

Abstract
This invited biographical review covers the career of Dr. David Julius and his discovery of thermosensitive TRP channels. Dr. Julius is currently the Morris Herzstein Chair in Molecular Biology and Medicine and Professor and Chair of Physiology at the University of California, San Francisco Medical School. He is a member of the National Academy of Sciences and has received many distinguished awards for his landmark discoveries of the molecular basis of pain and thermosensation.
Keywords: pain, somatosensation, sensory neuron, thermosensation, TRP channel
Abbreviations: TRP, transient receptor potential; TRPV1, transient receptor potential cation channel, subfamily V, member 1; TRPA1, transient receptor potential cation channel, subfamily A, member 1; TRPM8, transient receptor potential cation channel, subfamily M, member 8; DRG, dorsal root ganglia; GABAB, Gamma-Aminobutyric Acid (GABA) B Receptor; P2X, Purinergic Receptor P2X; 5-HT3 (also HTR3), 5-Hydroxytryptamine (Serotonin) Receptor 3; α-MSH, α-melanocyte stimulating hormone; GPCR, G protein-coupled receptor
Introduction
The ability to perceive changes in environmental temperature is essential for survival of all organisms. In humans, thermosensation is critical for homeostatic maintenance of core body temperature and also for appropriate behavioral responses to environmental temperature, such as wearing a coat outside when its snowing, using a pot holder to grab a red-hot frying pan, or wearing gloves to handle dry-ice in the laboratory. The quest to understand thermosensation and pain spans millennia. Indeed, artwork depicting the use of opium as an analgesic dates back to 5,000 BC.1 The Greek physician Galen proposed that sensations, including pain, were mediated by nerves connecting the organs to the brain.1 In 1664, Descartes supported Galen's view on sensory experiences and expanded further to provide the first description of how a stimulus, such as noxious heat, might trigger sensations. An illustration in the Treatise of Man (Descartes, 1664) depicts how particles from a hot flame tug on nerve tubules that connect the skin to spinal cord and brain, subsequently opening pores in the brain that trigger pain sensations and protective motor reflexes.
We now know that changes in environmental temperature in mammals are detected by primary afferent somatosensory neurons. These neurons have cell bodies in the dorsal root ganglia (DRG) for the body and the trigeminal ganglia for the face/head, and have a pseudo-unipolar axon that innervates peripheral target organs (e.g., skin) and the dorsal spinal cord. Nociceptors are a specialized subset of primary afferent neurons that mediate responses to noxious thermal, mechanical and/or chemical stimuli. Nociceptors are activated when temperatures reach levels that are capable of causing tissue damage; heat-sensitive nociceptors are activated by temperatures that exceed 43°C, while cold-sensitive nociceptors are activated by temperatures that fall below 15°C. Such neurons display little activity at normal body temperature but display robust action potential firing in response to noxious thermal stimuli that in turn activates central neurons to trigger protective reflexes and irritating or painful sensations.
A number of studies suggested that changes in environmental temperature triggered the opening of temperature-sensitive ion channels in primary afferent nerve endings. Heat-activated ionic currents were measured in cultured rodent primary afferents.2,3 Studies on capsaicin, the pungent compound in chili peppers that triggers the psychophysical sensation of heat, also supported the channel hypothesis. Capsaicin depolarized heat-sensitive sensory neurons4 to trigger excitability and electrophysiological experiments on cultured neurons revealed the activation of non-selective currents.5,6 Likewise, cold-activated currents and calcium signals were also observed in cultured neurons.7,8 However, other mechanisms were proposed for temperature-evoked excitability, ranging from a general perturbation of membrane properties to the activation or inhibition of a variety of voltage-gated channels or other conductances. Thus, it was not clear whether bona fide temperature-sensitive ion channels even existed.
In 1997, David Julius, Michael Caterina, and colleagues used expression cloning to identify TRPV1, a capsaicin- and heat-activated ion channel.9 This non-selective channel was activated by heat with a threshold of 43°C, similar to the threshold for action potential firing in sensory neurons and for noxious heat sensations in psychophysical experiments. Analyses of TRPV1-deficient animals have revealed a key role for TRPV1 in both acute heat detection and thermal hypersensitivity.10 Thus, TRPV1 became of great interest as a drug target for treating pain and inflammation.
TRPV1 was the first of many thermosensitive TRP channels to be identified by the Julius lab, and many other labs. Indeed, David Julius has gone on to identify the cold- and menthol-activated ion channel, TRPM8,11 and the wasabi receptor, TRPA1,12 both of which play key roles in acute and inflammatory pain. Most recently, Drs. Julius and Yifan Cheng and colleagues generated high-resolution structures of the TRPV113,14 and TRPA115 ion channels, defining regions that are conserved among TRP channels, as well as novel domains that give each channel their unique functions and roles in pain and inflammation. Importantly, these structures serve as a much-needed roadmap for the design of novel drugs to treat pain, itch and other inflammatory disorders linked to these channels.
Dr. Julius' many landmark discoveries have been discussed at length in a number of excellent, comprehensive reviews to which I refer the reader.16-18 Thus, what follows below is an excerpt of an interview with Dr. Julius where we discussed his discovery of TRPV1 and other TRP channels. I was fortunate to have been a postdoctoral fellow with David and can say that he was, and continues to be, an amazing mentor and role model. Indeed, Michael Caterina (Fig. 1), another former post-doctoral fellow of Dr. Julius, insists that, “Working with David was nothing short of awesome. As a scientist, David has a ‘sixth sense’ about what will constitute an interesting project and the best way to make that project come to fruition. Time after time, he finds a way to blow a field wide-open by cleverly and cleanly solving a longstanding problem in that field. As a mentor, David was incredibly supportive, in both good times and bad. Before we cloned TRPV1, I had been working on another project that was going nowhere. Even in the darkest days of that project, however, David provided immeasurable encouragement, patience and useful suggestions. When we began to realize that the TRPV1 project was going to be successful, David used that same enthusiasm and energy to deftly lead me and the rest of our team down the most efficient path to completing our work. That meant not only providing oversight, but also rolling up his sleeves and picking thousands of bacterial colonies late into the evening. Writing with David was also a joy. We would sit in his corner office overlooking the Pacific Ocean and bounce ideas back and forth for the next line of a paper. Those sessions provided a great opportunity to learn more broadly about David's philosophy of science and life. He is truly one of a kind.” Everyone I know who has had the pleasure to work with David shares these sentiments. His trainees also appreciate all the hands-on help David would provide in the lab; getting his hands dirty at the bench and doing some molecular biology, as evident in the photos below (Fig 2–3). This interview also includes details of David's early career in science, the events that led a yeast geneticist to neuroscience and some words of wisdom to others in the field.
Figure 1.

David Julius (left) with Michael Caterina (right, former postdoc; now Professor, Johns Hopkins School of Medicine) at an awards ceremony in 2014. Reproduced by permission of Michael J Caterina, MD, PhD, Johns Hopkins School of Medicine.
Figure 2.

David McKemy (back, former postdoc; now Professor at University of Southern California), Gunther Hollopeter (front, former graduate student; now postdoc, Jorgenson lab) with David Julius (middle) picking cDNA library clones that were candidate cold receptors. One of these clones encoded the cold and menthol receptor, TRPM8. 2001. Reproduced by permission of Sven E Jordt, Duke University School of Medicine.
Figure 3.
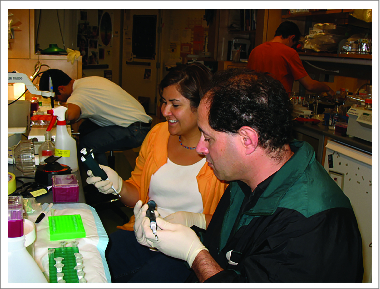
David Julius (right) and Diana Bautista (left, former postdoc; now Associate Professor at University of California, Berkeley) cloning TRPA1 mutants. 2003. Reproduced by permission of Gunther Hollopeter, PhD, University of Utah.
Interview recorded February 2015, Department of Physiology, UCSF
Diana: Tell me about your early career path and how you became interested in science.
David: Neither of my parents are scientists. But I became interested in science because I had a great physics teacher in high school, Irv Isaacson. He was really fun, and he made us think about a lot of stuff. He was fantastic.
I decided to go to MIT even though I didn't know much about it. Someone suggested I should I apply there if I was interested in science. You know, I was so naïve, so I went. I thought I'd be a physician, because at the time that's what I thought about science, that I'd be a medical doctor. Biology was a very small major at the time I was at MIT. Most people were studying engineering or physics. There were a few biology students and about 95% of them were pre-medical. But at some point that didn't interest me and I decided to start working in a lab.
Diana: Tell me about your undergraduate research experience with Alex Rich.
David: I went to Alex's lab and started working on tRNA amino acylation and protein synthesis and my research interests grew. Alex's lab was kind of a wild place, it was just chaos! But I learned a lot there. People were working on different things and there was a whole wing of people who were crystallographers doing tRNA structure. I met some fascinating people there, like Ned Zieman who is now considered one of the fathers of nano-technology, and Jeremy Nathans. Of course Jeremy was always precocious and he had this beautiful Nature paper looking at the structure of DNA with an intercalator.19
I started to appreciate the idea that you could put together models from doing experiments that seemed kind of arcane; that you can take this data and put it together into a physical idea of what was happening in the test tube. It fascinated me that you could start thinking about things in pictures even though you're really doing all these kind of complicated manipulations and reading them out just based on say counts coming out of the scintillation counter. And mostly I liked doing something where I could do something physical as well as intellectual and not just sit around reading and writing. I liked tinkering and I found that I was really good at that kind of work.
Diana: Tell me about graduate school at Berkeley
David: When I went to Berkeley, coming from MIT, I wanted to do something more molecular. I decided I wanted to stay in some sort of microorganisms system and do something related to development. I had talked to Jeremy Thorner. I heard he wasn't taking any students so I never really asked him about joining his lab. But luckily he asked me “how come you haven't talked to me about coming to my lab?” and I said, “I heard you're full.” And he said “there's always room for a good person.” At the time, I was in the Biochemistry Department and he was in Microbiology. The departments were separate and I was told that I would have to take my qualifying exam again if I joined Jeremy's lab. So Randy (Schekman) agreed to be my Biochemistry co-sponsor even though he didn't have room in his lab, and then I ended up doing a joint project between the 2 of them. Working on α factor and peptide secretion and processing was a happy accident. It turned into a great project and I really learned to love that area. I noticed at the time, some people in yeast were geneticists and some were biochemists and there were very few people who did both. But Randy and Jeremy were trained as biochemists in places where genetics was also emphasized and so they combined both. I think that's what made their lab so powerful and that was a big lesson for me.
Diana: So, how did you end up in the Axel Lab and begin your exploration of neuroscience?
David: Well, 2 things. About that time, Richard had published a paper that was on the cover of Cell, with Sheller and Kandel, about all these egg-laying hormones in Aplysia. They had these beautiful pictures of the Mag cells, which make egg-laying hormones, and it's basically a big precursor that gets cleaved up into many neuroactive peptides.20 And so that of course caught my eye because I was working on peptide hormone processing. At the time, there weren't that many molecular biologists working in the nervous system.
Then I started to think about what I wanted to do. It was a choice, should I stay in yeast? Because things were just rocketing and there were so many unanswered questions. In the end I decided I wanted to go into Neuroscience. But, I needed to go to a lab where they were doing some molecular biology and genetics. So I went to visit Richard's lab, and it was like Alex Rich's lab, it was this totally chaotic place that was exciting and where you can do what you want. There were some people who had already started to work on the nervous system. But everyone was working on something completely different.
Diana: How did you decide to work on cloning serotonin receptors?
David: I think it was studying peptide hormones in yeast and then thinking about mammalian equivalents in a way—endorphins, enkephalins, α-MSH, and wondering about how these things are working in the brain. That got me thinking about signaling in the brain. And I didn't stay with peptide hormones but I went to monoamines, because I was interested in things like natural products and hallucinogens. So I think that's where my interest in natural products came from.
I started reading some books by Sol Snyder (Fig. 4). Sol's always been kind of an influence on me. He approached science in such a different way than the people I've worked for. He has a paradigm that's all his own and unique. And at the time, as I read his work I realized he was the person who identified some opiate receptors using ligand binding assays and had a whole other take on neuroscience and molecular pharmacology that he really revolutionized. That influenced me as well, thinking about how Sol approached science using natural products like morphine to understand what their endogenous targets were.
Figure 4.

David Julius (left) with Sol Snyder (right, Professor at Johns Hopkins School of Medicine) at an awards ceremony in 2014. Reproduced by permission of Michael J Caterina MD, PhD, Johns Hopkins School of Medicine.
Diana: But you never worked with Sol?
David: I never worked with him. I've talked to him on a number of occasions. He's influenced me a lot. He's a really interesting guy and really fun to talk to. His discoveries are monumental. His discovery of μ-opioid receptor is one of the great achievements in modern neuropharmacology.21
Diana: Tell me about starting your own lab (Fig. 5). What was the first thing you did?
Figure 5.

The current members of the Julius lab, UCSF 2015.
David: The first thing I did was clone the 5-HT3 receptor.22 I planned to work on all the GPCRs and reclone the first couple of serotonin receptor subtypes. I started thinking, “What's the one quirky member of this family?” There was all this work on this 5-HT3 receptor that showed there were binding sites in the brain and it was this very mysterious thing. Was it really a channel? What relationship does it have to other serotonin receptors? What does it do? A lot of people were looking for this receptor, there were drug companies that were trying to affinity purify it because there were such good drugs. And so we expression cloned it. That was kind of our first big hit, and that got me interested in channels and GPCRs, at least for a while.
Diana: How did you begin working on pain?
David: There was a sort of a small fledgling effort in terms of people who wanted to look at the molecular biology of nociception. And the 5-HT3 receptor was known to be expressed in a subset of sensory neurons. And so I started getting more and more interested in what these cells were and what they do. I started reading about pain and I thought this was kind of an interesting area. Because I'd always been a little frustrated, working on neurotransmitter receptors in the brain, it seemed like such a distance between behavior and molecular biology. And it looked like in a sensory system you could do that a little bit better. You could give stimuli and ask what was happening. And of all the sensory systems, pain was the one that seemed to be the least picked over. But of everybody who was at all interested, and there weren't very many in molecular biology, they were all interested in things like capsaicin. And I knew enough to know that that was kind of a holy grail. And I thought, maybe it's one of these other channels that we've identified, some P2X or 5-HT3 subtype lurking in there. You have to remember that genomics was just starting, so it wasn't so easy to go in and pull out genes. People were pulling out different subtypes of P2Xs at the time and 5HT receptors, and it was hard work. And every time they get one, they asked is it a capsaicin receptor?
We tried that for a while, and then I just decided, maybe we shouldn't just look where we're shining the light, that we have to step back. But there were a lot of risks, obviously, nobody really knew. Up until the time we cloned it, there were still papers saying it wasn't really a real receptor. And one day I remember standing in the supermarket looking at all these rows of spices. Holly (Ingraham, professor at UCSF and David's spouse) was off somewhere, and she came around and I said, “This is such an interesting problem.” And she said, “So quit f**king around and do it!” And then I decided, I just got to do this! Mike (Caterina) was sniffing around GABA receptors, but then I heard this rumor that Benny Bentler had figured out what GABAB receptors were. So I said “You know what? We just oughta do this capsaicin thing. You can take a risk working on something you're going to get scooped on. Or you could just try this.” And so he was game for it.
Diana: Tell me about the cloning of TRPV1.9
David: We originally tried using oocytes to clone it, but decided it was too laborious, we'll never make enough RNA. This thing was supposed to be some sort of calcium channel, maybe. Whatever it does, it increases calcium in cells. We had this calcium imaging rig that I had bought, because I went to see Rich Lewis and tried to put together a little system. It took us awhile because we didn't know what we were doing. We were getting people from different labs to help us do different things, learn how to culture neurons. And then we just did it.
Diana: And so you saw calcium signals in DRG neurons first?
David: Yeah
Diana: What year was this?
David: I think 1996. And then I said you know, if we're going to do this, since we had never done expression cloning in mammalian cells before with calcium signals, we have to mock this up. We had to make a library. We had made libraries before, but never from DRG (dorsal root ganglia). Lucky for us, at the time, we had help from Hey-Young (Kong) who was post-doc with Moses Chao. She called me up and said, “We want to make a library from DRG from embryonic and adult mouse. But we don't know how to make libraries so can we come to your lab and do it?” And I said “Yeah, ok because we want to make those libraries too.” Then we just started screening, and, within a couple of weeks, we had some positives. I think it was fast. And I remember going away for like 2 d to give a seminar and then I came back and Mike had a big grin on his face and said, “I got to show you something!” So we went into the imaging room and he got the computer going and I could see there was nothing and then all of a sudden BOOM! Like four cells just went blazing with capsaicin. Then within 3 or 4 weeks we had a single clone.
Then the question after we got it was -what is this thing? What does it look like? I had never really spent any time thinking about TRP channels, at all. I mean, nobody had, really.
Diana: Right, because there wasn't very much out there on these channels at the time.
David: Nobody was really that interested in them. I remember that I wrote in a book, “For more general perspective, the cloning of the vanilloid receptor put a spotlight on TRP channels as important new players in vertebrate sensory systems. In retrospect this should not have come as a great surprise given the well established role of TRP channels in fly phototransduction, but for whatever reason, their relevance to sensory signaling in higher systems had not been fully appreciated.” And it was true.
David: So we got this sequence.
Diana: And it was related to Drosophila TRP?
David: Well they had these ankyrin repeats, and there's no other channel that had ankyrin repeats. And then Mike said “You know I think they're related to these things called TRP channels!” “Great, what's that?” And so we started looking at this. Then when we cloned it, the real question was, what does this thing normally do? You know, in retrospect it seems like Heat-capsaicin (go together), but no one had ever really enunciated that. Except to say that a lot of capsaicin-sensitive cells were heat sensitive. This was the legacy of Snyder in a way, that capsaicin was an exogenous agent mimicking the effects of an endogenous agent. Everyone thought there would be an endogenous capsicone. And you know, there's truth in that as well, with protons, anandamide, other bioactive lipids, prostaglandins. But these papers had come out from McNaughton and Cesare recording heat-activated currents,2 so you knew that there must have been some sort of heat-activated conductance. But things hadn't gelled enough to really know what was going on and the pharmacology wasn't good enough for people to put 2 and 2 together.
Diana: Yeah and I looked at some of the early current measurements too. And there weren't really good examples comparing the current triggered by capsaicin and the current activated by heat.
David: Yes, and put them together. There were some, Rang had sort of tiptoed around that a bit. And it was known that there were similar non-selective cation channels. Sometimes heat-evoked currents would be blocked by Ruthenium red and sometimes not. Of course, there were a lot of species differences and that complicated things. Because some things were done in rabbits and some in rats and mice, so there's a lot of species variation, which we now know to be the case.
So from our perspective, we literally just expressed the thing in oocytes and started throwing things at it that caused pain: bradykinin, serotonin and all this stuff. And then one day we decided “Hey, what about these physical stimuli? Like pressure and heat?” So one day we just heated up the chamber and all of a sudden we saw these currents. I mean, it was really exciting but it was also kind of worrying, because we had no experience in doing that kind of stuff. I just thought “Oh my god, what if this is some major artifact because we were changing the grounding or something?” We had convinced ourselves that, compared to background, we could really see these heat-evoked currents. But I was always worried about it. And we convinced ourselves it was real, but we didn't quite understand it yet. Nobody had that much experience with this. We thought we really had to put this as a major part of the paper, because we really think this is real and it made sense. We did as many controls as we could figure out. But (I thought) god, what if it's just some trivial thing? You put in this protein, and somehow it makes the cells heat sensitive. And so, I lost a lot of sleep over it but we decided that we had been as careful as we could reasonably be, and so we just got to plant our flag on that. And everybody else started looking at it, and collectively we all got more sophisticated at looking at heat-evoked currents.
Diana: Wow, that's amazing. So were you convinced that there was then going to be a big phenotype in the TRPV1 knockout?
David: Oh well, we didn't know, because I think there are multiple ways to sense heat. But it turned out there is a big phenotype in the tail-flick assay.10 And I don't know why that is, but it could be because the tail in the mouse is a thermoregulatory organ. It's very highly innervated by heat-sensitive fibers. What looked less profound, but it was still pretty robust, was the hot-plate test. And there the expectation was that we would see differences at a cooler threshold, closer to 43°C but I think naively we were equating the biophysical aspects of the channel with the psychophysical.
Diana: You saw it at higher temperatures.
David: Yeah, and we were scratching our heads thinking, “What does this mean?” And then we just thought that “Well, if there's any input to the (spinal) cord, maybe that's enough to discriminate noxious from innocuous.” We sort of figured that might be the case.
The most dramatic phenotype that we saw was looking at injury models where TRPV1 was required for thermal hypersensitivity. In addition, 2 observations using TRPV1 antagonists really solidified the role of this channel in acute thermosensation. First, drugs in human clinical trials decreased thermal acuity. Second, early phase TRPV1 inhibitors, increased body temperature by 0.6 to 2 degrees and induced hyperthermia,23 suggesting that the silencing of these peripheral receptors causes the body to think it's cold.
But it was only after the cloning and characterization of TRPM8 that I felt that TRP channels are bona fide heat sensors and play key roles in peripheral somatosensation.11 The TRPM8 phenotype was very dramatic. Like TRPV1, TRPM8 plays a key role in both acute temperature sensation and thermal hypersensitivity. Human studies using TRPM8 antagonists showed the opposite results of drugs that block TRPV1, namely hypothermia. And of course we have yet to see how effective drugs targeting these channels will be for treating pain, itch or conditions like cold allodynia.
Diana: Do you have words of advice for a successful career in science?
David: You have to be open-minded and objective and trust that you know what good data and science is. You need to be willing to be wrong, but don't be too quick to say you're wrong. Trust your data and construct a simple model. You have to constantly ask your self: are my findings advancing the field? Are people using your tools? Are there any therapeutic benefits?
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
I thank Shannan McClain for help with the photographs and transcription of the interview. I also thank David Julius for making time in his busy schedule for this interview and for being a wonderful mentor and role model.
References
- 1.Moayedi M, et al.. J Neurophysiol 2013; 109:5-12; PMID:23034364; http://dx.doi.org/ 10.1152/jn.00457.2012 [DOI] [PubMed] [Google Scholar]
- 2.Cesare P, et al.. Proc Natl Acad Sci U S A 1996; 93:15435-9; PMID:8986829; http://dx.doi.org/ 10.1073/pnas.93.26.15435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reichling DB, et al.. Proc Natl Acad Sci U S A 1997; 94:7006-11; PMID:9192682; http://dx.doi.org/ 10.1073/pnas.94.13.7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jancso G, et al.. Nature 1977; 270:741-3; PMID:593396; http://dx.doi.org/ 10.1038/270741a0 [DOI] [PubMed] [Google Scholar]
- 5.Bevan S, et al.. Trends Pharmacol Sci 1990; 11:330-3; PMID:2203194; http://dx.doi.org/ 10.1016/0165-6147(90)90237-3 [DOI] [PubMed] [Google Scholar]
- 6.Oh U, et al.. J Neurosci 1996; 16:1659-67; PMID:8774434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suto K, et al.. Neuroscience 1999; 92:1131-5; PMID:10426551; http://dx.doi.org/ 10.1016/S0306-4522(99)00063-9 [DOI] [PubMed] [Google Scholar]
- 8.Reid G, et al.. Nature 2001; 413:480; PMID:11586349; http://dx.doi.org/ 10.1038/35097164 [DOI] [PubMed] [Google Scholar]
- 9.Caterina MJ, et al.. Nature 1997; 389:816-24; PMID:9349813; http://dx.doi.org/ 10.1038/39807 [DOI] [PubMed] [Google Scholar]
- 10.Caterina MJ, et al.. Science 2000; 288:306-13; PMID:10764638; http://dx.doi.org/ 10.1126/science.288.5464.306 [DOI] [PubMed] [Google Scholar]
- 11.Bautista DM, et al.. Nature 2007; 448:204-8; PMID:17538622; http://dx.doi.org/ 10.1038/nature05910 [DOI] [PubMed] [Google Scholar]
- 12.Jordt SE, et al.. Nature 2004; 427:260-5; PMID:14712238; http://dx.doi.org/ 10.1038/nature02282 [DOI] [PubMed] [Google Scholar]
- 13.Cao E, et al.. Nature 2013; 504:113-8; PMID:24305161; http://dx.doi.org/ 10.1038/nature12823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao M, et al.. Nature 2013; 504:107-12; PMID:24305160; http://dx.doi.org/ 10.1038/nature12822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paulsen CE, et al.. Nature 2015; PMID:2585529725855297 [Google Scholar]
- 16.Julius D. Annual Rev Cell Devel Biol 2013; 29:355-84; http://dx.doi.org/ 10.1146/annurev-cellbio-101011-155833 [DOI] [PubMed] [Google Scholar]
- 17.Patapoutian A, et al.. Nature Rev Drug Discov 2009; 8:55-68; PMID:19116627; http://dx.doi.org/ 10.1038/nrd2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vriens J, et al.. Nat Rev Neurosci 2014; 15:573-89; PMID:25053448; http://dx.doi.org/ 10.1038/nrn3784 [DOI] [PubMed] [Google Scholar]
- 19.Wang AHJ, et al.. Nature 1979; 282:680-6; PMID:514347; http://dx.doi.org/ 10.1038/282680a0 [DOI] [PubMed] [Google Scholar]
- 20.Scheller RH, et al.. Cell 1982; 28:707-19; PMID:6284369; http://dx.doi.org/ 10.1016/0092-8674(82)90050-2 [DOI] [PubMed] [Google Scholar]
- 21.Pert CB, et al.. Science 1973; 179:1011-4; PMID:4687585; http://dx.doi.org/ 10.1126/science.179.4077.1011 [DOI] [PubMed] [Google Scholar]
- 22.Maricq AV, et al.. Science 1991; 254:432-7; PMID:1718042; http://dx.doi.org/ 10.1126/science.1718042 [DOI] [PubMed] [Google Scholar]
- 23.Wong GY, et al.. Brain Res Rev 2009; 60:267-77; PMID:19150372; http://dx.doi.org/ 10.1016/j.brainresrev.2008.12.006 [DOI] [PubMed] [Google Scholar]