetic background |
|
| 2. Incorporation of environmental factors |
|
|
| 3. Computational modeling |
|
|
| 4. Genome engineering |
|
|
| 5. Phenotyping |
|
Figure 1.
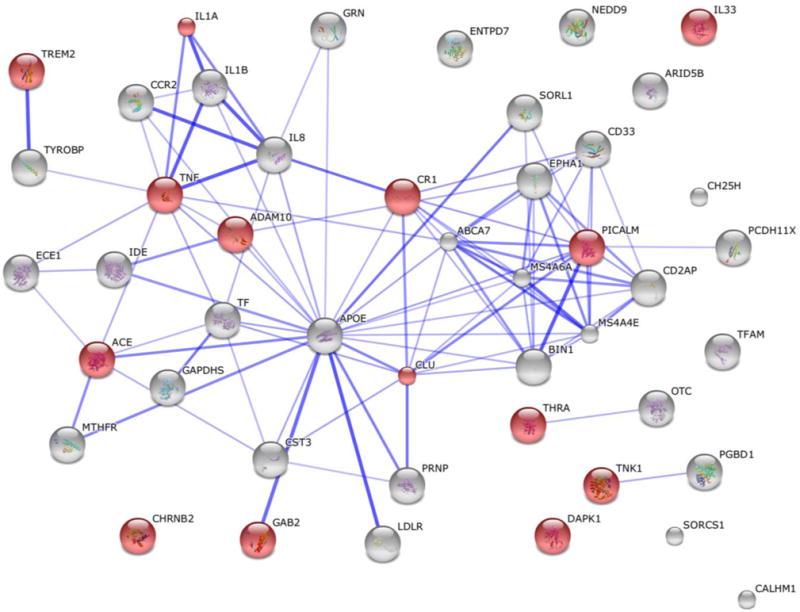
Known and Predicted Protein-Protein Interactions (STRING) of the top 48 LOAD genes. The current top 48 LOAD genes compiled by ALZFORUM [1]. Highlighted red nodes are related with the key word ‘neuroinflammation’.
2. Modeling early-onset Alzheimer’s disease
Genetic mouse models of early-onset Alzheimer’s disease have been reviewed recently [2] and are summarized in Table 2; we focus here on developments since then. A summary of existing AD mouse models is compiled and maintained by the Alzforum (http://www.alzforum.org/research-models). This important resource includes updated information regarding genetic construct, phenotype, and availability of each mouse model.
Table 2.
Summary of prominent mouse models of Alzheimer’s disease
| Type | Model | Promoter | Allele(s) | Allele Type | Lab | # PubMed citations | Repository availability; relative interest |
|---|---|---|---|---|---|---|---|
| Amyloid, tau | 3XTg-AD | Thy1 | APP(Swe), Mapt(P301L); Psen1(M146V) | Transgenic (cDNA); KI |
LaFerla | 702 | JAX #4807 *** |
| Amyloid | APP/PS1 | Prnp | APP(Swe); Psen1dE9 | Transgenic (cDNA) |
Borchelt | 207 | JAX #5864, #4462 *** |
| Amyloid | 5XFAD | Thy1 | APP(SweFlLon); PSEN1(M146L; L286V) | Transgenic (cDNA) |
Vasser | 277 | JAX #8730, #6554 *** |
| Amyloid | J20 | PDGFB | APP(SweInd) | Transgenic (cDNA) |
Mucke | 394 | JAX #6293 ** |
| Amyloid | Tg-SwDI | Thy1 | APP(SweDutIowa) | Transgenic (cDNA) |
Van Nostrand | 90 | JAX #7027 * |
| Amyloid | Tg-SwDI/Nos2 | Thy1 | APP(SweDutIowa); Nos2 KO | Transgenic (cDNA) |
Van Nostrand | 20 | JAX #9126 * |
| Amyloid | Tg2576 | Prnp | APPswe | Transgenic (cDNA) |
Ashe | 800 | Taconic; Charles River |
| Amyloid | R1.40 | APP | APP(Swe) | Transgenic (YAC) |
Lamb | 18 | JAX #5300 * |
| Amyloid | APPPS1 | Thy1 | APP(Swe) | Transgenic (cDNA) |
Jucker | 101 | NA |
| Amyloid | APP23 | Thy1 | APP(Swe) | Transgenic (cDNA) |
Novartis | 216 | Novartis |
| Amyloid | PDAPP | PDGF | APP(Ind) | Transgenic (cDNA) |
Games | 270 | NA |
| Amyloid | APP NL-G-F | APP | APP(SweIbeArc) | Knock-in | Saido | 14 | NA |
| Amyloid | TgCRND8 | Prnp | APP(SweInd) | Transgenic | Westaway | 182 | NA |
| tau | rTg4510 X Camk2a-tTA | Camk2a | MAPT(P301L) | Inducible transgenic | Ashe/Hutton/Lewis | 363 | JAX #24854 *** |
| tau | hTau | MAPT | MAPT; Mapt KO | Transgenic | Davies | 128 | JAX #5491 ** |
| tau | PS19 | Prnp | MAPT(P301S) | Transgenic (cDNA) |
V. Lee | 250 | JAX #8169 ** |
| tau | rTg4510 X Nop-tTA | Nop | MAPT(P301L) | Inducible transgenic | Hyman | 207 | JAX #15815 * |
| Other | GFAP-apoE4 | GFAP | APOE | Transgenic (cDNA); KO |
Holtzman | 29 | JAX #4631 * |
| Other | Apoetm3(APOE*4) | Apoe | APOE | Targeted replacement | Maeda | 53 | Taconic |
PubMed citations is the number of papers that have cited the initial paper describing the strain; it is not meant to indicate how many times a strain has been used.
Relative interest is a representation of the number of orders received by the JAX mouse repository.
KO, Knock-out
KI, Knock-in
NA, not applicable
YAC, yeast artificial chromosome
While traditional transgenic mouse models have been essential to our understanding of AD, they suffer from a variety of drawbacks: mis- or over-expression of transgenically expressed protein relative to the endogenous protein; developmental compensation for knocked-out or over-expressed genes; and inadvertent and unknown disruption of an endogenous gene by the transgenic construct. Most transgenic lines have been created on standard genetic backgrounds, which may not be the most suitable for expressing disease phenotypes (see discussion below). Perhaps most critically, the timing of expression off the transgenic promoter will not mimic the disease condition, so that mechanisms of disease onset cannot be studied in a realistic context. In addition, the utility of many of these models to the research community is limited due to legal restrictions on their availability and use, particularly for therapy development projects by for-profit companies[3].
Over the past few years, a variety of techniques have been used to attempt to generate improved mouse models of EOAD. In order to get away from the inherent technical issues with using traditional transgenic models, some groups have used a knock-in strategy. This has the advantage of providing more realistic expression patterns and levels, and avoids the complication of disrupting an unknown genomic locus. Perhaps due to the lack of over-expression of APP and/or tau or the relatively short lifespan of the mouse, these models have shown relatively mild, late onset phenotypes [4–6].
Another recent approach to improving mouse models of EOAD has been to use systems that enable inducible expression of transgenic protein to study the half-life and reversibility of AD endophenotypes. When mutant APP expression was turned off after the formation of initial amyloid deposits, performance in some cognitive tasks improved [7]. Likewise, suppression of transgenic mutant tau expression demonstrated that tau-induced impairments are reversible [8]. These studies provide justification for drug trials in AD patients, at least early in progression of the disease. A similar approach has been used to distinguish the effects of soluble Aβ relative to deposited amyloid [9].
In addition, a recent paper using genetic mouse models [10] presents evidence that cleavage of the APP protein by other than the well-known beta and gamma secretase pathways results in forms of Aβ that may be important in APP pathology. This highlights the fact that we still do not fully understand which fragments of APP cause AD, and that genetic models are useful to dissect the physiological cleavage events.
Various labs now are using a transcriptomics approach to compare transcriptional profiles of mouse models to patient tissue, with varying conclusions [11–14].
One of the major benefits to the mouse model is that it is relatively easy to assay the effect of other genes/pathways to see if they impinge on an established AD endophenotype. There have been hundreds of publications showing that combining an existing AD model with an established genetic knockout can modify a specific phenotype (e.g. plaque load, performance in Morris water maze, etc.). Most prominently, genetic ablation of the tau (Mapt) locus in AD models has enabled dissection of the relative contributions of the Aβ and tau pathways to AD pathophysiology[15]. While this approach may be useful for identifying relevant disease pathways and even targets, it has not led to a model that recapitulates all aspects of the human disease.
3. Creating animal models for late-onset Alzheimer’s disease
In contrast to creating mouse models for EOAD, generating models for late-onset Alzheimer’s disease (LOAD) is more challenging. This is mainly due to the complex genetic and environmental factors (diet, physical activity, microbiome, etc.) that interact to cause LOAD, many of which remain undiscovered. Currently, very few specific genetic variations are known to increase risk for LOAD. The two that have garnered the most interest for AD are the ε4 allele of apolipoprotein E (APOE), a common variant and the R47H allele of the triggering receptor expressed on myeloid cells 2 (TREM2) gene, a rare variant. Both variants greatly increase the risk of developing AD.
3.1 APOE – the greatest genetic risk factor for AD
The ε4 allele of the apolipoprotein E (APOE4) gene is the greatest genetic risk factor for AD, with the ε3 allele (APOE3) considered neutral and the ε2 allele (APOE2) considered protective. In human brains, APOE is normally synthesized and secreted by astrocytes and microglia and binds to high-density lipoproteins to facilitate cholesterol and phospholipid transport to LDL receptors. Low plasma APOE and APOE4 genotype are associated with morphological changes to the hippocampus, specifically reductions in size [16, 17]. Mouse models for studying both Apoe function and the human APOE variants have been created and have highlighted a complex role for APOE in the aging and diseased brain. Mice either deficient in Apoe or carrying the APOE4 ‘humanized’ allele in place of the mouse Apoe gene show a variety of phenotypes including deficits in cholesterol trafficking, amyloid clearance and the blood brain barrier. Importantly, mice heterozygous or homozygous for human APOE4 do not develop the complete AD phenotype showing that additional genetic and/or environmental factors are required.
3.2 TREM2 – elevating the significance of immune responses in AD
A genome-wide association study (GWAS) identified the R47H variation in TREM2 that conferred increased risk for AD [18], and more recently, a group at Brigham and Women’s Hospital found higher cortical TREM2 RNA expression was associated with increased amyloid pathology, suggesting a pathogenic role of TREM2 in AD susceptibility [19]. Highlighting the importance of TREM2 in neurodegenerative diseases more generally, some studies have also associated variations in TREM2 with Parkinson’s disease [20], frontotemporal dementia [21, 22] and amyotrophic lateral sclerosis [23]. These discoveries led to a series of studies in mice to understand the relevance of TREM2 variations to AD. First, APP/PS1 mice haploinsufficient for TREM2 (carrying only one copy) showed an altered microglial response without impacting plaque load [24]. Second, deficiency of TREM2 prevented infiltration of blood-derived myeloid cells and ameliorated plaque burden in APP/PS1 mice [25]. Finally, deficiency or haploinsufficiency of TREM2 augment amyloid accumulation due to a dysfunctional microglial response [26]. By generating R47H expressing reporter cells, the studied showed that the R47H mutation impairs the ability of TREM2 to recognize lipid ligands. There is much still to learn about the role of TREM2 in AD and so the generation of mice carrying human forms of TREM2 (including the R47H variant) is essential.
3.3 The strengths and limitations of genome-wide association studies
Beyond APOEε 4 and TREM2R47H, although much effort has been directed towards identifying additional specific genetic variants, little progress has been made, making modeling genes relevant to LOAD particularly challenging. However, large GWAS have identified multiple loci that increase risk for AD. For instance, one study that included approximately 75,000 individuals identified as many as 21 loci. The limitations of GWAS are that they commonly identify loci that are predicted to confer a small increase in risk and do not normally identify the causative variant, merely the haplotype block in which the causative risk variant is located. For each locus, the gene(s) that lie closest to the most strongly associated variant(s) become associated with the disease and for AD include complement receptor 1 (CR1), bridging integrator 1 (BIN1), clusterin (CLU) and Phosphatidylinositol-binding clathrin assembly protein (PICALM). A recent paper sought to control for this confound by using targeted sequencing of GWAS loci and identified an excess burden of deleterious coding mutations, specifically in relation to ATP-binding cassette sub-family A member 7(ABCA7) and BIN1[27].
Genes associated with LOAD are anticipated to function in pathways thought to be important in AD including cholesterol metabolism, endocytosis and immune responses. The lack of causative variants for these genes provides particular challenges for modeling in mice. Using traditional resources, the mouse is continuing to play a critical role in understanding the biology of genes associated with LOAD. Commonly, mutations in associated genes are introduced to AD mouse models and the impact of either haploinsufficiency or complete deficiency on AD-relevant phenotypes determined. While incredibly informative in terms of associating genes/pathways with different aspects of AD, these types of experiments do not necessarily get at the underlying causes of why variations in genes/loci confer increased risk for AD.
The challenges of interpreting GWAS ‘hits’ are not unique to AD, but are being discussed for other genetically complex diseases including diabetes. Given the majority of GWAS hits lie outside coding regions, it is anticipated that these small effects are due to variations in non-coding regions such as regulatory elements that may impact the dosage of transcripts rather than the protein function directly. This has led researchers to propose systematic haploinsufficiency studies, whereby one copy of each GWAS gene is mutated and the effects on AD phenotypes evaluated. Given susceptibility to LOAD is likely due to variations affecting multiple genes it will be necessary to assess perturbations in pairs or groups of genes to understand how genetic variations interact to increase risk for AD.
3.4 Developing human-relevant mouse models for LOAD
Ultimately, major breakthroughs in modeling LOAD in mice will come from the identification of additional specific genetic variations that increase (or decrease) risk for AD. Therefore, major efforts to identify causative variants continue. These have been greatly facilitated by the development of high throughput sequencing, allowing for large scale sequencing of exomes and genomes from AD patients and unaffected controls. For instance, the Alzheimer’s disease sequencing project (ADSP) is sequencing more than 600 whole genomes from a family-based study and more than 10,000 exomes from AD cases and controls. Coupled with large-scale sequencing studies are large-scale clinical based studies such as the Alzheimer’s disease neuroimaging initiative (ADNI) that aims to develop clinical, imaging, genetic and biochemical biomarkers for the early detection and tracking of AD. Information coming out of projects such as ADSP, ADNI and the Accelerating Medicines Partnership (AMP) should greatly facilitate the development of the next generation of mouse models for LOAD, ensuring that new models are as human-relevant as possible.
4. Maximizing the power of genome engineering and genetics to model AD in mice
We are in the middle of a revolution in mouse-based research because of the development of (i) high-throughput sequencing promising the identification of increased numbers of candidate causative variants for human diseases, (ii) genome engineering technologies allowing for development of engineered mice in months not years at a fraction of the cost, and (iii) the development of new generations of inbred and outbred mice that allow for more precise identification of genes and variants that impact disease phenotypes. Collectively, these advances are likely to play a crucial role in understanding AD pathogenesis and in generating improved mouse models for preclinical testing.
4.1. Genome editing with CRISPR
The discovery that naturally existing nucleases such as CRISPRs [28] can alter individual bases, often termed ‘genome editing’, means that genetically engineered mice can be created efficiently and cost effectively. Further advances are expected that will include incorporating larger stretches of DNA enabling replacement of human gene sequences to the corresponding mouse locus (humanizing the mouse) and generating conditional alleles and new cre driver lines to study the temporal and spatial function(s) of genes. Thus, candidate variants can readily be assessed in mice by genome editing. Given that LOAD is likely caused by variations in multiple genes, it is critical that genome editing allows the introduction of multiple variants simultaneously. Therefore, future mouse models for LOAD based on human sequence variation should have greatly improved utility for preclinical and translational studies. These strategies can complement genetic manipulation strategies in human tissues/cells including the ability to assess variants in specific cell types derived using induced pluripotent stem cells (IPSCs) [29, 30].
4.2. Identifying genetic modifiers using inbred and outbred mouse populations
The majority of mouse models for AD exist on one or only a few genetic backgrounds. Previous studies have shown that modifying the genetic background can have significant impact on AD-relevant phenotypes. However, systematic assessment of AD mutations on the most genetically diverse mouse strains (including wild-derived strains such as CAST/EiJ) has not been performed. Also, a major limitation of using quantitative trait loci (QTL) mapping is the lack of power to identify specific variations that affect the phenotype. More often, a large genetic interval was identified. This disadvantage has led to the development of new, genetically diverse inbred mouse strains (Collaborative Cross, CC) and genetically unique outbred strain (Diversity Outbred, DO) [31]. CC and DO mice capture the greatest amount of genetic diversity currently available in mice, so studies using DO mice greatly reduce the size of QTL loci and therefore the number of candidate variants for further testing [32, 33]. As mouse models for AD improve, DO and CC strains hold great promise for identifying genetic modifiers that can be tested as novel therapeutic targets.
5. Improving reproducibility of existing and the next generation of mouse models
The majority of current animal models of AD develop amyloidosis and tauopathy and therefore much of the molecular and physiological phenotyping methods surround APP and tau pathology. Indeed, the most stable and commonly used biomarkers used in human patients today are CSF measurements of Aβ42/Aβ40 and p-Tau/t-Tau; these have been reviewed previously ([34, 35]. However, additional pathologies may be critical in the development of AD and include neuroinflammation, neurogenesis disruption, and vascular dysfunction that should be considered in the phenotypic assessment of AD models. Also, due to the recent concerns over the use of behavioral endpoints with animal models (https://www.nia.nih.gov/research/recommendations-nih-ad-research-summit-2015), we conclude this section with recommendations for best practices in behavioral testing to address issues of reproducibility and clinical translatability.
5.1 Amyloidosis and tau pathology
Current mouse models of AD have been successful at separately modeling amyloidosis and tau pathology. As discussed earlier, these models are necessary to understand early drivers of amyloidosis and tau pathology, especially when study design takes into account disease relevant changes in the context of normal aging. This is particularly highlighted with the early success of aducanumab, a drug developed by Biogen that binds to and reduces amyloid plaques in human cases. Much of the success of aducanumab trials compared with other failed amyloid targeting drugs may be related to better identification of individuals in the early stages of AD and early treatment with the drug. Regardless, this has caused renewed enthusiasm for amyloidosis inhibiting therapies.
However, the relationship between amyloidosis and tau pathology remains elusive, and improved models in this domain are essential. The amyloid cascade hypothesis is not limited to the accumulation of Aβ plaques, but includes oligomeric aggregates as these appear to be better correlated to early memory impairments and severity of dementia [36]. Recent research has shown that Aβ oligomers prompt tau oligomerization [37]. One such species, Aβ*56, has been hypothesized to be a molecular trigger of disease onset and is shown to be correlated with levels of total tau and phosphorylated tau in those with an elevated risk for developing AD ([38]. Presence of Aβ*56 in AD mouse models has been variable, though when present or injected it is correlated with strong memory deficits. Mice treated with anti-tau oligomer-specific monoclonal antibody show reduced levels of Aβ*56 and show improved performance on memory tasks, hinting at a direct interaction between Aβ and tau [39]. The diffuse dispersal of Aβ*56 throughout many brain regions may lead to a more substantial impact on synaptic function and morphology in comparison with the high degree of plaque localization of Aβ dimers [40]. This highlights the importance of characterization and localization of processed and unprocessed Aβ aggregates in current and future AD models. In order to understand the true potential of amyloidosis-inhibiting therapies it is not only critical to target Aβ in preclinical stages, but equally important to target the appropriate Aβ species. The use of more diverse mouse genetic backgrounds in amyloidosis modeling may uncover yet unknown forms of Aβ that may also contribute to disease onset and severity.
Amyloid reduction therapies have been shown to lead to the undesired increase in tau phosphorylation as a consequence, and the inverse is also true: antagonism of tau phosphorylation also results in an increase in amyloidosis [39, 41]. Earlier this year Calafate and colleagues (2015) demonstrated in vitro neuron-to-neuron transmission of tau across the synapse[42]. Decreases in synaptic density or activity appeared to weaken this propagation and this suggests that therapies focused on promoting synaptogenesis in later stages of AD should be discouraged. While it remains to be confirmed in vivo, this study emphasizes importance of understanding the dynamic kinetics of neurodegenerative proteins. The decrease or downregulation of a particular cell product or type may in fact be a protective mechanism rather than simply the consequence of disease.
In a similar vein, much research has focused on how the brain normally clears toxic byproducts. The pathways through which Aβ and tau are removed from the brain constitute the glymphatic system-cerebral arteries create a convective force allowing CSF to circulate inside the parenchyma along arterial walls where it encounters interstitial fluid and flushes interstitial proteins. As demonstrated using a genetic knock-out in a mouse model, this system is strongly dependent on the water channel aquaporin-4 located in astrocytic end feet [43]. Reductions in the efficiency of the glymphatic system are seen with age as pulsatility of arteries wanes and localization of the aquaporin-4 receptor becomes more erratic [44]. A strong relationship between glymphatic system function and sleep and wake cycles has also been established. In a now seminal paper, researchers at the Center for Translational Neuromedicine were able to use two-photon imaging in live mice to demonstrate a 60% increase in interstitial space while the animals were engaged in sleep or under the influence of anesthesia, suggesting that clearance of neuronal waste is enhanced during sleep [45]. This is further supported by work that shows that levels of Aβ follow a diurnal pattern: Aβ40 and Aβ42 levels are higher in the CSF in non-diseased young individuals during wakefulness and lower during sleep [46]. Thus, changes to sleep duration could impact the ability of the brain to effectively expel protein aggregates. Disruptions in sleep patterns are also a common component of aging [47, 48], and this paired with the decreased efficiency of the glymphatic system could contribute to disease pathology. This relationship becomes more complex as recent work demonstrated that Aβ exerts direct effects on sleep, specifically on non-rapid eye movement slow wave sleep (NREM SWS). NREM SWS is associated with long-term hippocampus-dependent memory consolidation; therefore the accumulation of Aβ may indirectly contribute to memory impairments through disruption of NREM SWS [49]. Sleep disturbances also trigger a neuroinflammatory response, which with age-related alterations in immune function can lead to persistent neuroinflammation [50].
5.2. The emergence of neuroinflammation as a key component of Alzheimer’s disease
Convincing evidence from both pathological and genetic studies shows neuroinflammation plays a key role in the susceptibility, onset and progression of AD pathology. At the forefront of this response is the activation of astrocytes and microglia. While the short-term release of inflammatory cytokines, chemokines and activation of the complement system by these cells is beneficial and neuroprotectant, long-term activation and proliferation can lead to neuronal damage. Recent studies suggest chronic neuroinflammation could be a key contributing factor to neurodegenerative features of AD. However, it is important to note that an increase in neuroinflammation with age is normal and as AD is tightly coupled to aging, it is important to elucidate these processes in parallel [51].
Of late there has been renewed interest in the role of microglial receptors such as cannabinoid receptor 2 (CB2) in AD. Studies have focused on the utility of CB2 as a biomarker of Aβ-related inflammation, particularly in early stages of disease progression prior to significant neuronal loss [52]. Increased expression of CNR2, the gene that encodes CB2 receptor, is present in AD brains along with increased CB2 expression in plaque-associated microglia, and expression levels are also correlated with cognitive decline [53, 54]. This is also seen in patients with Down’s syndrome-associated Aβ amyloidosis [55]. This phenotype is present in AD mouse models. CRN2 KO mice show an increase in amyloidosis and alterations in tau processing that led to a decrease in measures of total tau [41]. Taken together, this suggests that CB2 should be considered as a marker of neuroinflammation.
5.3 Neuronal dysfunction/loss – a key hallmark missing in current Alzheimer’s disease models
As was mentioned previously, while AD is a disorder characterized by neuronal degeneration, there are currently no animal models that demonstrate significant neuron loss. Therefore, a major goal is to generate mouse models that develop significant neuronal pathology such as synaptic and axonal dysfunction/loss. Regions of the cortex, particularly the entorhinal cortex, and the hippocampus are some of the earliest brain regions to be affected in human AD. The hippocampus includes the dentate gyrus, CA1, CA2, and CA3 fields and subiculum. A plethora of animal lesion and inactivation work along with human case studies have established the critical role of this structure in learning and memory processes; thus, early AD symptoms of memory loss can be attributed to hippocampal disruption. In fact, two of the earliest changes reported are to the two major underlying sources of hippocampal plasticity: neurogenesis and long-term potentiation (LTP) [56].
Impairments in adult neurogenesis have been reported in human AD cases [57, 58], and alterations appear to be mutation-specific in murine models. Some models such as the J20 line show increased neurogenesis but significant impairments in LTP. Other models such as the APP/PS1 mice showed a decrease in both neurogenesis and LTP [59]. It is important to note that while neurogenesis and LTP appear to be intimately linked, blockade of neurogenesis impairs dentate gyrus LTP only for a limited time- restoration of LTP is observed within 6 weeks despite the continued absence of neurogenesis [60].
Many of the newly identified genes for LOAD are involved in neuroinflammation, which directly impacts neurogenesis and LTP [61]. Work by Moriyama and colleagues (2011) localized complement receptor 2 to the surface of neural progenitor cells[62]. Cr2−/− mice have a significant increase in immature neuroblasts and 40% increase in mature neurons, suggestive that the role of Cr2 is to dampen adult hippocampal neurogenesis. Neural stem cell populations are also influenced by endocannabinoid signaling; upregulation of CB1 and CB2 activity stimulate adult hippocampal neurogenesis, whereas inhibition reduces it [63]. More research is necessary to elucidate the role of endocannabinoids in AD-related impairments in neurogenesis. Regardless, markers of neurogenesis such as bromodeoxyridine (BrdU), doublecortin (DCX) and calretinin (CALB2) should be considered in the characterization of all AD mouse models.
5.4 Vascular dysfunction
Historically, vascular dysfunction has not been included as part of the pathology of AD, with vascular dementia being considered a different form of dementia to AD. More recently, strong evidence supports a key role of vascular disruption in AD and therefore is an important area to study in mouse models. The double hit hypothesis for AD has been proposed whereby vascular dysfunction precedes and promotes amyloid toxicity. In support of this, Apoe deficiency or mice carrying a human APOE4 transgene can show a disrupted blood brain barrier [64, 65]. APOE4 is proposed to induce the activation of the proinflammatory cyclophilin A (CypA)–matrix metalloproteinase 9 (MMP-9) pathway in brain pericytes leading to the breakdown of the BBB and neurodegeneration. A recent study from our lab shows that vascular dysfunction, including basement membrane breakdown, pericyte loss and vascular leakage, occurs in the cortex and hippocampus of aging mice[66]. Although more work is needed, data support a role of astrocytic APOE whose expression declines with age, possibly as a result of astrocyte senescence. Vascular dysfunction was prevented by exercise, suggesting a possible mechanism by which these types of treatment paradigms may delay AD onset or slow progression.
Further evidence for vascular dysfunction in AD came from GWAS study that showed that copy number variations in mesenchyme homeobox 2 (MEOX2), a gene involved in vascular development, are associated with severe forms of AD [67]. Mice carrying only one copy of Meox2 also have altered vasculature. Therefore, blood-brain barrier disruption needs to be considered in current and future models of AD.
The neuro-vascular unit comprises multiple cell types including astrocytes, pericytes and endothelial cells. Complex communications between these cell types are required for the development and maintenance a healthy neurovascular unit [68] and disruption of even some of them can lead to a dysfunctional vasculature. Importantly, there is an intimate relationship between neurovascular health and neuroinflammation, including immune cell infiltration, and mouse models can facilitate a better understanding of these processes.
5.5 Improved Translation of mouse models for Alzheimer’s disease
A tiered phenotyping approach should be employed for evaluating mouse models of Alzheimer’s disease that are not solely limited to the primary endpoint (e.g. cognitive measures) but also includes evaluations of general locomotor and exploratory behaviors, measures of disease progression which encompass typical age-related impairments such as vision, hearing, fine motor skills, and metabolism, and co-morbidity phenotypes related to AD such as anxiety, depression, motivation, and sleep disturbances (Table 3 and Figure 2). Importantly, behavioral endpoints that could be influenced by another competing behavior should be well characterized; hence the need for activity measures independently (e.g., open field assessment) and as a measurable endpoint within assays that require an activity dependent measure. For example, the typical battery of cognition tests used to evaluate mouse models of AD (i.e., water maze, fear conditioning, y-maze) are motor based assays and it is crucial to ensure that alterations in activity levels in the disease model relative to the WT control is not driving any perceived cognitive deficit. Specifically, hyperactivity which has been reported to be observed in a number of AD mouse models[69] may be the main reason for mice not being capable of the freezing response required to demonstrate intact memory in a fear conditioning assay. Further, mice should be tested for visual impairments to ensure that deficits in cognitive tests that employ visual cues are not confounded by impaired visual acuity that may be a known factor dependent upon background strain, but should not be ruled out in the mutant relative to its WT control (reviewed in [2]).
Table 3.
Relevant assays for comprehensive phenotyping of mouse models of Alzheimer’s disease
| Behavioral Domain | Assay |
|---|---|
| Motor, Coordination, & Strength | Locomotor Activity – Open Field |
| Gait - Treadmill or spontaneous walking | |
| Coordination – accelerating rotarod | |
| Strength – grip strength | |
| Cognition | Learning & Memory – Water Maze |
| Learning & Memory – Contextual Fear Conditioning | |
| Learning & Memory - Visual Discrimination & Reversal Task (Touchscreen) | |
| Learning & Memory- Paired Associates Learning Task (Touchscreen) | |
| Learning & Memory – Baited Holeboard Task | |
| Memory – Novel Object or Novel Spatial Recognition Task | |
| Spatial Working Memory – Spontaneous Alternation | |
| Episodic Memory - Episodic Memory Task | |
| Attention – 5 Choice Serial Reaction Time Task | |
| Disease Progression | Vision- electroretinography |
| Visual acuity – optokinetics | |
| Hearing – Auditory Brainstem Responding | |
| Hearing – Acoustic Startle Response | |
| Fine Motor – adhesive removal test | |
| Metabolism – Indirect Calorimetry | |
| Blood Pressure Monitoring | |
| Bone Densitometry (DEXAscan) | |
| Co-morbidity Phenotypes | Anxiety – Light/Dark Test |
| Anxiety - Stress-Induced Hyperthermia Assay | |
| Anxiety – Elevated Zero or Plus Maze | |
| Depression – Forced Swim Test | |
| Depression – Tail Suspension Test | |
| Social Behavior | |
| Motivation – Progressive Ratio Responding | |
| Motivation – sexual behavior | |
| Circadian Activity– Wheel Running | |
| Sleep - EEG |
Figure 2.
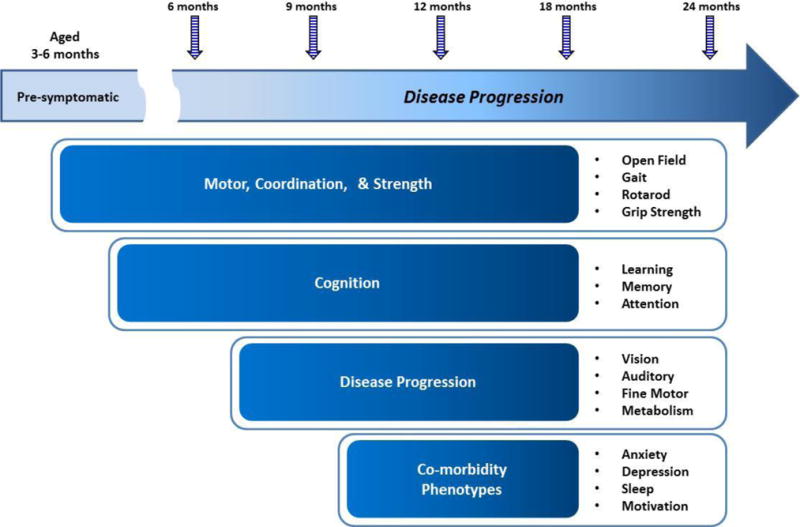
A tiered longitudinal phenotypic assessment
In order to improve the translational utility of animal models, the preclinical strategy should be designed in parallel to the clinical strategy. For example, if clinical trials will not be employing spatial learning memory as part of the functional assessment in patients, then results in the water maze may not be useful. It has been reported that some of the earliest deficits observed in AD patients when pathology is likely minimal are impairments in episodic memory (reviewed in [70]). Recent development of behavioral assays that specifically assess episodic memory may be of greater value than traditional rodent learning and memory tasks such as fear conditioning and water maze, which may only be impaired as disease has progressed significantly in the mouse model, to the point that deficits in these tasks may be confounded by deficits in motor and physiology. New technologies for cognitive assessments in rodents using touchscreen technology provide improved translation from mouse to man and include tests for attention, learning, and memory. This advanced technology for assessing cognitive measures in animal models not only includes similar visual cues that can be used across species, but also the similar tactile responses used in the clinic (i.e. touchscreen tablets or iPads) [71–73]. These technologies are not without their limitations however, as the test subjects require weeks to months of testing, food restriction for reward motivation, and to date it remains unknown whether the touchscreen tests have predictive validity.
5.5 Reproducibility
The ongoing criticisms of the lack of ability to reproduce scientific data has been a frustrating reality; in particular to those who are experts in the field of behavioral neuroscience and who can recognize the lack of important environmental and experimental details not described in the initially reported datasets [74, 75]. While criticisms have often focused on those that are unable to reproduce the initial discovery, it is just as likely that the originally published datasets were not necessarily produced under the optimal conditions or with the appropriate controls that deem it reproducible in the first place; notwithstanding the lack of critical details published in the methods sections [75–79]. Poorly conducted behavioral studies often performed by minimally experienced scientists with limited training in behavioral pharmacology, driven by pressures to provide functionally translational and relevant endpoints for molecular findings often leads to rushed experiments without proper controls and optimized testing conditions [80]. Furthermore, similar to the way clinical trials are carefully planned, preclinical studies for drug discovery need to follow a similar planning process including predetermined sample size, blinding, randomization, counterbalancing, and the inclusion of appropriate controls. The piecing together of several small underpowered experiments to achieve the desired results, typically by adding in subjects to treatment groups retrospectively when the initial experiments did not in themselves yield statistical significance independently of each other, is not an acceptable practice for a clinical trial design and is also not acceptable for preclinical studies [78] [80]. While pilot studies with small sample sizes are helpful to inform power calculations for follow up experiments, separate appropriately powered experiments should be planned and then reproduced to confirm positive outcomes in an independent cohort [81]. Importantly, behavioral tests should be conducted by highly experienced technicians proficient in conducting sensitive behavioral studies under blinded conditions and considerations should be made for second site or cross laboratory confirmation to ensure confidence and reproducibility.
5.6 Improving Clinical Translation
To date there remains a gap in the translatability of preclinical data. While more rigorous experimental designs are an important requirement, a clear understanding of the experimental compound being evaluated, and more specifically its pharmacokinetic and pharmacodynamic properties will inform the selection of a relevant and target specific dose range, as well as an optimal pretreatment time and route of administration (reviewed in [82]). It is also important to not overly interpret the data generated in animal models. As previously noted, many AD mouse models have been reported to demonstrate “cognitive impairments” which may be confounded by hyperactivity in those models. A dose of an experimental compound that is mildly sedative may normalize the hyperactivity in the mouse, unmasking its ability to successfully perform the cognitive task. Data should therefore be carefully interpreted, ensuring that the dose range is specific to the target (e.g. based on receptor occupancy studies or correlative biomarker data). Furthermore, in addition to the dose range of the compound that produces the therapeutic effect, it is critically important to also identify the compound’s therapeutic window, and the dose range at which non-specific effects occur that may be adverse or which could confound the interpretation of the behavioral response (Figure 3).
Figure 3.
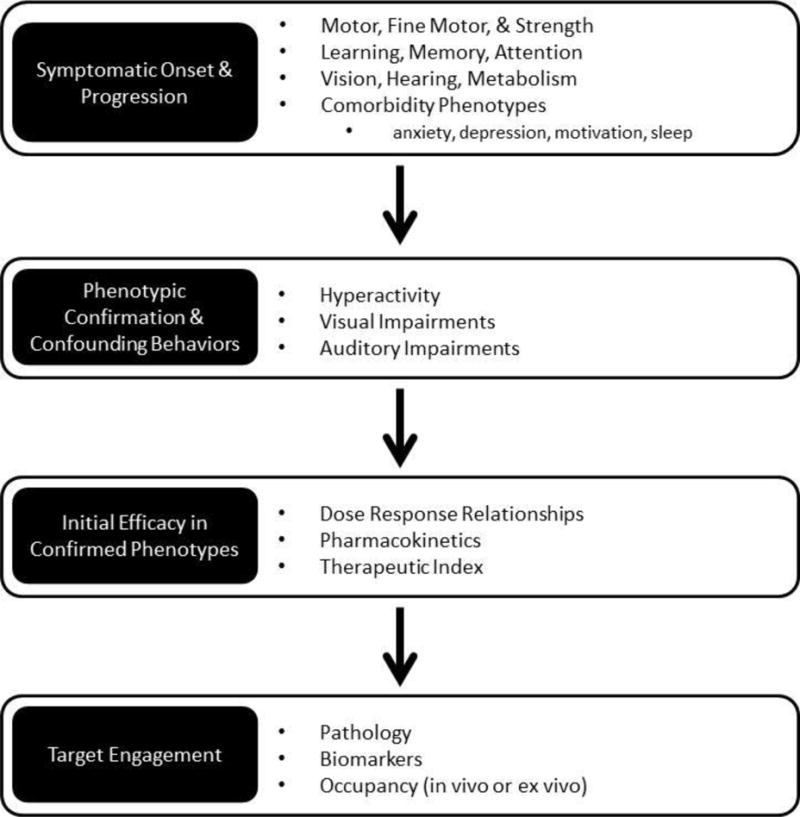
Strategy for improving the translational utility of preclinical models
6. Conclusions
While results in AD mouse models to date have not been shown to have high predictive validity, there are reasons to believe that could be changing. Improved understanding of the genetics of sporadic AD along with better tools for manipulating the mouse genome should lead to mouse models with improved construct validity, and the use of assays that more closely replicate those used in clinical trials (along with the use of biomarkers in common between the models and patients) should lead to better measures of face validity.
Highlights.
Mouse models have improved our understanding of early-onset Alzheimer’s disease.
The field lacks useful models of the more common late-onset Alzheimer’s disease.
Genomic tools are giving us a better understanding of the genetics of AD.
Improved animal modeling techniques make models easier to make and characterize.
These new technologies should lead to more predictive models in the near future.
Acknowledgments
KDO is supported by a T32 training grant. GRH is supported by NIH RF1 AG051496 and the Jane B. Cook Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that there are no conflicts of interest.
References
- 1.Bertram L, et al. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39(1):17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 2.Hall AM, Roberson ED. Mouse models of Alzheimer’s disease. Brain Res Bull. 2012;88(1):3–12. doi: 10.1016/j.brainresbull.2011.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bubela T, Cook-Deegan R. Keeping score, strengthening policy and fighting bad actors over access to research tools. Nat Biotechnol. 2015;33(2):143–7. doi: 10.1038/nbt.3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Malthankar-Phatak GH, et al. Amyloid deposition and advanced age fails to induce Alzheimer’s type progression in a double knock-in mouse model. Aging Dis. 2012;3(2):141–55. [PMC free article] [PubMed] [Google Scholar]
- 5.Saito T, et al. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17(5):661–3. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- 6.Plucinska K, et al. Knock-in of human BACE1 cleaves murine APP and reiterates Alzheimer-like phenotypes. J Neurosci. 2014;34(32):10710–28. doi: 10.1523/JNEUROSCI.0433-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Melnikova T, et al. Reversible pathologic and cognitive phenotypes in an inducible model of Alzheimer-amyloidosis. J Neurosci. 2013;33(9):3765–79. doi: 10.1523/JNEUROSCI.4251-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van der Jeugd A, et al. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012;123(6):787–805. doi: 10.1007/s00401-012-0987-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fowler SW, et al. Genetic modulation of soluble Abeta rescues cognitive and synaptic impairment in a mouse model of Alzheimer’s disease. J Neurosci. 2014;34(23):7871–85. doi: 10.1523/JNEUROSCI.0572-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willem M, et al. eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature. 2015;526(7573):443–7. doi: 10.1038/nature14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson HM, et al. Clustering of transcriptional profiles identifies changes to insulin signaling as an early event in a mouse model of Alzheimer’s disease. BMC Genomics. 2013;14:831. doi: 10.1186/1471-2164-14-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landel V, et al. Temporal gene profiling of the 5XFAD transgenic mouse model highlights the importance of microglial activation in Alzheimer’s disease. Mol Neurodegener. 2014;9:33. doi: 10.1186/1750-1326-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morihara T, et al. Transcriptome analysis of distinct mouse strains reveals kinesin light chain-1 splicing as an amyloid-beta accumulation modifier. Proc Natl Acad Sci U S A. 2014;111(7):2638–43. doi: 10.1073/pnas.1307345111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burns TC, et al. Mouse models rarely mimic the transcriptome of human neurodegenerative diseases: A systematic bioinformatics-based critique of preclinical models. Eur J Pharmacol. 2015;759:101–17. doi: 10.1016/j.ejphar.2015.03.021. [DOI] [PubMed] [Google Scholar]
- 15.Vossel KA, et al. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol. 2015;209(3):419–33. doi: 10.1083/jcb.201407065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi L, et al. Cumulative effects of the ApoE genotype and gender on the synaptic proteome and oxidative stress in the mouse brain. Int J Neuropsychopharmacol. 2014;17(11):1863–79. doi: 10.1017/S1461145714000601. [DOI] [PubMed] [Google Scholar]
- 17.Teng E, et al. Low plasma ApoE levels are associated with smaller hippocampal size in the Alzheimer’s disease neuroimaging initiative cohort. Dement Geriatr Cogn Disord. 2015;39(3–4):154–66. doi: 10.1159/000368982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerreiro R, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368(2):117–27. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan G, et al. CD33 modulates TREM2: convergence of Alzheimer loci. Nat Neurosci. 2015;18(11):1556–1558. doi: 10.1038/nn.4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benitez BA, Cruchaga C, G. United States-Spain Parkinson’s Disease Research TREM2 and neurodegenerative disease. N Engl J Med. 2013;369(16):1567–8. doi: 10.1056/NEJMc1306509#SA4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rayaprolu S, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borroni B, et al. Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging. 2014;35(4):934 e7–10. doi: 10.1016/j.neurobiolaging.2013.09.017. [DOI] [PubMed] [Google Scholar]
- 23.Cady J, et al. TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol. 2014;71(4):449–53. doi: 10.1001/jamaneurol.2013.6237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ulrich JD, et al. Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. doi: 10.1186/1750-1326-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jay TR, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212(3):287–95. doi: 10.1084/jem.20142322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 2015;160(6):1061–71. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vardarajan BN, et al. Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol. 2015;78(3):487–98. doi: 10.1002/ana.24466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–55. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Q, et al. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ Res. 2014;115(5):488–92. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tucker BA, et al. Duplication of TBK1 Stimulates Autophagy in iPSC-derived Retinal Cells from a Patient with Normal Tension Glaucoma. J Stem Cell Res Ther. 2014;3(5):161. doi: 10.4172/2157-7633.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Churchill GA, et al. The Diversity Outbred mouse population. Mamm Genome. 2012;23(9–10):713–8. doi: 10.1007/s00335-012-9414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang H, et al. Subspecific origin and haplotype diversity in the laboratory mouse. Nat Genet. 2011;43(7):648–55. doi: 10.1038/ng.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svenson KL, et al. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics. 2012;190(2):437–47. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosen C, Zetterberg H. Cerebrospinal fluid biomarkers for pathological processes in Alzheimer’s disease. Curr Opin Psychiatry. 2013;26(3):276–82. doi: 10.1097/YCO.0b013e32835f6747. [DOI] [PubMed] [Google Scholar]
- 35.Blennow K, et al. Amyloid biomarkers in Alzheimer’s disease. Trends Pharmacol Sci. 2015;36(5):297–309. doi: 10.1016/j.tips.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Broersen K, Rousseau F, Schymkowitz J. The culprit behind amyloid beta peptide related neurotoxicity in Alzheimer’s disease: oligomer size or conformation? Alzheimers Res Ther. 2010;2(4):12. doi: 10.1186/alzrt36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lasagna-Reeves CA, et al. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49(47):10039–41. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- 38.Handoko M, et al. Correlation of specific amyloid-beta oligomers with tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol. 2013;70(5):594–9. doi: 10.1001/jamaneurol.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castillo-Carranza DL, et al. Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J Neurosci. 2015;35(12):4857–68. doi: 10.1523/JNEUROSCI.4989-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zahs KR, Ashe KH. beta-Amyloid oligomers in aging and Alzheimer’s disease. Front Aging Neurosci. 2013;5:28. doi: 10.3389/fnagi.2013.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koppel J, et al. CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer’s disease. Mol Med. 2014;20:29–36. doi: 10.2119/molmed.2013.00140.revised. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calafate S, et al. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015;11(8):1176–83. doi: 10.1016/j.celrep.2015.04.043. [DOI] [PubMed] [Google Scholar]
- 43.Iliff JJ, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jessen NA, et al. The Glymphatic System: A Beginner’s Guide. Neurochem Res. 2015 doi: 10.1007/s11064-015-1581-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xie L, et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science. 2013;342(6156):373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang Y, et al. Effects of age and amyloid deposition on Abeta dynamics in the human central nervous system. Arch Neurol. 2012;69(1):51–8. doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crowley K. Sleep and sleep disorders in older adults. Neuropsychol Rev. 2011;21(1):41–53. doi: 10.1007/s11065-010-9154-6. [DOI] [PubMed] [Google Scholar]
- 48.Pace-Schott EF, Spencer RM. Age-related changes in the cognitive function of sleep. Prog Brain Res. 2011;191:75–89. doi: 10.1016/B978-0-444-53752-2.00012-6. [DOI] [PubMed] [Google Scholar]
- 49.Mander BA, et al. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18(7):1051–7. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wisor JP, Schmidt MA, Clegern WC. Evidence for neuroinflammatory and microglial changes in the cerebral response to sleep loss. Sleep. 2011;34(3):261–72. doi: 10.1093/sleep/34.3.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochemical Pharmacology. 2014;88(4):594–604. doi: 10.1016/j.bcp.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Savonenko AV, et al. Cannabinoid CB2 Receptors in a Mouse Model of Abeta Amyloidosis: Immunohistochemical Analysis and Suitability as a PET Biomarker of Neuroinflammation. PLoS One. 2015;10(6):e0129618. doi: 10.1371/journal.pone.0129618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Solas M, et al. CB2 receptor and amyloid pathology in frontal cortex of Alzheimer’s disease patients. Neurobiol Aging. 2013;34(3):805–8. doi: 10.1016/j.neurobiolaging.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Grunblatt E, et al. Gene expression as peripheral biomarkers for sporadic Alzheimer’s disease. J Alzheimers Dis. 2009;16(3):627–34. doi: 10.3233/JAD-2009-0996. [DOI] [PubMed] [Google Scholar]
- 55.Nunez E, et al. Glial expression of cannabinoid CB(2) receptors and fatty acid amide hydrolase are beta amyloid-linked events in Down’s syndrome. Neuroscience. 2008;151(1):104–10. doi: 10.1016/j.neuroscience.2007.10.029. [DOI] [PubMed] [Google Scholar]
- 56.Poirier R, et al. Enhanced dentate gyrus synaptic plasticity but reduced neurogenesis in a mouse model of amyloidosis. Neurobiol Dis. 2010;40(2):386–93. doi: 10.1016/j.nbd.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 57.Ziabreva I, et al. Altered neurogenesis in Alzheimer’s disease. J Psychosom Res. 2006;61(3):311–6. doi: 10.1016/j.jpsychores.2006.07.017. [DOI] [PubMed] [Google Scholar]
- 58.Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur J Neurosci. 2011;33(6):1139–51. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- 59.Crews L, Rockenstein E, Masliah E. APP transgenic modeling of Alzheimer’s disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Struct Funct. 2010;214(2–3):111–26. doi: 10.1007/s00429-009-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singer BH, et al. Compensatory network changes in the dentate gyrus restore long-term potentiation following ablation of neurogenesis in young-adult mice. Proc Natl Acad Sci U S A. 2011;108(13):5437–42. doi: 10.1073/pnas.1015425108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fuster-Matanzo A, et al. Role of neuroinflammation in adult neurogenesis and Alzheimer disease: therapeutic approaches. Mediators Inflamm. 2013;2013:260925. doi: 10.1155/2013/260925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Moriyama M, et al. Complement receptor 2 is expressed in neural progenitor cells and regulates adult hippocampal neurogenesis. J Neurosci. 2011;31(11):3981–9. doi: 10.1523/JNEUROSCI.3617-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di Marzo V, Stella N, Zimmer A. Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci. 2015;16(1):30–42. doi: 10.1038/nrn3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Methia N, et al. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. 2001;7(12):810–5. [PMC free article] [PubMed] [Google Scholar]
- 65.Bell RD, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Soto I, et al. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. Plos Biology. 2015;13(10) doi: 10.1371/journal.pbio.1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu Z, et al. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11(9):959–65. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- 68.Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med. 2013;19(12):1584–96. doi: 10.1038/nm.3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rodgers SP, et al. Transgenic APP expression during postnatal development causes persistent locomotor hyperactivity in the adult. Mol Neurodegener. 2012;7:28. doi: 10.1186/1750-1326-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Webster SJ, et al. Using mice to model Alzheimer’s dementia: an overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front Genet. 2014;5:88. doi: 10.3389/fgene.2014.00088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bussey TJ, et al. New translational assays for preclinical modelling of cognition in schizophrenia: the touchscreen testing method for mice and rats. Neuropharmacology. 2012;62(3):1191–203. doi: 10.1016/j.neuropharm.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Romberg C, Bussey TJ, Saksida LM. Paying more attention to attention: towards more comprehensive cognitive translation using mouse models of Alzheimer’s disease. Brain Res Bull. 2013;92:49–55. doi: 10.1016/j.brainresbull.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 73.Romberg C, et al. Impaired attention in the 3xTgAD mouse model of Alzheimer’s disease: rescue by donepezil (Aricept) J Neurosci. 2011;31(9):3500–7. doi: 10.1523/JNEUROSCI.5242-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van der Staay FJ, Steckler T. The fallacy of behavioral phenotyping without standardisation. Genes Brain Behav. 2002;1(1):9–13. doi: 10.1046/j.1601-1848.2001.00007.x. [DOI] [PubMed] [Google Scholar]
- 75.Steckler T. Preclinical data reproducibility for R&D–the challenge for neuroscience. Psychopharmacology (Berl) 2015;232(2):317–20. doi: 10.1007/s00213-014-3836-3. [DOI] [PubMed] [Google Scholar]
- 76.Kilkenny C, et al. Survey of the quality of experimental design, statistical analysis and reporting of research using animals. PLoS One. 2009;4(11):e7824. doi: 10.1371/journal.pone.0007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kilkenny C, et al. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. J Pharmacol Pharmacother. 2010;1(2):94–9. doi: 10.4103/0976-500X.72351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Landis SC, et al. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490(7419):187–91. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Steward O, Balice-Gordon R. Rigor or mortis: best practices for preclinical research in neuroscience. Neuron. 2014;84(3):572–81. doi: 10.1016/j.neuron.2014.10.042. [DOI] [PubMed] [Google Scholar]
- 80.Unger EF. All is not well in the world of translational research. J Am Coll Cardiol. 2007;50(8):738–40. doi: 10.1016/j.jacc.2007.04.067. [DOI] [PubMed] [Google Scholar]
- 81.Sukoff Rizzo SJ. In: Mouse Models for Drug Discovery. Wiles Pa., editor. 2015. [Google Scholar]
- 82.Rizzo SJ, et al. Future viable models of psychiatry drug discovery in pharma. J Biomol Screen. 2013;18(5):509–21. doi: 10.1177/1087057113475871. [DOI] [PubMed] [Google Scholar]