

Abstract
There are many opportunities to use macromolecules, such as peptides and oligonucleotides, for intracellular applications. Despite this, general methods for delivering these molecules to the cytosol in a safe and efficient manner are not available. Efforts to develop a variety of intracellular drug delivery systems such as viral vectors, lipoplexes, nanoparticles, and amphiphilic peptides have been made, but various challenges such as delivery efficiency, toxicity, and controllability remain. A central challenge is the ability to selectively perturb, not destroy, the membrane to facilitate cargo introduction. Herein, we describe our efforts to design and characterize peptides that form pores inside membranes at acidic pH, so-called pH-switchable pore formation (PSPF) peptides, as a potential means for facilitating cargo translocation through membranes. Consistent with pore formation, these peptides exhibit low-pH-triggered selective release of ATP and miRNA, but not hemoglobin, from red blood cells. Consistent with these observations, biophysical studies (tryptophan fluorescence, circular dichroism, size-exclusion chromatography, analytical ultracentrifugation, and attenuated total reflectance Fourier transformed infrared spectroscopy) show that decreased pH destabilizes the PSPF peptides in aqueous systems while promoting their membrane insertion. Together, these results suggest that reduced pH drives insertion of PSPF peptides into membranes, leading to target-specific escape through a proposed pore formation mechanism.
Graphical Abstract
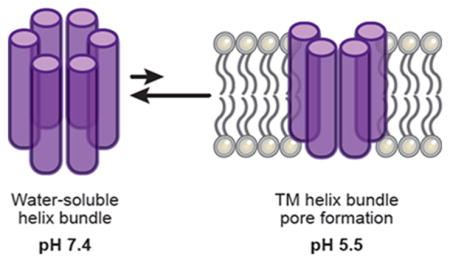
Despite years of dedicated research, efficient delivery of macromolecular substances to the cytosol of target cells or intracellular compartments is still a major challenge.1,2 For certain macromolecules cellular uptake can be spontaneous. However, the general task, known as the delivery problem, is largely unsolved.1,3 This is primarily because biological membranes serve as effective barriers that prevent most substances from freely flowing in and out of cells and between organelles. Many approaches have been developed to deliver nonpermeable molecules to intracellular compartments, including encapsulation of the desired molecule in a lipid or polymer framework, attachment to a molecule capable of facilitating uptake, or coadministration with cell permeable peptides.4 While powerful for in vitro and some in vivo applications, they suffer from either poor efficacy or unacceptable toxicity. In general, lipid nanoparticle and polymer-based systems are the most active and rely upon a membrane lysis mechanism. Although effective, these systems typically exhibit unacceptable toxicity, which is thought, in part, to be a result of unselective membrane perturbation. Thus, there is still a critical need to develop systems that facilitate macromolecular cargo translocation across membranes in a controlled and minimally disruptive manner.
In nature, various pathogens must reach the cytosol to replicate or better exert their biological activity. Commonly, this is achieved by first hijacking pre-existing entry mechanisms (e.g., endocytosis) followed by translocation into the cytoplasm. Critically, this latter endosomal escape occurs before reaching the degradative environment of the lysosome. Since the intracellular membrane also presents a strong barrier for translocation of larger molecules, different mechanisms for cytosolic delivery have evolved. Some macromolecules, like Shiga and Ricin toxins, use the cell’s own translocation machinery in the endoplasmic reticulum,5 whereas others take advantage of molecular cues within the cells.6 One such cue is the difference in pH between the neutral pH in the extracellular space and the lower pH within endosomes. For enveloped viruses like influenza A, endosomal escape is mediated by fusion of viral lipids with endosomal membrane lipids at low pH.7,8 Other pathogens do not require lipid–lipid fusion events but form defined pores in the endosomes. One example is the escape mechanism of the Anthrax toxin.9,10 This toxin forms defined pores in endosomes, allowing the enzymatic subunits of the toxin (lethal factor and edema factor) to be translocated into the cytosol. Importantly, because the pore formation is efficiently triggered by the low pH in the endosome, a catastrophic loss of cell integrity due to disruption of the plasma membrane is avoided. Endosomal pore formation is more likely to be tolerated by the cell since a few pores at the plasma membrane may quickly lead to complete cell destruction. It is important to note that antimicrobial peptides derived from nature also form pores as a defense mechanism.11,12
From a delivery perspective, pore formation provides an opportunity to create a well-defined system to mimic these powerful biological examples and thereby potentially overcome limitations with current synthetic approaches. On that premise, we aim here to design peptides that bind nonspecifically to biological membranes at pH 7.4 (mimicking the extracellular environment13), selectively form pores only at pH 5.5 (resembling the environment of an endosome or lysosome), and do not rupture membranes at any pH. This concept is illustrated schematically in Figure 1. We envision that following endocytosis a drop in pH would trigger the formation of pores. This is in contrast to a detergent mechanism, which would destroy the endosomal integrity.11,12 From a cargo delivery perspective, this pore formation would then facilitate release of the cargo into the cytosol. A pore-driven mechanism also has a practical advantage in that it requires a lower concentration of an appropriately designed peptide; a nonselective detergent mechanism generally requires higher peptide density across the entire membrane surface. As such, a lower dose of pore-forming peptide would potentially be required for successful application of cargo delivery. Inspired by nature and the potential advantages conferred by pore formation, we focused on rationally designing and characterizing peptides that form pores in membranes at endosomal pH. If these design criteria are ultimately understood and defined, then we envision applying this strategy to therapeutic macromolecular cargo delivery via selective pore formation.
Figure 1.
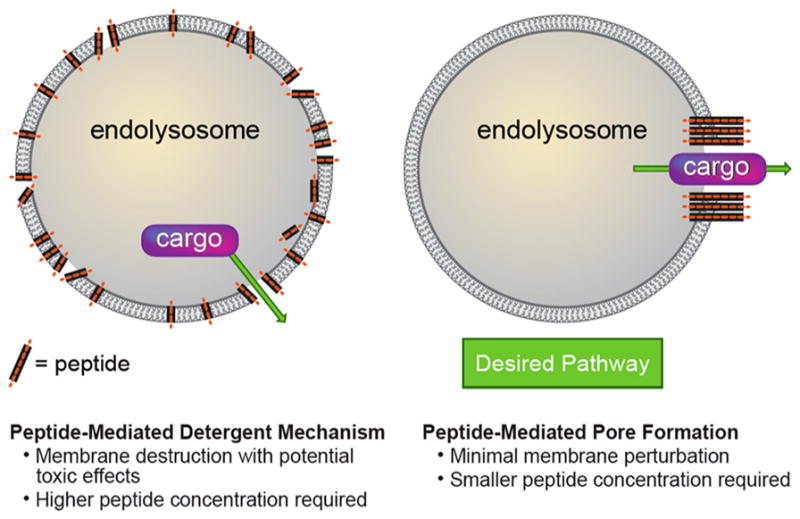
Comparison of a detergent mechanism for membrane disruption versus pore formation.12
RESULTS
Design Strategy
Our initial focus was to design 28-residue peptides composed of four seven-residue repeats. The overall length of a 28-residue linear and helical peptide is approximately 42 Å, which is sufficient to span the hydrophobic and headgroup region of the bilayer.14 A seven-residue repeat was chosen to encourage coiled-coil formation and to simplify the design process.15–21 Finally, each amino acid was chosen for its ability to facilitate a controlled interaction with membranes and water at different pH values.
To realize this pH-switchable behavior, we considered three thermodynamic states in our design process, as illustrated in Figure 2. At physiological pH, the peptide should be stored in a water-soluble form that does not interact with the membrane (State 1). A good way to encode this is to ensure the formation of a stable water-soluble helical bundle at high pH. Upon lowering the pH, this state must destabilize and allow individual peptides to interact with the membrane (State 2). Here, we consider either a surface-adsorbed form, in which helical individual peptides are engaged with the membrane surface, or a fully inserted state capable of self-associating to form a channel. Because insertion and channel formation are thermodynamically linked, the relative stability of the inserted versus surface-adsorbed states will have a concentration dependence with higher peptide concentrations favoring insertion and channel formation (State 3).
Figure 2.
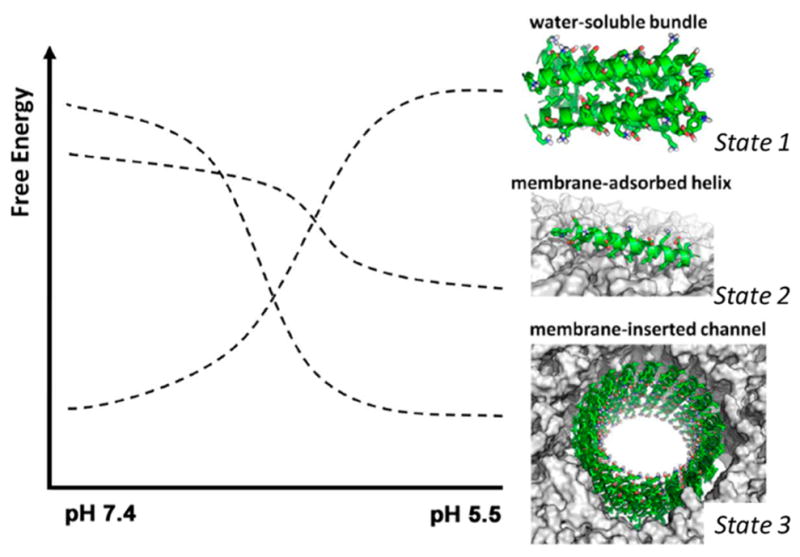
Desired free energy diagram of the designed peptide as a function of pH. Lowering pH should destabilize the water-soluble bundle (State 1) and stabilize first the membrane-associated individual peptide (State 2) and then, in a concentration-dependent manner, the membrane-inserted channel (State 3).
We postulate that we can achieve this pH modulation of stability and hydrophobicity by including amino acids in the peptide sequence whose charge state and hydrophobicity are pH-dependent, such as Asp, Glu, and His, and by considering the stability of the water-soluble coiled-coil-like bundle. In addition, we must also consider the specific inter-residue interactions of the membrane-inserted pore in selecting the design sequence, as we are interested in stabilizing a specific pore-forming state at low pH rather than simply ensuring membrane insertion. Importantly, there are already numerous examples of natural and synthetic peptides that insert into membranes or those that insert and form indiscriminately large pores.22 While informative, these motifs would constitute unsuccessful end points in our design efforts either because of lack of pore formation or potential toxicity. Thus, our overall design procedure combined the use of pH-switchable residues with the consideration of inter-residue contacts and stabilities of both the water-soluble as well as membrane-inserted pore states.
Amino Acid Selection Strategy
The design goal was to create a water-soluble peptide that associates into a stable coiled-coil bundle at neutral pH while preferring a membrane-inserted channel state at low pH. This means that upon pH decrease the nonpolar residues facing inward in the soluble bundle should invert and face the lipid phase in the membrane-inserted channel (Table 1 and Figure 3). Canonical coiled coils have only seven environmentally distinct positions, which are referred to as the heptad and designated with letters a–g, as shown in Figure 3A.23,24 We therefore focused on choosing the appropriate amino acids for each of these seven sites. Furthermore, each site in our design must play two roles: stabilizing the water-soluble hydrophobic-inside state at high pH and changing to the membrane channel hydrophobic-outside state at low pH. To impart stability on the water-soluble bundle, we chose to adhere to the canonical Leu-zipper coiled-coil motif, meaning that coiled-coil positions a and d were set to Leu. As illustrated in Figure 3B, these same residues face the lipid phase in the membrane channel state, and Leu residues are ideal for this task as well. The solvent-exposed b, c, and f positions in the water-soluble bundle should be polar to impart solubility and fold specificity, and these can also be used to modulate bundle stability through their innate helix-forming propensities. In the membrane-channel state, these positions point into the center of the channel and therefore remain water-facing, so their polar nature is appropriate here as well. However, unlike in the water-soluble state, the b and c positions are also located at the interhelical interface of the channel. Thus, the importance of these positions goes beyond their physicochemical character and includes potential interactions stabilizing specific interfacial conformations of channel helices. The interhelical geometry in the channel state is important, as it ultimately defines the shape and size of the entire channel.
Table 1.
Amino Acid Design Elementsa
| position in water | function in water, high pH | function in membrane, low pH | amino acid choice |
|---|---|---|---|
| a | helical bundle hydrophobic core | membrane-facing | Leu |
| b | solvent-exposed, imparts solubility | small residue for helical interface, potential interhelical hydrogen bonding | Ser |
| c | solvent-exposed, imparts solubility | trigger residue, changes protonation state/hydrophobicity at low pH; potential interhelical hydrogen bonding | Asp, Glu, His |
| d | helical bundle hydrophobic core | membrane-facing | Leu |
| e | modulation of helical propensity | small residue for helical interface | Ala |
| f | solvent-exposed, imparts solubility | solvent-exposed in channel state (inner channel lining); imparts folds specificity by encoding helical orientation preference | Lys, Gln |
| g | modulation of helical propensity | small residue for helical interface | Ala, Gly |
Refer to Figure 3 for corresponding helical description.
Figure 3.
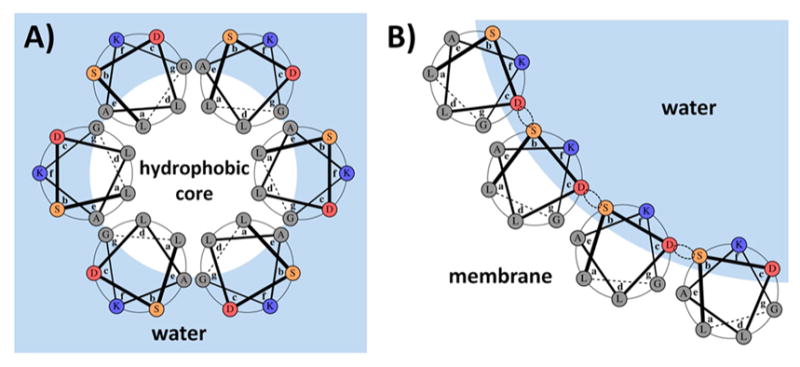
Design concept illustrated using one of the designed sequences (PSPF-DKG). Hydrophobic residues are either lining the core of the bundle in the water-soluble state (A) or are facing the lipid membrane in the membrane channel state (B). Dotted circles illustrate potential hydrogen bonding in the channel state. Heptad positions in both panels are labeled according to the water-soluble state. The amino acid choices at each position are shown in Table 1.
At the f position, we chose to consider Lys or Gln, which have favorable helix propensities and are common at this position in coiled coils.25 At position b, we considered Ser because of its slightly polar nature and intermediate helix propensity. The packing of Ser and other small residues provides a potent driving force for helix–helix association in transmembrane proteins.26–29 The c position was chosen as the pH-sensing switch. We considered amino acids Glu and Asp at this position, as their ionization state is dependent on pH, causing them to become neutral and thus more hydrophobic at lower pH. Nature uses this strategy in pH-triggered fusion proteins such as the hemagglutinins from influenza virus. These proteins have an N-terminal fusion peptide domain rich in negatively charged Glu and Asp residues that stabilize the water-soluble form at neutral pH. As the pH is lowered, these interactions are disrupted, and the affinity of the peptides for membranes is increased.30,31
Although the pKa of the carboxylic side chain groups of Glu and Asp in water are around 4 (somewhat lower than the typical endosomal pH of ~5.5), significant shifting in protonated populations would still be expected relative to neutral pH, and the collective effect of having multiple closely spaced acidic groups on one face of a helix will likely increase the effective pKa of the side chains. An additional significance of Glu and Asp residues is their potential ability to participate in interhelical hydrogen bonding, as shown in Figure 3B, thus further dialing in a specific, closely packed interhelical geometry in the membrane-channel state. As a way of testing the importance of the pH-switch residue, we also considered the amino acid His at the c position. The side chain of His has a pKa of ~6.1 and is positively charged at acidic pH. Because of the opposite charge state as compared to that of Asp and Glu, His provides a convenient point of reference.
Positions e and g are located along the helix–helix interface in both the water-soluble and the membrane-channel states. Because the primary driver of the water-soluble bundle stability is the canonical leucine-zipper motif, we opted to choose small hydrophobic residues at e and g with the primary purpose of stabilizing a closely packed transmembrane helical interface.29 On the basis of the criteria listed above, a group of sequences has been generated for the pH-switchable pore formation (PSPF) peptide, as listed in Table 2. Finally, a tryptophan residue was added to the N-terminus to help stabilize the membrane-bound state through interactions with the head-group region of the bilayer while also providing a convenient fluorescent probe of membrane interactions.
Table 2.
Sequences and Molecular Weights of PSPF Peptides
| peptide | sequence | MW (g/mol) |
|---|---|---|
| heptad in membrane | cdefgab cdefgab cdefgab cdefgab | |
| PSPF-DQA | WSDLAQA LSDLAQA LSDLAQA LSDLAQA | 2886.3 |
| PSPF-DQG | WSDLAQG LSDLAQG LSDLAQG LSDLAQG | 2830.1 |
| PSPF-DKA | WSDLAKA LSDLAKA LSDLAKA LSDLAKA | 2886.3 |
| PSPF-DKG | WSDLAKG LSDLAKG LSDLAKG LSDLAKG | 2830.3 |
| PSPF-EQA | WSELAQA LSELAQA LSELAQA LSELAQA | 2942.3 |
| PSPF-EQG | WSELAQG LSELAQG LSELAQG LSELAQG | 2886.2 |
| PSPF-EKA | WSELAKA LSELAKA LSELAKA LSELAKA | 2942.5 |
| PSPF-EKG | WSELAKG LSELAKG LSELAKG LSELAKG | 2886.3 |
| PSPF-HQA | WSHLAQA LSHLAQA LSHLAQA LSHLAQA | 2974.4 |
| PSPF-HQG | WSHLAQG LSHLAQG LSHLAQG LSHLAQG | 2918.3 |
| PSPF-HKA | WSHLAKA LSHLAKA LSHLAKA LSHLAKA | 2974.6 |
| PSPF-HKG | WSHLAKG LSHLAKG LSHLAKG LSHLAKG | 2918.4 |
| heptad in water | abcdefg abcdefg abcdefg abcdefg |
Cellular Release Examination
Red Blood Cell Hemolysis Assay
For initial characterization, the designed peptides were tested in a red blood cell (RBC) lysis assay, which is commonly used to determine membrane disruption properties of peptides via quantification of hemoglobin release.32 In our hands, this assay was carried out at pH 5.4 and 7.4 to mimic endosomal and extracellular pH, respectively. While total cell rupture was assessed by measuring the amount of hemoglobin released, the assay was expanded to include miRNA and ATP. We reasoned that the different hydrodynamic volumes of hemoglobin, miRNA, and ATP would offer a potential method (albeit crude) for inferring pore formation, as it would be expected that diffusion rates from RBCs would be different depending on the species being released. As shown in Table 3, none of the peptides listed in Table 2 resulted in the release of hemoglobin under our assay conditions at either pH, indicating that these peptides do not alter the overall integrity of the plasma membrane. Encouragingly, relatively high percentages of release of miRNA (>10%) and ATP (>20%) were observed for some of the peptides under the same assay conditions.
Table 3.
Release of Hemoglobin, ATP, and miRNA from Red Blood Cells in the Presence of PSPF Peptides
| peptide | RBC lysis assay (% relative to Triton X-100 controls)
|
|||||
|---|---|---|---|---|---|---|
| hemoglobin release
|
% ATP at 5 μM
|
% miRNA at 5 μM
|
||||
| pH 7.5 | pH 5.4 | pH 7.5 | pH 5.4 | pH 7.5 | pH 5.4 | |
| PSPF-DQA | none | none | 3.81 | 17.91 | 0.81 | 18.61 |
| PSPF-DQG | none | none | 3.21 | 8.61 | 0.06 | 0.16 |
| PSPF-DKA | none | none | 4.64 | 5.79 | 4.13 | 0.79 |
| PSPF-DKG | none | none | 7.54 | 24.1 | 5.54 | 7.47 |
| PSPF-EQA | none | none | 3.69 | 3.61 | 0.46 | 0.02 |
| PSPF-EQG | none | none | 3.36 | 9.52 | 0.15 | 2.64 |
| PSPF-EKA | none | none | 2.02 | 3.66 | 0.51 | 0.21 |
| PSPF-EKG | none | none | 3.38 | 27.3 | 0.14 | 12.54 |
| PSPF-HQA | none | none | 6.17 | 11.72 | 0.02 | 1.43 |
| PSPF-HQG | none | none | 5.69 | 10.55 | 0.2 | 0.64 |
| PSPF-HKA | none | none | 0.93 | 5.7 | 0.43 | 0.02 |
| PSPF-HKG | none | none | 32.1 | 39.22 | 72.28 | 0.44 |
Peptide Binding to Lipid Bilayers Measured by Tryptophan Fluorescence
As some of the peptides triggered a pH-dependent release of molecules from red blood cells, we next tested membrane–peptide interactions more directly via tryptophan fluorescence. Single unilamellar vesicles were prepared as described in the Material and Methods, and tryptophan fluorescence was measured for selected peptides at different pH values in the presence of a large molar excess of phospholipid. As summarized in Table 4, the extent of environmental change around the N-terminal Trp was assessed by measuring the fluorescence emission profile and tabulating intensities and shifts in maxima. Given the position of the Trp at the N-terminus of the peptide and its expected location within the headgroup of the bilayer, we expected that binding to membranes would give rise to an increase in intensity and a small shift in the emission profile.14
Table 4.
Tryptophan Fluorescence Emission of PSPF Peptides at pH 7.4 and 5.5 with and without Lipid Vesicles
| peptide | pH 7.4
|
pH 5.5
|
||||||
|---|---|---|---|---|---|---|---|---|
|
λmax (nm)
|
Δλmax (nm)a | % intensity increaseb |
λmax (nm)
|
Δλmax (nm)a | % intensity increaseb | |||
| 0 μM lipid | 200 μM lipid | 0 μM lipid | 200 μM lipid | |||||
| PSPF-DQA | 352 | 351 | −1 | 32 | 348 | 347 | −1 | 38 |
| PSPF-DQG | 354 | 353 | −1 | 18 | 350 | 358 | +8 | 33 |
| PSPF-DKA | 354 | 353 | −1 | 18 | 349 | 358 | +9 | 38 |
| PSPF-DKG | 355 | 351 | −4 | 36 | 350 | 347 | −3 | 38 |
| PSPF-EQA | 352 | 352 | 0 | 6 | 349 | 348 | −1 | 21 |
| PSPF-EQG | 355 | 354 | −1 | 15 | 349 | 346 | −3 | 42 |
| PSPF-EKA | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| PSPF-EKG | 354 | 352 | −2 | 28 | 350 | 347 | −3 | 52 |
| PSPF-HQA | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| PSPF-HQG | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| PSPF-HKA | N/A | N/A | N/A | N/A | 349 | 347 | −2 | 34 |
| PSPF-HKG | N/A | N/A | N/A | N/A | 351 | 341 | −10 | 72 |
Despite different experimental conditions between the two assays, increases in Trp emission intensities and shifts in emission maxima values are strongly correlated with increasing ATP release at pH 5.5, as illustrated in Figure 4. At pH 5.5, a blue-shift and larger increase in total Trp fluorescence (attributable to shielding of the Trp residue from water molecules and therefore indicative of insertion into the membrane) corresponds to greater release of ATP. This suggests that the peptides are acting in a similar manner in both experimental assays and is consistent with pH-sensitive insertion with concomitant release of small molecules from the cells.
Figure 4.
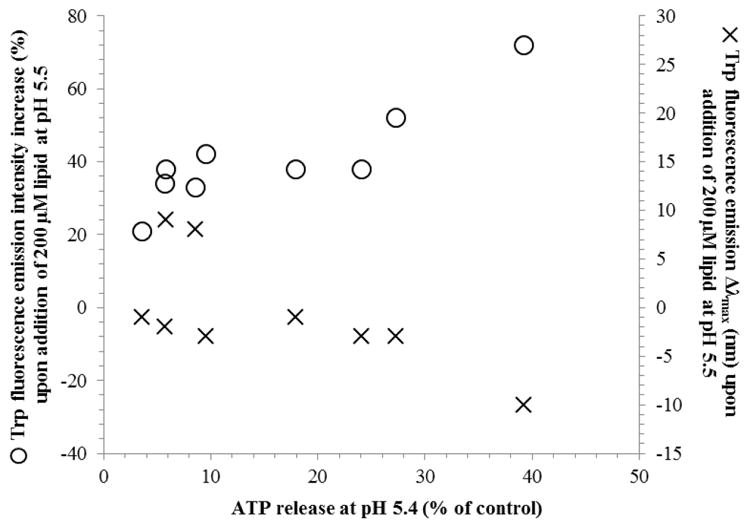
Correlation between ATP release by PSPF peptides and the degree of lipid engagement as assessed by the relative increase of Trp fluorescence emission and Δλmax upon addition of 200 μM lipid vesicles. The correlation coefficient for the increase in Trp florescence intensity as a function of ATP release is 0.88 (significant at P = 0.001 using the ρ-test).
The Association Properties of PSPF Peptides in an Aqueous System
At this point in our approach, we opted to focus resources on a more in-depth characterization of the most promising peptides: PSPF-EKG and PSPF-DKG. These peptides exhibited the desired pH-switch behavior, acceptable miRNA and ATP release, and strong membrane association characteristics.
Size-Exclusion Chromatography
The association states of the designed peptides PSPF-EKG and PSPF-DKG were investigated by size-exclusion chromatography (SEC)33 using a GE Healthcare Superdex 75 column eluted with aqueous buffers containing 150 mM NaCl and either 50 mM Tris adjusted to pH 7.4 or 50 mM MES adjusted to pH 5.5. Figure 5 summarizes the SEC results for both PSPF peptides under each condition. For comparison, the chromatograms for PSPF-DKG (red) and PSPF-EKG (green) are overlaid with the standard mixture (blue). PSPF-EKG produced well-defined chromatographic peaks with apparent molecular weights approximately 6- and 5-fold higher than the known single peptide molecular weight at pH 7.4 and 5.5, respectively (apparent molecular weights were 19 000 and 15 000 Da). As compared to the pH 7.4 conditions, the peak for PSPF-EKG at pH 5.5 was somewhat broader and more tailed, indicating that the lower pH increases nonspecific interactions with the column. Dissociation equilibria might also contribute to the poorer peak shape at pH 5.5, consistent with lower stability of the water-soluble helical bundle at the lower pH. Similarly, PSPF-DKG produced a well-defined chromatographic peak having an apparent molecular weight approximately 5-fold higher than the known peptide molecular weight at pH 7.4. Under the pH 5.5 conditions, PSPF-DKG produces a very broad, tailing peak, again attributable to interaction with the column. To rule out the possibility that electrostatic interactions with the stationary phase were responsible for the asymmetric peaks at low pH, the chromatography was repeated in the presence of 2 M NaCl, which failed to significantly improve the peak shape (data not shown).
Figure 5.

Size-exclusion chromatography of PSPF-EKG and PSPF-DKG at pH 5.5 and 7.4. At pH 7.4, both PSPF-EKG and PSPF-DKG produce a single well-behaved peak with an apparent molecular weight corresponding to an aggregation state of n ≈ 6 (B). At pH 5.5, PSPF-EKG produces a single major peak corresponding to n ≈ 5, whereas PSPF-DKG produces only a poorly defined peak (A).
Sedimentation Equilibrium of Analytical Ultracentrifugation
Analytical ultracentrifugation (AUC) was employed as a complementary technique to further probe the association state and affinity of the water-soluble bundles of both PSPF-EKG and PSPF-DKG.33,34 The peptides were subjected to AUC at a constant 100 μM starting level in pH 7.4 and 5.5 media and at multiple rotor speeds (35, 40, 45, and 50 krpm). Finally, sedimentation and equilibrium parameters were fitted globally to the data collected. Fitting the curves to a single MW species gave apparent molecular weights for PSPF-EKG of 18 000 ± 300 Da and 16 000 ± 300 Da at pH values 7.4 and 5.5, respectively. These values agree well with the data from size-exclusion chromatography (apparent molecular weights were 19 000 and 15 000 Da) and suggest a hexameric association state across this pH range for PSPF-EKG. The data can be further fit to a monomer–hexamer equilibrium, as summarized in Table 5, resulting in association energies of −6.3 and −5.6 kcal/mol at pH 7.4 and 5.5, respectively. To illustrate the relationship between pH and the association state, overlaid AUC data, curve fits, residuals, and weight fraction distributions were assembled, as shown in Figure 6. As seen in panels B and D, dissociation is shifted to lower concentrations at pH 7.4 versus pH 5.5, consistent with decreasing pH destabilizing a water-soluble helical bundle.
Table 5.
Analytical Ultracentrifugation (AUC) Sedimentation Equilibrium Parameters for PSPF-EKG and PSPF-DKG at pH 5.5 and 7.4
| PSPF-EKG
|
PSPF-DKG
|
|||
|---|---|---|---|---|
| pH 7.4 | pH 5.5 | pH 7.4 | pH 5.5 | |
| apparent MW | 18 000 | 16 000 | 17 000 | 24 000 |
| aggregation statea | 6.2 | 5.5 | 6.0 | N/A |
| –log(Kdissociation) | 28.0 ± 0.4 | 24.8 ± 0.1 | 28.7 ± 0.4 | N/A |
| association ΔGb (kcal/mol peptide) | −6.3 | −5.6 | −6.5 | N/A |
Figure 6.

AUC sedimentation data, fitted curves, and residuals for PSPF-EKG at pH 5.5 (A) and 7.4 (C). Single species fitting suggests an association state of n = 6 at both pH 7.4 (apparent MW = 18 000) and pH 5.5 (apparent MW = 16 000). The corresponding weight fraction distributions are plotted for pH 5.5 (B) and pH 7.4 (D), and the free energies of association are listed in Table 5.
As shown in Figure 7, a global fit of AUC data for PSPF-DKG resulted in a single-species apparent molecular weight of 17 000 at pH 7.4, which is, again, 6-fold higher than the known molecular weight of the peptide and once more agrees well with size-exclusion chromatography. AUC data for PSPF-DKG at pH 7.4 have also been fit to a simple monomer–hexamer equilibrium, producing an association energy of −6.5 kcal/mol peptide (Table 5). The single-species apparent molecular weight for PSPF-DKG at pH 5.5 was 24 000 (consistent with higher order aggregates); however, we did not attempt to fit the data to an association equilibrium owing to the poor SEC peak shape for this peptide under these conditions.
Figure 7.

AUC sedimentation data, fitted curves, and residuals for PSPF-DKG at pH 5.5 (A) and pH 7.4 (B). Single-species fitting suggests an association state of n = 7 at pH 7.4 (apparent MW = 17 000), whereas the apparent molecular weight at pH 5.5 is approximately 24 000.
Circular Dichroism and Thermal Denaturation
Circular dichroism (CD) was employed to characterize the secondary structure of the PSPF-EKG peptide. CD data suggest that PSPF-EKG adopts an α-helical secondary structure at both pH values, as seen in Figure 8. Thermal denaturation and thermodynamic stability of the PSPF-EKG peptide were assessed by collecting CD data as a function of temperature35 and over a range of concentrations (2, 4, and 20 μM) at both pH values. A summary of the results is given in Figure 9 and Table 6. For each pH and concentration, the raw data were fitted to a curve according to the Gibbs–Helmholtz equation and using ΔHm, Tm, and baselines as global parameters:
Figure 8.

Circular dichroism of PSPF-EKG at pH 5.5 (A) and 7.4 (B). α-Helical secondary structure is clear at both pH values.
Figure 9.

Thermal denaturation of PSPF-EKG at pH 7.4 (A) and pH 5.5 (B). The raw data are fitted to the Gibbs–Helmholtz equation.
Table 6.
Fitting Results for Thermal Denaturation of PSPFEKG at pH 7.4 and 5.5 by CD
| pH | ΔH (kcal/mol peptide) | Tm (K) | [PSPF-EKG] at 50% folded and 300 K |
|---|---|---|---|
| 7.4 | 22.0 ± 0.1 | 339.0 ± 0.1 | 0.14 μM |
| 5.5 | 19.6 ± 0.1 | 333.4 ± 0.1 | 0.31 μM |
Here, ΔG refers to the unfolding energy upon thermal denaturation, T refers to temperature, Tm refers to the melting temperature at which ΔG equals to zero, ΔHm refers to the enthalpy at Tm, and ΔCp refers to the change in the heat capacity over the temperature range. For the purposes of curve fitting, Tm was defined with a reference concentration of 4 μM; ΔCp was also included, but, over the range of experimental data examined, this parameter was not well-defined. Given these inputs, the enthalpy was found to be approximately 12% higher at pH 7.4 than at pH 5.5 (22.0 and 19.6 kcal/mol peptide, respectively). These values are comparable to those of other water-soluble helical peptide bundles in the literature.36 The melting temperatures (Tm) are found to be 339.0 and 333.4 K at pH 7.4 and 5.5, respectively, a decrease of 5.6 K. The cumulative effect of these observed differences can be summarized by calculating the concentration of PSPF-EKG required to have 50% of the total amount of peptide remain folded at a given temperature and for each pH value. At 300 K, these values are 0.31 and 0.14 μM at pH 5.5 and 7.4, respectively, approximately a 2-fold difference. Taken together with the SEC and AUC data, these results suggest that a decrease in pH destabilizes not only the water-soluble associated state but also the helical structure of individual PSPF-EKG peptides.
The Structural Properties of PSPF Peptides in a Membrane Micelle System
To assess the relative strength of the self-association of the PSPF-EKG peptide, AUC experiments were carried out in a membrane-mimicking micellar medium at both pH values. PSPF-EKG was first dissolved in a micellar solution of N-tetradecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate (C-14 betaine) in water. The density of the peptide solution was then adjusted with D2O to precisely match the known density of the betaine alone at both pH 7.4 and 5.5 so that only the peptide component contributed to the sedimentation equilibrium.28,37 For each pH, three samples were then prepared at different peptide-to-betaine ratios (1:50, 1:100, 1:200), and each was subjected to AUC at four rotor speeds (35, 40, 45, 50 krpm). The raw data could be fit equally well into monomer–trimer, monomer–tetramer, and higher order aggregate equilibria, suggesting that PSPF-EKG weakly associates in detergent micelles. Figure 10 shows an example in which a monomer–trimer equilibrium is fit to the data at pH 7.4 (panel A) and pH 5.4 (panel C); the corresponding weight fraction distributions are shown in panels B and D.
Figure 10.

Single-species fitting of AUC data for PSPF-EKG in detergent micelles at pH 5.5 (A) and pH 7.4 (C). Weight fraction distributions for monomer–trimer equilibria at pH 5.5 and 7.4 are shown in panels B and D, respectively.
The Orientation of PSPF-EKG in a Lipid Bilayer
The secondary structure and orientation of PSPF-EKG in deuterium oxide (D2O) hydrated bilayers were evaluated using attenuated total reflection IR spectroscopy (ATR-IR).38–40 As shown in Figure 11, the IR spectra of PSPF-EKG in the amide I region showed a single peak at 1656 cm−1, indicative of a dehydrated helical conformation within a bilayer. The dichroic ratio for parallel versus perpendicularly polarized light was 1.5, corresponding to an order parameter of −0.42. This order parameter would correspond to an orientation of approximately 75° relative to the membrane normal, assuming that the bilayers are well-ordered, that the entire peptide is fully helical, and that it adopts only a single conformation. Alternatively, the majority of the peptide might be predominantly at a 90° orientation with a small population adopting the transmembrane orientation. In either case, the data indicate that the majority of peptide lies roughly parallel to the lipid surface and rules out the possibility of the peptide being oriented predominantly perpendicular to the bilayer surface.
Figure 11.
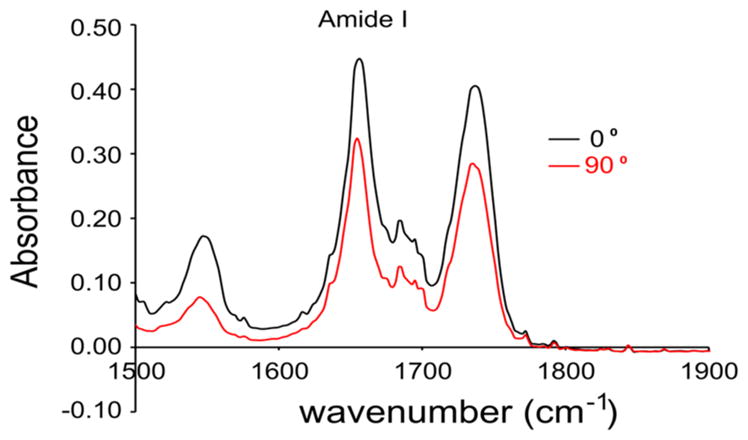
ATR-IR spectra of PSPF-EKG in phospholipids (POPC) bilayers. The peak at 1656 cm−1 is indicative of α-helical secondary structure. The orientation is demonstrated by the ratio of areas for the 1656 cm−1 peak for incident light polarized parallel (0°) and perpendicular (90°) to the membrane normal.
DISCUSSION
Therapeutic macromolecules such as peptides and proteins are easily cleared from the bloodstream. Furthermore, delivery to the cytosol to achieve desirable therapeutic effects is often hindered by a limited ability to cross cellular membranes. Decades of research effort have been devoted to develop delivery agents with high efficiency and low toxicity in order to overcome these obstacles.41,42
Most nonviral carriers are synthetic chemical conjugates. Active ingredients (drugs) are usually linked or enclosed into a vehicle and delivered into the cell via endocytosis, membrane fusion, or another unspecified mechanism.43,44 These vehicles are typically designed as liposomes/lipoplexes,45 cationic polymers,46 polypeptides,47 proteins,48 amphiphilic polymers/ peptides,49 nanoparticles,50–52 and cell-penetrating peptides.53 Native sequences such as fusogenic peptides from viral fusion proteins54 have also been manipulated as a cargo carrier to cross the barrier of cell membranes. A number of these approaches have progressed as far as clinical trials in humans, but most of them have failed due to high toxicity or lack of manipulability.44
Here, we have designed a series of pH-switchable pore-forming peptides as potential candidates for intracellular drug delivery. One of the top candidates (PSPF-EKG) stands out on the basis of relatively higher levels of miRNA and ATP from RBCs at pH 5.4. Importantly, a lack of hemoglobin release in this same assay and at both pH values suggests that PSPF-EKG does not cause membrane rupture. Taken together with the strong correlation observed between ATP release and tryptophan fluorescence properties for the series (Figure 4), membrane insertion appears to play a key role in the mechanism of action.
RBC lysis data also provide a means to compare how amino acid choice in our design strategy impacts activity. First, we have three options of pH-trigger residues in this peptide series. Asp and Glu residues both presented expected pH-switchable ATP and miRNA release in peptides PSPF-DQA and PSPF-DKG, indicating the carboxyl side chain groups respond efficiently to environmental pH change, with intrinsic pKa values of the unperturbed side chains around 4. The third trigger candidate, His, failed to show significant pH preferences in terms of ATP or miRNA release. However, PSPF-HKG induced high ATP release percentage at both pH values. Presumably, His will induce pore formation in a pH-independent manner. Nevertheless, the poor solubility of the His variants prevented more detailed biophysical characterization and thus were not further studied.
Second, Lys and Gln were placed in the f position in order to promote helix formation in aqueous systems and to provide a solvent-exposure surface in membrane systems. The RBC lysis results did not discriminate between these two residues when comparing the performance of the aspartate and glutamate peptide variants (PSPF-EKG versus PSPF-EQG and PSPF-EKA versus PSPF-EQA).
The third screened parameter is the choice between Ala or Gly for residues packed in the helix interface. This element of the design follows from the fact that small residues are known to stabilize the final folded state in transmembrane helix interfaces.28,29 In the case of PSPF-EKG versus PSPF-EKA, Gly resulted in a much higher pH-switchable ATP and miRNA release. The results collected here agree with the previous conclusion that Gly in a transmembrane helical interface drives stronger association than Ala, presumably because Gly stabilizes the helix interaction via weak Cα–H interactions.55
A variety of biophysical assays have been applied in order to obtain a comprehensive view of the mechanism of pH-dependent pore formation for the PSPF-EKG peptide, a model of which is depicted in Figure 12. We first looked at the structural conformation and folding stability of PSPF-EKG in aqueous solutions. CD, AUC, and SEC data all suggest that PSPF-EKG forms a stable helical bundle at both pH values, which is expected due to the designed canonical Leu-zipper coiled-coil motif. AUC and thermal denaturing have been further used to study the folding stability difference between pH 7.4 and 5.5. The free energy of the helical bundle increases by 0.7 kcal/mol peptide as the pH decreases from 7.4 to 5.5. Likewise, both ΔCp and Tm decrease at pH 5.5, suggesting that PSPF-EKG is better packed at higher pH. The relative stability of the PSPF-EKG aggregates as a function of pH was also reflected empirically in the SEC data. While this peptide produced a sharp peak at pH 7.4, the pH 5.5 conditions resulted in a broad and tailing peak. Because the peak shape at pH 5.5 did not improve as the NaCl concentration was increased from 150 mM to 2 M, we conclude that a drop in pH destabilizes the self-association of PSPF-EKG in aqueous systems. Thus, we considered the first design element to be validated for this peptide.
Figure 12.
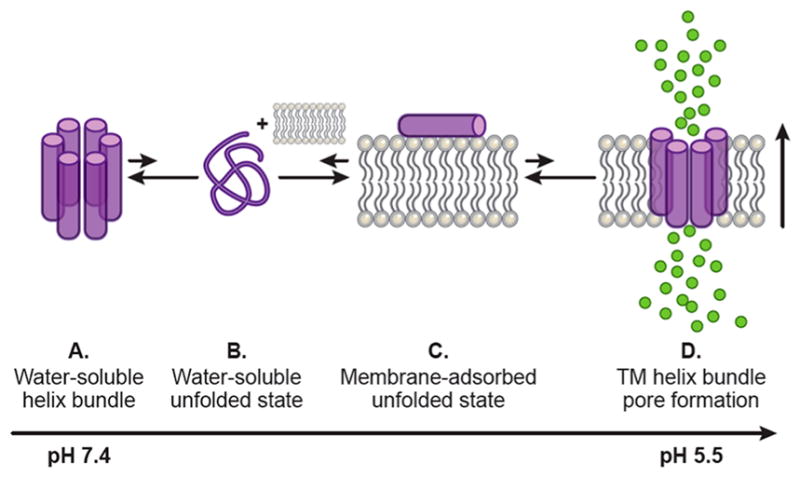
Model for PSPF peptide membrane insertion and pore formation upon pH decrease.
We also characterized PSPF-EKG in micelles and bilayers. Equilibrium sedimentation AUC suggests that PSPF-EKG is in equilibrium with multiple associated states in C14-betaine micelles at both pH values. Furthermore, the orientation of PSPF-EKG has been studied by ATR-FTIR in POPC lipid bilayers. The average dichroic angle is about 75° with respect to the lipid normal, revealing that the majority of peptides are in a membrane-surface-absorbed state. Conceivably, this state corresponds to the AUC results in the presence of C14-betaine. We postulate that some of the peptides adopt a transmembrane orientation, which might reflect a weakly associated aggregated form. This dynamic equilibrium between vertical individual peptides in the membrane-surface-absorbed and transmembrane associated states is consistent with transient membrane pore formation and consequently can play a role in ATP and miRNA release.
The biophysical and red blood cell data are also consistent with a pH-controlled pore formation. The factor that determines the fraction of peptide that is inserted in a given orientation depends on the free energy between the states. In the inserted fully assembled state, the peptides would bury their hydrophobic residues within the membrane and engage in favorable helix–helix interactions, and their polar side chains would be hydrated in the pore. Additionally, the transmembrane voltage might enhance unidirectional insertion. The primary future direction for enhancing the designs at this point is to increase the population of transmembrane peptide relative to the surface-absorbed state. To our minds, there are two avenues to pursue toward this aim. First, the interpeptide interactions in the transmembrane state should be strengthened to favor self-association. Second, the energetic cost of insertion into the bilayer should be decreased by modulating the hydrophobicity of these peptides. It is our belief that understanding the properties that control aggregation, pore formation, and, ultimately, size will allow for the rational design of selective membrane-permeating tools that can be specifically designed to allow release of bioactive cargo from endosomal membranes.
MATERIALS AND METHODS
Peptides and Reagents
The PSPF peptides were all obtained from New England Peptide and used as received. All other chemicals and reagents were commercially available and used as received without further purification.
RBC Lysis Assay and Analysis
The human red blood cell hemolysis assay was by carried out as described elsewhere56 with some modifications. Briefly, 5 mL aliquots of human blood from healthy individuals were dispensed into 50 mL centrifuge tubes and resuspended in 35 mL of buffer containing 150 mM NaCl and either 20 mM MES adjusted to pH 5.4 or 20 mM HEPES adjusted to pH 7.4. Red blood cells (RBCs) were washed 3 times via centrifugation with buffer and finally resuspended in a total of 50 mL of buffer. For the final assay, 175 μL of buffer solution was dispensed into each well of a clear-bottom 96-well plate followed by 50 μL of final resuspended RBCs (approximately 2.5 × 107 cells). For transfers of RBCs, wide-bore pipet tips were used to avoid cell damage. Test PSPF peptides at the appropriate concentration were first diluted in 25 μL of buffer and then added to the cells. All steps prior to incubation were carried out with chilled buffers and on ice. Finally, the suspension was mixed reciprocation with a wide-bore tip, and the plate was covered and incubated at 37 °C for the indicated time. After incubation, the cells were centrifuged for 5 min at 500 rcf, and 150 μL of the supernatant was transferred into a new 96-well clear-bottom plate. Absorbance was measured at 541 nm, and the resulting raw hemolysis figures were normalized to a matching set of RBCs incubated in the presence of 1% Triton X-100 (100% hemolysis control). Alternatively, ATP and micro-RNA release measurements were performed on samples of the final supernatant as described below.
Micro-RNA (mir-16)
The release of micro-RNA mir-16 from RBCs was determined using stem-loop PCR as described elsewhere.57 Briefly, 5 μL of final supernatant was processed with TaqMan MicroRNA cells-to-CT kit (Applied Biosystems) according to the manufacturer’s protocol, and quantitative PCR reaction was performed on an ABI (Life Technologies; Carlsbad, CA) 7500 fast real time PCR system using standard cycling conditions.58 The derived Ct values for mir-16 (Applied Biosystems cat. no. 4373121) in each experiment were transformed into copy numbers using a linear equation derived from a standard curve that was run in parallel.
ATP
To quantitatively determine the amount of adenosine triphosphate (ATP) in the supernatant, the ATPLite assay kit (PerkinElmer; Waltham, MA) was used according to the manufacturer’s instructions. A 100 μL aliquot of supernatant was employed for each assay per reaction point.
Tryptophan Fluorescence
Fluorescence emission spectra for each peptide were collected with constant excitation on a Fluorolog spectrofluorometer at both pH 5.5 and 7.4 with and without lipid titration.59 The lipid stock was prepared with 90% POPC and 10% POPG, and the final concentration of lipid after titration was 200 μM. The peptide concentration in each measurement was 2 μM.
CD Measurement and Thermal Denaturation
CD spectra were collected with a Jasco J-810 spectropolarimeter using a 1 nm step at 4 °C at both pH 5.5 and 7.4.49 The PSPF-EKG peptide concentration was 2 μM, and the final CD spectrum was obtained by averaging over three scans. To assess thermal denaturation of the helix, the ellipticity at 222 nm was monitored for the peptide at 2, 4, and 20 μM concentrations as the temperature was increased from 4 to 96 °C in 2 °C steps at both pH 5.5 and 7.4.33 The parameters from the Gibbs–Helmholtz equation were fit to the data as demonstrated elsewhere.36
Size-Exclusion Chromatography
Size-exclusion chromatography (SEC) was carried out for the PSPF-EKG and PSPF-DKG peptides with the aid of an AKTA FPLC (GE) fitted with a Superdex 75 column (GE) and eluted at 25 °C with aqueous media containing 150 mM NaCl and either 50 mM Tris adjusted to pH 7.4 or 50 mM MES adjusted to pH 5.5.33 Peptide samples were prepared at 100 μM. Four size standards were employed for calibration: blue dextran (2 000 000 g/mol), carbonic anhydrase (29 000 g/mol), cytochrome C (12 400 g/mol), and aprotinin (6500 g/mol). In order to test the effect of salt concentration upon peptide elution, SEC data were also collected in media containing 2 M NaCl (data not shown).
Sedimentation Equilibrium of Analytical Ultracentrifugation (AUC)
Analytical ultracentrifugations (AUC) for PSPF-EKG were performed at 25 °C using a Beckman XL-I analytical ultracentrifuge operated at 35, 40, 45, and 50 krpm. Solutions containing 100 μM PSPF-EKG were prepared in 150 mM NaCl containing either 50 mM Tris adjusted to pH 7.4 or 50 mM MES adjusted to pH 5.5. The data were globally fit via a nonlinear least-squares algorithm with IGOR Pro (WaveMetrics) as discussed previously.28,60–62 The errors quoted in the text are standard deviations associated with the fitting procedure. The largest contribution to the error of the computed MW arises from the use of the group-additivity method to compute partial specific volumes, which we estimate to lead to 5–10% error. AUC data for PSPF-EKG were also collected in the presence of 8 mM N-tetradecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate (C-14 betaine) in media containing 150 mM NaCl and 50 mM sodium phosphate adjusted to pH 7.4 or 5.5. To precisely match the known density of the micellar phase, 17% D2O was required at pH 7.4 and 22% D2O was required at pH 5.5. Three groups of samples at each pH were prepared with peptide/betaine molar ratios of 1:50, 1:100, and 1:200. AUC data were collected for each of the three peptide/betaine ratios and at each pH at four rotor speeds (35, 40, 45, and 50 krpm) and were fit as before.49,60–62
Attenuated Total Reflection IR Spectroscopy (ATR-IR)
ATR-IR spectra for the PSPF-EKG peptide were collected with the aid of a Nicolet Magna IR 4700 spectrometer at 1 cm−1 resolution.38–40 A sample containing 0.5 μmol PSPF-EKG in trifluoroethanol (TFE) was mixed with a 20-fold molar amount of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and dried into a thin film on the surface of the ATR Ge crystal by means of dry N2 gas. The film was rehydrated by D2O-saturated air overnight in the closed environment of a D2O bath. During data acquisition, the polarized mirror was adjusted to 0° and 90°, creating incident light oriented parallel and perpendicular to the lipid normal, respectively. The infrared spectra under each condition were collected with 256 scans. The dichroic ratio of the 1656 cm−1 amide I bond absorption was computed for parallel (0°) versus perpendicular (90°) polarized incident light relative to the membrane normal and was employed to calculate the peptide orientation as discussed previously.38
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Whittlesey KJ, Shea LD. Delivery systems for small molecule drugs, proteins, and DNA: the neuroscience/ biomaterial interface. Exp Neurol. 2004;190:1–16. doi: 10.1016/j.expneurol.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 2.Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- 3.Veldhoen S, Laufer SD, Restle T. Recent developments in peptide-based nucleic acid delivery. Int J Mol Sci. 2008;9:1276–1320. doi: 10.3390/ijms9071276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stanton MG, Colletti SL. Medicinal chemistry of siRNA delivery. J Med Chem. 2010;53:7887–7901. doi: 10.1021/jm1003914. [DOI] [PubMed] [Google Scholar]
- 5.Sandvig K, van Deurs B. Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett. 2002;529:49–53. doi: 10.1016/s0014-5793(02)03182-4. [DOI] [PubMed] [Google Scholar]
- 6.Gruenberg J, van der Goot FG. Mechanisms of pathogen entry through the endosomal compartments. Nat Rev Mol Cell Biol. 2006;166:495–504. doi: 10.1038/nrm1959. [DOI] [PubMed] [Google Scholar]
- 7.Colman M, Lawrence MC. The structural biology of type I viral membrane fusion. Nat Rev Mol Cell Biol. 2003;4:309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 8.Weissenhorn W, Dessen A, Calder LJ, Harrison SC, Skehel JJ, Wiley DC. Structural basis for membrane fusion by enveloped viruses. Mol Membr Biol. 1999;16:3–9. doi: 10.1080/096876899294706. [DOI] [PubMed] [Google Scholar]
- 9.Abrami L, Reig N, van der Goot FG. Anthrax toxin: the long and winding road that leads to the kill. Trends Microbiol. 2005;13:72–78. doi: 10.1016/j.tim.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 10.Krantz BA, Finkelstein A, Collier RJ. Protein translocation through anthrax toxin’s transmembrane pore is driven by a proton gradient. J Mol Biol. 2006;355:968–979. doi: 10.1016/j.jmb.2005.11.030. [DOI] [PubMed] [Google Scholar]
- 11.Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim Biophys Acta. 1999;1462:55–70. doi: 10.1016/s0005-2736(99)00200-x. [DOI] [PubMed] [Google Scholar]
- 12.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 13.Mellman I, Fuchs R, Helenius A. Acidification of the endocytic and exocytic pathways. Annu Rev Biochem. 1986;55:663–670. doi: 10.1146/annurev.bi.55.070186.003311. [DOI] [PubMed] [Google Scholar]
- 14.White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- 15.Grigoryan G, DeGrado WF. Probing designability via a generalized model of helical bundle geometry. J Mol Biol. 2011;405:1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen C, Parry DAD. α-Helical coiled coils and bundles: how to design an α-helical protein. Proteins. 1990;7:1–15. doi: 10.1002/prot.340070102. [DOI] [PubMed] [Google Scholar]
- 18.Harbury PB, Tidor B, Kim PS. Repacking protein cores with backbone freedom: structure prediction for coiled coils. Proc Natl Acad Sci USA. 1995;92:8408–8412. doi: 10.1073/pnas.92.18.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lupas A. Coiled coils: new structures and new functions. Trends Biochem Sci. 1996;21:375–382. [PubMed] [Google Scholar]
- 20.Oakley MG, Hollenbeck JJ. The design of antiparallel coiled coils. Curr Opin Struct Biol. 2001;11:450–457. doi: 10.1016/s0959-440x(00)00232-3. [DOI] [PubMed] [Google Scholar]
- 21.Offer G, Hicks MR, Woolfson DN. Generalized Crick equations for modeling noncanonical coiled coils. J Struct Biol. 2002;137:41–53. doi: 10.1006/jsbi.2002.4448. [DOI] [PubMed] [Google Scholar]
- 22.Lacy DB, Stevens RC. Unraveling the structures and modes of action of bacterial toxins. Curr Opin Struct Biol. 1998;8:778–784. doi: 10.1016/s0959-440x(98)80098-5. [DOI] [PubMed] [Google Scholar]
- 23.Crick FHC. The packing of alpha-helices: simple coiledcoils. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- 24.Talbot JA, Hodes RS. Tropomyosin: a model protein for studying coiled-coil and α-helical stabilization. Acc Chem Res. 1982;15:224–230. [Google Scholar]
- 25.O’Neil KT, DeGrado WF. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]
- 26.Adamian L, Liang J. Interhelical hydrogen bonds and spatial motifs in membrane proteins: polar clamps and serine zippers. Proteins. 2002;47:209–218. doi: 10.1002/prot.10071. [DOI] [PubMed] [Google Scholar]
- 27.Eilers M. Comparison of helix interactions in membrane and soluble alpha-bundle proteins. Biophys J. 2002;82:2720–2736. doi: 10.1016/S0006-3495(02)75613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Kulp DW, Lear JD, DeGrado WF. Experimental and computational evaluation of forces directing the association of transmembrane helices. J Am Chem Soc. 2009;131:11341–11343. doi: 10.1021/ja904625b. [DOI] [PubMed] [Google Scholar]
- 29.Walters RF, DeGrado WF. Helix-packing motifs in membrane proteins. Proc Natl Acad Sci USA. 2006;103:13658–13663. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lear JD, DeGrado WF. Membrane binding and conformational properties of peptides representing the NH2 terminus of influenza HA-2. J Biol Chem. 1987;262:6500–6505. [PubMed] [Google Scholar]
- 31.Rafalski M, Ortiz A, Rockwell A, van Ginkel LC, Lear JD, DeGrado WF, Wilschut J. Membrane fusion activity of the influenza virus hemagglutinin: interaction of HA2 N-terminal peptides with phospholipid vesicles. Biochemistry. 1991;30:10211–10220. doi: 10.1021/bi00106a020. [DOI] [PubMed] [Google Scholar]
- 32.Boeckle S, Fahrmeir J, Roedl W, Ogris M, Wagner E. Melittin analogs with high lytic activity at endosomal pH enhance transfection with purified targeted PEI polyplexes. J Controlled Release. 2006;112:240–248. doi: 10.1016/j.jconrel.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 33.Bryson JW, Desjarlais JR, Handel TM, DeGrado WF. From coiled coils to small globular proteins: design of a native-like three-helix bundle. Protein Sci. 1998;7:1404–1414. doi: 10.1002/pro.5560070617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slovic AM, Kono H, Lear JD, Saven JG, DeGrado WF. Computational design of water-soluble analogues of the potassium channel KcsA. Proc Natl Acad Sci USA. 2004;101:1828–1833. doi: 10.1073/pnas.0306417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Betz SF, Liebman PA, DeGrado WF. De novo design of native proteins: characterization of proteins intended to fold into antiparallel, rop-like, four-helix bundles. Biochemistry. 1997;36:2450–2458. doi: 10.1021/bi961704h. [DOI] [PubMed] [Google Scholar]
- 36.Raleigh DP, Betz SF, DeGrado WF. A de novo designed protein mimics the native state of natural proteins. J Am Chem Soc. 1995;117:2. [Google Scholar]
- 37.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 38.Menikh A, Saleh MT, Gariepy J, Boggs JM. Orientation in lipid bilayers of a synthetic peptide representing the C-terminus of the A1 domain of shiga toxin. A polarized ATR-FTIR study. Biochemistry. 1997;36:15865–15872. doi: 10.1021/bi970944+. [DOI] [PubMed] [Google Scholar]
- 39.Tucker MJ, Getahun Z, Nanda V, DeGrado WF, Gai F. A new method for determining the local environment and orientation of individual side chains of membrane-binding peptides. J Am Chem Soc. 2004;126:5078–5079. doi: 10.1021/ja032015d. [DOI] [PubMed] [Google Scholar]
- 40.Donald JE, Zhang Y, Fiorin G, Carnevale V, Slochower DR, Gai F, Klein ML, DeGrado WF. Transmembrane orientation and possible role of the fusogenic peptide from parainfluenza virus 5 (PIV5) in promoting fusion. Proc Natl Acad Sci USA. 2011;108:3958–3963. doi: 10.1073/pnas.1019668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang H, Wang G, Yang H. Drug delivery systems for differential release in combination therapy. Expert Opin Drug Delivery. 2011;8:171–190. doi: 10.1517/17425247.2011.547470. [DOI] [PubMed] [Google Scholar]
- 42.Lammers T, Kiessling F, Hennink WE, Storm G. Nanotheranostics and image-guided drug delivery: current concepts and future directions. Mol Pharmaceutics. 2010;7:1899–1912. doi: 10.1021/mp100228v. [DOI] [PubMed] [Google Scholar]
- 43.Shim MS, Kwon YJ. Efficient and targeted delivery of siRNA in vivo. FEBS J. 2010;277:4814–4827. doi: 10.1111/j.1742-4658.2010.07904.x. [DOI] [PubMed] [Google Scholar]
- 44.Pathak A, Patnaik S, Gupta KC. Recent trends in non-viral vector-mediated gene delivery. Biotechnol J. 2009;4:1559–1572. doi: 10.1002/biot.200900161. [DOI] [PubMed] [Google Scholar]
- 45.Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, Judge AD, Lam K, McClintock K, Nechev LV, Palmer LR, Racie T, Rohl I, Seiffert S, Shanmugam S, Sood V, Soutschek J, Toudjarska I, Wheat AJ, Yaworski E, Zedalis W, Koteliansky V, Manoharan M, Vornlocher HP, MacLachlan I. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441:111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]
- 46.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe K, Harada-Shiba M, Suzuki A, Gokuden R, Kurihara R, Sugao Y, Mori T, Katayama Y, Niidome T. In vivo siRNA delivery with dendritic poly(L-lysine) for the treatment of hypercholesterolemia. Mol BioSyst. 2009;5:1306–1310. doi: 10.1039/b900880b. [DOI] [PubMed] [Google Scholar]
- 48.Cronican JJ, Thompson DB, Beier KT, McNaughton BR, Cepko CL, Liu DR. Potent delivery of functional proteins into mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem Biol. 2010;5:747–752. doi: 10.1021/cb1001153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruan L, Zhang H, Luo H, Liu J, Tang F, Shi YK, Zhao X. Designed amphiphilic peptide forms stable nanoweb, slowly releases encapsulated hydrophobic drug, and accelerates animal hemostasis. Proc Natl Acad Sci USA. 2009;106:5105–5110. doi: 10.1073/pnas.0900026106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hasadsri L, Kreuter J, Hattori H, Iwasaki T, George JM. Functional protein delivery into neurons using polymeric nanoparticles. J Biol Chem. 2009;284:6972–6981. doi: 10.1074/jbc.M805956200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan JM, Zhang L, Tong R, Ghosh D, Gao W, Liao G, Yuet KP, Gray D, Rhee JW, Cheng J, Golomb G, Libby P, Langer R, Farokhzad OC. Spatiotemporal controlled delivery of nanoparticles to injured vasculature. Proc Natl Acad Sci USA. 2010;107:2213–2218. doi: 10.1073/pnas.0914585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qiu LY, Wang RJ, Zheng C, Jin Y, Jin le Q. Beta-cyclodextrin-centered star-shaped amphiphilic polymers for doxorubicin delivery. Nanomedicine. 2010;5:193–208. doi: 10.2217/nnm.09.108. [DOI] [PubMed] [Google Scholar]
- 53.An M, Wijesinghe D, Andreev OA, Reshetnyak YK, Engelman DM. pH-(low)-insertion-peptide (pHLIP) translocation of membrane impermeable phalloidin toxin inhibits cancer cell proliferation. Proc Natl Acad Sci USA. 2010;107:20246–20250. doi: 10.1073/pnas.1014403107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kwon EJ, Bergen JM, Pun SH. Application of an HIV gp41-derived peptide for enhanced intracellular trafficking of synthetic gene and siRNA delivery vehicles. Bioconjugate Chem. 2008;19:920–927. doi: 10.1021/bc700448h. [DOI] [PubMed] [Google Scholar]
- 55.Senes A, Ubarretxena-Belandia I, Engelman DM. The Cα—H···O hydrogen bond: a determinant of stability and specificity in transmembrane helix interactions. Proc Natl Acad Sci USA. 2001;98:9056–9061. doi: 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henry SM, El-Sayed ME, Pirie CM, Hoffman AS, Stayton PS. pH-responsive poly(styrene-alt-maleic anhydride) alkylamide copolymers for intracellular drug delivery. Biomacromolecules. 2006;7:2407–2414. doi: 10.1021/bm060143z. [DOI] [PubMed] [Google Scholar]
- 57.Bartz R, Fan H, Zhang J, Innocent N, Cherrin C, Beck SC, Pei Y, Momose A, Jadhav V, Tellers DM, Meng F, Crocker LS, Sepp-Lorenzino L, Barnett SF. Effective siRNA delivery and target mRNA degradation using an amphipathic peptide to facilitate pH-dependent endosomal escape. Biochem J. 2011;435:475–487. doi: 10.1042/BJ20101021. [DOI] [PubMed] [Google Scholar]
- 58.Abrams MT, Koser ML, Seitzer J, Williams SC, DiPietro MA, Wang W, Shaw AW, Mao X, Jadhav V, Davide JP, Burke PA, Sachs AB, Stirdivant SM, Sepp-Lorenzino L. Evaluation of efficacy, biodistribution, and inflammation for a potent siRNA nanoparticle: effect of dexamethasone co-treatment. Mol Ther. 2010;18:171–180. doi: 10.1038/mt.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tang J, Signarvic RS, DeGrado WF, Gai F. Role of helix nucleation in the kinetics of binding of mastoparan X to phospholipid bilayers. Biochemistry. 2007;46:13856–13863. doi: 10.1021/bi7018404. [DOI] [PubMed] [Google Scholar]
- 60.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Computational design of peptides that target transmembrane helices. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 61.Cristian L, Lear JD, DeGrado WF. Determination of membrane protein stability via thermodynamic coupling of folding to thiol–disulfide interchange. Protein Sci. 2003;12:1732–1740. doi: 10.1110/ps.0378603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kochendoerfer GG, Salom D, Lear JD, Wilk-Orescan R, Kent SB, DeGrado WF. Total chemical synthesis of the integral membrane protein influenza A virus M2: role of its C-terminal domain in tetramer assembly. Biochemistry. 1999;38:11905–11913. doi: 10.1021/bi990720m. [DOI] [PubMed] [Google Scholar]