

Abstract
The mismatch negativity (MMN) evoked potential, a preattentive brain response to a discriminable change in auditory stimulation, is significantly reduced in psychosis. Glutamatergic theories of psychosis propose that hypofunction of NMDA receptors (on pyramidal cells and inhibitory interneurons) causes a loss of synaptic gain control. We measured changes in neuronal effective connectivity underlying the MMN using dynamic causal modeling (DCM), where the gain (excitability) of superficial pyramidal cells is explicitly parameterised. EEG data were obtained during a MMN task—for 24 patients with psychosis, 25 of their first‐degree unaffected relatives, and 35 controls—and DCM was used to estimate the excitability (modeled as self‐inhibition) of (source‐specific) superficial pyramidal populations. The MMN sources, based on previous research, included primary and secondary auditory cortices, and the right inferior frontal gyrus. Both patients with psychosis and unaffected relatives (to a lesser degree) showed increased excitability in right inferior frontal gyrus across task conditions, compared to controls. Furthermore, in the same region, both patients and their relatives showed a reversal of the normal response to deviant stimuli; that is, a decrease in excitability in comparison to standard conditions. Our results suggest that psychosis and genetic risk for the illness are associated with both context‐dependent (condition‐specific) and context‐independent abnormalities of the excitability of superficial pyramidal cell populations in the MMN paradigm. These abnormalities could relate to NMDA receptor hypofunction on both pyramidal cells and inhibitory interneurons, and appear to be linked to the genetic aetiology of the illness, thereby constituting potential endophenotypes for psychosis. Hum Brain Mapp 37:351–365, 2016. © 2015 The Authors Human Brain Mapping Published by Wiley Periodicals, Inc.
Keywords: psychosis, schizophrenia, unaffected relatives, genetic risk, effective connectivity, dynamic causal modeling, DCM, cortical excitability, cortical gain, NMDA receptor
INTRODUCTION
Psychotic disorders are among the most severe and enduring mental illnesses, characterised by a distorted sense of reality; an inability to distinguish subjective experiences from the objective world. Disorders where psychosis is commonly experienced include, amongst others, schizophrenia, bipolar disorder, and schizoaffective disorder [NICE, 2014; WHO, 2008].
The mismatch negativity (MMN) event related potential is a pre‐attentive brain response to a discriminable change in auditory stimulation [Duncan et al., 2009; Näätänen, 1992; Todd et al., 2013; Umbricht et al., 2005]. Reduced MMN amplitude is one of the most reliable findings in schizophrenia research, and since the first publication by Shelley et al [1991] over 100 papers have commented on this reduced amplitude [e.g., Baldeweg and Hirsch, 2015; Shaikh et al., 2012; Todd et al., 2013], with a mean effect size of 0.99 [Umbricht et al., 2005]. The MMN is abnormal in clinical risk groups as well as in patients, and is a promising biomarker for psychosis prediction [Bodatsch et al., 2014; Nagai et al., 2013]. Furthermore, the MMN has been proposed as a potential endophenotype or a biological marker of genetic risk for psychosis, because it is heritable [Hall et al., 2006, 2009; Hong et al., 2012], and abnormal in first degree relatives of patients, who have an increased genetic risk for psychosis [Jessen et al., 2001; Michie et al., 2002]. However, not all studies in unaffected relatives have found MMN abnormalities [Bramon et al., 2004; Hong et al., 2012; Kim et al., 2014].
Most previous studies of the MMN use classical electroencephalogram (EEG) analysis methods that investigate the observed amplitude of the event related potential at the sensor level. However, abnormal functional integration among brain regions or “dysconnection,” has been proposed as a core pathology of psychosis [Friston, 1998; Stephan et al., 2006]. Motivated by this hypothesis, we investigated the MMN in terms of the underlying neuronal connectivity. We used dynamic causal modeling (DCM), which explains EEG data using a hierarchical network of dynamically coupled sources, and estimates effective connectivity—the influence that one neuronal system exerts over another—using Bayesian model comparison and inversion [David et al., 2006; Friston et al., 2003]. Several previous DCM studies have found abnormal effective connectivity in psychosis, both using EEG/MEG [Dima et al., 2012, 2010; Fogelson et al., 2014; Roiser et al., 2013] and fMRI methods [Crossley et al., 2009; Deserno et al., 2012; Dima et al., 2009; Mechelli et al., 2007; Schmidt et al., 2014]. However, this is the first DCM study investigating the MMN paradigm in patients as well their unaffected relatives, with a view to examining whether abnormal effective connectivity (and its modulation) could act as an endophenotype for psychosis.
Our hypothesis is based on current theories of psychosis that implicate the neuromodulation of postsynaptic excitability or cortical gain control [Harrison et al., 2011; Lisman et al., 2008; Phillips and Silverstein, 2013; Stephan et al., 2006]. The most ubiquitous neurotransmitter receptor involved in gain modulation is the glutamatergic N‐methyl‐D‐aspartate receptor (NMDA‐R), which is expressed more densely in superficial cortical layers [Friston, 1998; Gonzalez‐Burgos and Lewis, 2012; Lakhan et al., 2013]. NMDA‐R hypofunction is known to be associated with psychosis; it is for example well established that NMDA‐R antagonists such as ketamine or phencyclidine produce psychotomimetic symptoms in healthy individuals and worsen symptoms in patients with schizophrenia [Gilmour et al., 2012; Javitt and Zukin, 1991; Kantrowitz and Javitt, 2010; Krystal et al., 1994; Lahti et al., 1995; Malhotra et al., 1996; Pilowsky et al., 2006]. Recent genetic association studies also implicate the NMDA‐R and its postsynaptic signaling cascade in the disorder [Purcell et al., 2014; Ripke et al., 2014]. Furthermore, the hypofunctioning of NMDA‐Rs on inhibitory GABAergic interneurons is also thought to contribute to a loss of balance between excitation and inhibition, which has been implicated in the neuropathology of psychosis [Gonzalez‐Burgos and Lewis, 2012]. Lastly, reduced MMN amplitudes have been observed in healthy volunteers after NMDA‐R blockade, for example by administration of ketamine [Javitt et al., 1996; Näätänen et al., 2012; Schmidt et al., 2012a; Umbricht et al., 2000]. From a theoretical perspective, this loss of gain control or excitation‐inhibition balance fits comfortably with hierarchical predictive coding models of psychosis and false inference–that rest on the abnormal encoding of uncertainty or precision by the gain of (superficial pyramidal) cells reporting prediction errors [Adams et al., 2013].
Given the prominence of NMDA‐Rs in superficial cortical layers, it is unsurprising that the gain of superficial pyramidal cell populations is strongly affected by NMDA‐R function [Fox et al., 1990; Pinotsis et al., 2014]. In DCM, this gain is parameterized as the inhibitory self‐connectivity (or “intrinsic connectivity”) of superficial pyramidal cells within a cortical source [Friston, 2008]. Our aim in this study was to investigate group differences in MMN responses of patients with psychosis, their unaffected relatives, and healthy controls, and test whether these are best explained by modulations of synaptic gain at different levels of the cortical hierarchy. We hypothesised that, compared to controls, we would see abnormal cortical gain control in both individuals with psychosis and (to a lesser extent) in their first degree relatives.
MATERIALS AND METHODS
Sample and Clinical Assessment
The total sample of 84 participants included 24 patients with a psychotic illness (75% schizophrenia, no comorbid diagnoses; see breakdown in Table 1), 25 unaffected first degree relatives of psychosis sufferers (without any personal history of a psychotic illness), and 35 unrelated controls (without any personal or family history of psychotic illnesses).
Table 1.
Sample demographics (N = 84)
| Patients with psychosis N = 24 | Unaffected relatives N = 25 | Controls N = 35 | |
|---|---|---|---|
| Mean age (years, SD) | 34.6 (±9.3) | 43.7 (±14.5) | 41.8 (±14.5) |
| Age range (years) | 23–54 | 16–62 | 19–69 |
| Gender (N male/female, % female) | 18/6 (25%) | 12/13 (52%) | 17/18 (51%) |
| Education (mean years, SD) | 13.6 (±2.8) | 14.0 (±3.1) | 14.4 (±3.7) |
| Diagnosis (N, %) | |||
| Schizophrenia | 18 (75%) | – | – |
| Schizoaffective disorder | 3 (13%) | – | – |
| Psychosis NOS | 1 (4%) | – | – |
| Bipolar I disorder (w. psychosis) | 2 (8%) | – | – |
| Major Depression | – | 3 (12%) | 1 (3%) |
| No psychiatric illness | – | 22 (88%) | 34 (97%) |
| Illness duration (mean years, SD) | 12.1 (8.4) | NA | NA |
| Psychotropic medication (N, %) | 23 (95.8%) | NA | NA |
| CPZ equivalent (mean, min‐max)* | 549.4 (30‐1100) | NA | NA |
| Years medicated (mean, SD) | 10.6 (±8.6) | NA | NA |
| First medicated (mean years, SD) | 24.4 (±7.2) | NA | NA |
| PANSS (mean, SD)** | |||
| Positive | 12.5 (±4.6) | 7.2 (±0.6) | 7.0 (±0.0) |
| Negative | 14.9 (±5.5) | 7.2 (±0.6) | 7.0 (±0.0) |
| General | 24.3 (±4.9) | 17.5 (±2.0) | 16.1 (±0.4) |
| Relationship to proband (N, %) | |||
| Mother | NA | 4 (16.0%) | NA |
| Father | NA | 9 (36.0%) | NA |
| Sister | NA | 8 (32.0%) | NA |
| Brother | NA | 3 (12.0%) | NA |
| Daughter | NA | 1 (4.0%) | NA |
NA = not applicable; SD = standard deviation; NOS = not otherwise specified; * CPZ equivalent = average chlorpromazine equivalent dosage (mg) for those taking antipsychotic medication (N = 18); ** PANSS positive and negative scores range from 7 to 49, PANSS general scores range from 16 to 112
A personal history of nonpsychotic psychiatric illnesses did not constitute an exclusion criterion for relatives or controls, provided they were well and not taking any psychotropic medication at the time of testing and for the preceding 12 months. This was to avoid recruiting biased control groups, unrepresentative of the general and local populations. Three relatives (12%) and one control (3%) had a history of major depressive disorder.
Patients with psychosis and relatives were recruited through voluntary organisations, advertisements in the local press and from clinical teams at the South London and Maudsley NHS Foundation Trust. Controls were recruited by advertisements in the local press and job centres. Participants were excluded if they had a diagnosis of alcohol or substance dependence in the last 12 months, neurological disorders or a previous head injury with loss of consciousness longer than a few minutes.
All participants were clinically interviewed to confirm or exclude a Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition [DSM‐IV; APA, 1994] diagnosis. Instruments used included the Schedule for Affective Disorders and Schizophrenia—Lifetime version [SADS‐L; Endicott and Spitzer, 1978] and the Positive and Negative Syndrome Scale [PANSS; Kay et al., 1987]. Information regarding psychiatric diagnoses of family members not directly assessed was collected from the most reliable informant(s) with the Family Interview for Genetic Studies [FIGS; Maxwell, 1992].
All participants gave informed written consent to participate, and the study was approved by the Institute of Psychiatry Research Ethics Committee, conforming to the standards set by the Declaration of Helsinki. This sample is part of the larger Maudsley Family Study of Psychosis [e.g., Dutt et al., 2012; Ranlund et al., 2014; Schulze et al., 2008; Shaikh et al., 2013].
EEG Data Acquisition
Electroencephalogram (EEG) was collected from 17 scalp sites according to the 10/20 International system (FP1, FP2, F7, F8, F3, F4, C3, C4, P3, P4, FZ, CZ, PZ, T3, T4, T5, T6), grounded at Fpz using silver/silver‐chloride electrodes [Jasper, 1958]. Vertical, horizontal, and radial electro‐oculographs monitored eye movements, and the left ear lobe served as reference. Data were continuously digitised at 500 Hz with a 0.03–120 Hz band‐pass filter (24 dB/octave roll‐off). Impedances were kept below 5 kΩ [Bramon et al., 2004, 2005].
MMN paradigm
This was a duration‐deviant auditory two tone paradigm. The stimuli were 1,200 tones (80 dB, 1,000 Hz, 5 ms rise/fall time), with a 300 ms inter‐stimulus interval, presented in three blocks of 400 stimuli through bilateral intra‐aural earphones. 85% of the tones were “standards” (25 ms duration), and 15% were “deviants” (50 ms duration) [Hall et al., 2009; Shaikh et al., 2012]. The total duration of the experiment was about 10 min.
Participants were sitting comfortably in an armchair, and were instructed to keep their eyes open, fixate on a point in front of them, and disregard the sounds presented.
The classical group comparisons of the MMN amplitude in this sample have been reported in a previous study [Bramon et al., 2004]. Here we undertake a new analysis of effective connectivity during the MMN task.
EEG Data Preprocessing
Signal processing was conducted using SPM 12b (http://www.fil.ion.ucl.ac.uk/spm/software/spm12) [Litvak et al., 2011] and FieldTrip (http://www.fieldtrip.nl) [Oostenveld et al., 2011] in MATLAB R2013b (http://www.mathworks.co.uk).
The raw EEG data were converted to SPM format, and re‐referenced to the common average. A high‐pass filter of 0.5 Hz was applied, followed by a low‐pass 70 Hz filter. A stop‐pass (49–50 Hz) filter was also applied, to remove line noise. The data were then downsampled to 200 Hz, and epoched with a peristimulus window of −100 to 300 ms. Baseline correction was performed using the 100 ms before stimulus onset.
Independent Component Analysis was used to correct for ocular artefacts in the data. The EEG activity was decomposed into 17 independent components, of which a maximum of two that clearly corresponded to eye blinks were removed from the data. Additional automatic artefact rejection was then conducted, removing any trials whose activity exceeded ±70 μV across all channels. This resulted in an average of 45 trials (3.7%) being rejected per participant, which did not differ between the three groups (F(2,81)=1.1, P = 0.3).
The EEG data were then averaged using robust averaging in SPM. This procedure produces the best estimate of the average by weighting data points as a function of their distance from the sample mean, so that outlier values have less influence on the overall mean [Wager et al., 2005]. This was followed by an additional low‐pass filter of 70 Hz, as recommended with robust averaging [Litvak et al., 2011].
The grand average event related potential waveforms across subjects were computed for patients, relatives and controls separately. The use of grand average waveforms ensures cleaner (almost noiseless) data for each group and condition. Grand averages retain features that are conserved within groups, and suppress individual differences. These grand averages constitute six event related potentials—one for each group and stimulus condition (standard and deviant tones)—that were characterised in the subsequent DCM analysis [Fogelson et al., 2014].
Dynamic Causal Modeling
Dynamic causal modeling (DCM) explains measured data using a hierarchical network of dynamically interacting sources, and estimates effective connectivity (the influence that one neuronal system exerts over another), using Bayesian model inversion [Friston et al., 2007]. DCM was originally developed for fMRI [Friston et al., 2003] and was subsequently generalised to other modalities, including evoked responses measured by EEG [David et al., 2006].
DCM permits source reconstruction whilst incorporating biological constraints on neuronal dynamics and coupling [David et al., 2005; Kiebel et al., 2009; Pinotsis et al., 2012]. The neuronal model makes predictions about the dynamics of each source based on the underlying anatomy and biology. We used the canonical microcircuit neural mass model [Bastos et al., 2012], in which each neural source comprises four cell populations: Superficial and deep pyramidal cells, spiny stellate cells and inhibitory interneurons. Each source is connected to other sources via extrinsic excitatory connections, and cell populations within sources are connected to each other via intrinsic connections [Pinotsis et al., 2013]. In this study, we focused on the self‐inhibition of superficial pyramidal cell populations (see Supporting Information Fig. S1), because the strength of this connection reflects the gain (or excitability) of this population, which is linked to NMDA‐R function.
Each source (i.e., each node in the network) was modeled with a single equivalent current dipole under bilateral symmetry assumptions [Kiebel et al., 2006]. We used a boundary elements head model [Fuchs et al., 2001] to approximate the brain, cerebrospinal fluid, skull and scalp surfaces. A canonical MRI head model was used, and coregistration of electrode positions and head model was performed for each subject to map the Montreal Neurological Institute coordinates to points on the head.
Following standard practice, the EEG data were projected onto eight spatial modes to ensure more robust model inversion and dynamical stability. These are the eight principal components or modes of the prior predictive covariance in sensor space [Fastenrath et al., 2009]. We modeled responses from 0 to 250 ms post stimulus onset, to ensure selective modeling of the MMN response per se, rather than later components [Garrido et al., 2008].
DCM specification
In DCM, Bayesian inference is used to optimise neural source dipoles based on a priori information about their locations. This information is available from studies investigating the sources underlying the MMN—using fMRI [Molholm et al., 2005; Rinne et al., 2005; Schönwiesner et al., 2007], PET [Dittmann‐Balçar et al., 2001; Müller et al., 2002], EEG/MEG [Deouell et al., 1998; Fulham et al., 2014; Jemel et al., 2002; Rinne et al., 2000; Tiitinen et al., 2006], and DCM [Garrido et al., 2007, 2009a, 2008]—showing that the MMN is generated by temporal and frontal sources. Using DCM, the model with the most evidence consists of a three‐level hierarchy comprising bilateral primary auditory cortices (Heschl's gyrus, A1), bilateral superior temporal gyri (STG), and the right inferior frontal gyrus (rIFG). The frontal source is lateralised to the right hemisphere for auditory paradigms [Garrido et al., 2009a; Levanen et al., 1996].
Following Garrido et al. [2008], we included the following five sources, with prior source locations in our DCM analysis (in Montreal Neurological Institute coordinates): Left A1 (−42, −22, 7), right A1 (46, −14, 8), left STG (−61, −32, 8), right STG (59, −25, 8), and right IFG (46, 20, 8), illustrated in Figure 1A. DCM incorporates source reconstruction, and the inversion algorithm provides efficient Bayesian estimates of dipole sources that optimise these [David et al., 2005; Kiebel et al., 2009].
Figure 1.
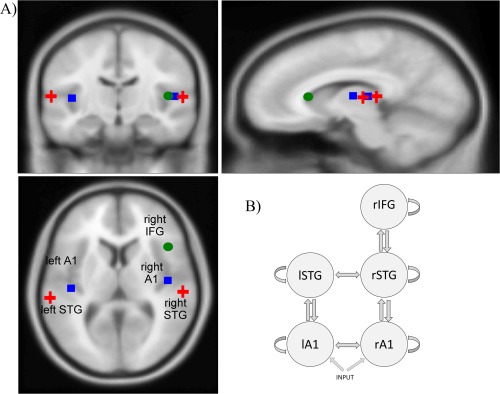
Image showing (A) the prior source locations (overlaid on an MRI image of a standard brain) and (B) the structural model used for dynamic causal modeling. The sources are linked by extrinsic (forward, backward, and lateral) connections, and each source has intrinsic inhibitory self‐connections. A1 = primary auditory cortex; STG = superior temporal gyrus; IFG = inferior frontal gyrus; l = left hemisphere; r = right hemisphere. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Our DCM assumes the existence of extrinsic (forward and backward) connections between, and intrinsic (interlaminar and intralaminar) connections within the specified sources. This has been supported by previous MMN research [Dietz et al., 2014; Garrido et al., 2007, 2009a, 2008]. We also included lateral connections linking left and right A1 and STG [Schmidt et al., 2012b]. Auditory stimuli were modeled as direct input, entering bilateral A1. This model is shown in Figure 1B.
Experimental effects
We used condition‐specific grand averaged data over all subjects within each group, allowing us to test for the effect of group directly, as well as the effect of condition by group interactions [e.g., Fogelson et al., 2014; Kiebel et al., 2007]. In other words, the grand averages were treated as the six cells of a 2 × 3 factorial design, with two levels of “condition” (standard and deviant tones) and three levels of “group” (controls, relatives and patients with psychosis).
Group effects were defined as (i) having a genetic risk for psychosis (controls versus relatives and patients combined) and (ii) having a diagnosis of a psychotic illness, irrespective of genetic risk (relatives versus patients). We tested for a main effect of diagnosis and genetic risk on effective connectivity, and the interactions with the effect of condition (standard versus deviant tones). The interactions reflect a diagnosis or risk effect on deviant‐related changes in effective connectivity or postsynaptic sensitivity.
Bayesian model selection was used to find the model with the largest (free energy approximation to the) log model evidence, among the models tested, where models are penalised for increased complexity [Penny et al., 2004]. A difference in log evidence of three or more is considered strong evidence in favour of a model, corresponding to an odds ratio of about 20:1 [Friston and Penny, 2011].
Before testing for the effects of genetic risk and diagnosis, we established the best model to explain the effect of the deviant stimulus across all three groups. We considered eight candidate models with modulations of forward, backward and/or intrinsic connections. The model that allowed for modulations of intrinsic connections (self‐inhibition of superficial pyramidal populations) only had the highest evidence, and was used in all subsequent analyses (see Supporting Information Figs. S2 and S3).
To study the effects of genetic risk and diagnosis we used Bayesian model selection to establish where in the hierarchy synaptic gain—intrinsic (self‐inhibitory) connectivity—was modulated. Our model space consisted of models with modulations of intrinsic connections at each of the hierarchical levels (A1, STG, rIFG), and all combinations of these. A total of 8 models were thus compared, shown in Figure 2.
Figure 2.
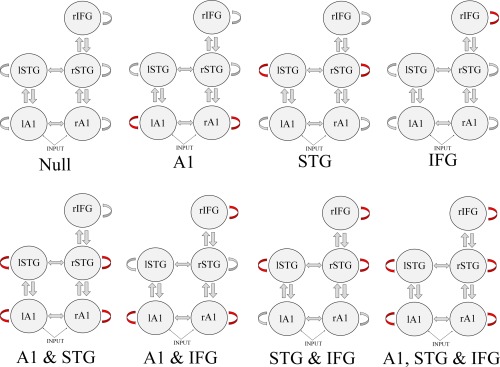
Dynamic causal modeling model space; identifying group differences in intrinsic (self‐inhibitory) connectivity. Red arrows indicate a modulated connection. A1 = primary auditory cortex; STG = superior temporal gyrus; IFG = inferior frontal gyrus; l = left hemisphere; r = right hemisphere. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Having established the model with the greatest evidence, we examined the posterior estimates of the effective connectivity under this model [Friston and Penny, 2011]. We focused on changes in intrinsic connectivity induced by the mismatch negativity, to identify any differences between patients with psychosis, unaffected relatives, and controls.
RESULTS
Sample Demographics
The demographic and clinical characteristics of the sample are detailed in Table 1. All participants were of European Caucasian ethnicity. Patients were significantly younger than controls (t = 2.14, P = 0.04) and relatives (t = 2.60, P = 0.01), and this group also contained more males compared to controls (χ 2 = 4.1, P = 0.04) and relatives (χ 2 = 3.8, P = 0.05). Controls and relatives did not differ significantly in age (t = 0.51, P = 0.61) or gender (χ 2 = 0.002, P = 0.97) distributions. Importantly, patients and relatives together (i.e., the genetic risk group) did not differ from controls in age (t = −0.83, P = 0.41) or gender (χ 2 = 1.33, P = 0.27) distributions. Years in education did not differ between groups (F = 0.40, P = 0.67).
The sample comprised 63 families, each including between 1 and 4 individuals. 49 participants (58.3%) were singletons, 18 (21.4%) were part of families with two members in the study, 9 (10.7%) were in three‐person families, and 8 (9.5%) were part of families with four members participating. All unaffected relatives had a first‐degree relative with a psychotic illness, although 8 (32%) did not have a proband participating in this study.
Mismatch Negativity Group Differences
The grand averaged event related potential waves for patients, relatives, and controls are shown in Figure 3. Group differences in the amplitude of the MMN wave of this sample have been reported in a previous paper [Bramon et al., 2004]: Patients with psychosis had significantly reduced MMN amplitude compared to both relatives and controls. The relatives did not differ significantly in MMN amplitude compared to the controls.
Figure 3.
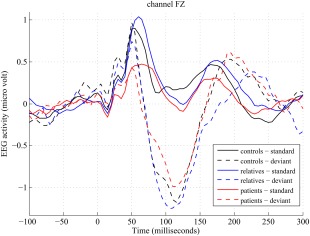
EEG activity to standard and deviant tones for each group (grand averages across subjects), at channel FZ. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Dynamic Causal Modeling Results
The Bayesian model selection results are presented in Figure 4A, showing model evidences relative to the null model (with no intrinsic modulations). The model that best explained the differences between groups allowed modulations of intrinsic connectivity in bilateral A1 and rIFG. The difference in model evidence between the winning model and the runner‐up was 80. This is significant seeing as a difference of 3 (corresponding to an odds ratio of 20:1) is considered strong evidence in favour of the winning model [Friston and Penny, 2011].
Figure 4.
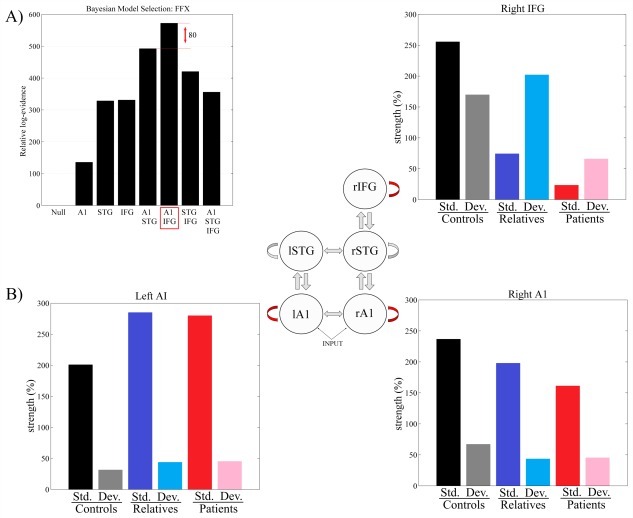
(A) Bayesian model selection results investigating intrinsic (inhibitory) modulations at different levels of the hierarchy. Log model evidences relative to the null model are shown. The winning model has modulations at A1 and IFG, and the difference in log evidence between this and the runner‐up is 80. (B) Changes in intrinsic connectivity strengths under the winning model, at each source, for patients, relatives and controls, and for standard (std.) and deviant (dev.) trials. A1 = primary auditory cortex; STG = superior temporal gyrus; IFG = inferior frontal gyrus; l = left hemisphere; r = right hemisphere. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Figure 4B shows the posterior estimates of the modulations of intrinsic connectivity in the winning model for each group (controls, relatives, and patients) and condition (standard and deviant trials). Note that because the intrinsic self‐connectivity is inhibitory, increased values correspond to reduced neural excitability, and vice versa. Posterior estimates of the modulations are also shown in Figure 5, for each source and experimental effect.
Figure 5.
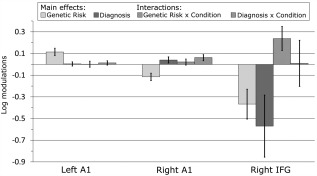
Posterior estimates of the (log scaling of) intrinsic connection parameters and their 95% confidence intervals, for each source and experimental effects investigated. A1 = primary auditory cortex; IFG = inferior frontal gyrus.
The largest effects are observed at the high‐level frontal source (rIFG), where there are striking group differences. First, both relatives and patients show reduced self‐inhibition (increased excitability) across task conditions compared to controls (i.e., a main effect of having a genetic risk for psychosis). Second, patients with psychosis show an additional reduction in self‐inhibition compared to relatives, across task conditions (i.e., a main effect of diagnosis).
Third, there is a clear interaction between having a genetic risk for psychosis and task condition in rIFG; both relatives and patients show the opposite pattern of responses to the task compared to controls. While controls demonstrate reduced inhibition (i.e., increased excitability) in response to deviants compared to standard tones, the two groups with a genetic risk showed decreased excitability in response to changes in stimulus regularities.
At the sensory level (left and right primary auditory cortices, A1), all three groups show similar responses to the MMN task conditions: Increased excitability in response to deviant compared to standard tones.
DISCUSSION
The aim of this study was to investigate whether, compared to controls, patients with psychosis and/or their unaffected relatives show altered cortical gain control (intrinsic connectivity) within cortical sources using the mismatch negativity (MMN) paradigm. We used DCM, where intrinsic connectivity is a parameterisation of the (to some extent NMDA‐R mediated) excitability of superficial pyramidal cells, which is thought to be abnormal in psychosis [Stephan et al., 2006].
Our main findings were that; (i) the largest differences in cortical responses between controls and the other groups were expressed at the top of the cortical hierarchy in the right inferior frontal gyrus (rIFG), rather than in primary sensory areas (A1); (ii) in rIFG, both groups with an increased genetic risk for psychosis (patients and their relatives) demonstrated an increase in cortical excitability across task conditions (with an additional increase in patients compared to relatives); and (iii) the two groups with a genetic risk for psychosis also showed a reversal of the normal pattern of increased excitability to deviant tones in rIFG.
Our finding of reduced self‐inhibition within rIFG across task conditions in those with a genetic risk for psychosis—as well as an additional reduction in patients with psychosis compared to relatives—is in line with theories of NMDA‐R hypofunction in psychosis [Abi‐Saab et al., 1998; Corlett et al., 2011; Goff and Coyle, 2001; Olney et al., 1999; Stephan et al., 2006]. Specifically, NMDA‐R hypofunction on parvalbumin positive inhibitory interneurons results in decreased inhibitory γ‐aminobutyric acid (GABA) input to (and therefore disinhibition of) pyramidal cells and hence a loss of balance between excitation and inhibition in prefrontal cortex [Lewis et al., 2012; Murray et al., 2014; Pinotsis et al., 2014]. These abnormalities may be linked to neurophysiological disorganisation [Díez et al., 2014], cognitive dysfunction and the development of symptoms of psychosis [Ahn et al., 2011; Lewis et al., 2008; Spencer et al., 2004].
Crucially, patients with psychosis and relatives show the opposite pattern of rIFG responses to deviant and standard tones, compared to controls. Controls show reduced self‐inhibition (increased excitability) in response to deviants, whereas both patients and relatives show a reduction in excitability in this condition. This indicates that those with an increased genetic risk for psychosis (including both relatives and patients) fail to adjust or optimise the excitability of superficial pyramidal cells in response to changes of stimulus regularities.
In a visual target detection task, in which subjects had to respond to target appearances that were either predictable or unpredictable, Fogelson et al. [2014] also investigated differences in intrinsic connectivity in patients with schizophrenia and healthy controls using EEG and DCM. They found that changes in intrinsic self‐inhibition in response to predictable stimuli were significantly attenuated in patients; this is further evidence that patients with schizophrenia fail to adjust neuronal connectivity in response to the context of incoming stimuli.
Our results can be interpreted in the context of predictive coding theories of brain function, in which the brain infers the causes of its sensory data using Bayesian inference by minimizing prediction errors throughout the cortical hierarchy [Friston, 2008; Rao and Ballard, 1999]. Predictive coding can be implemented neurobiologically by deep pyramidal cells sending top‐down predictions about lower level representations, and superficial pyramidal cells sending bottom‐up prediction errors (the difference between the actual and predicted activity) back up the hierarchy, in order to update the higher level representations [Friston, 2008]. These neurobiological details are important, because superficial pyramidal cells—that is, prediction error units—make the primary contribution to event related potentials [Garrido et al., 2009b; Lieder et al., 2013]. Crucially, the influence of ascending prediction errors on higher representations depends upon their precision, which is thought to be encoded by the gain or excitability of superficial pyramidal cells. In this setting, precision (inverse variance) corresponds to the confidence or reliability attributed to prediction errors at each level of the cortical hierarchy [Adams et al., 2013; Feldman and Friston, 2010].
In our MMN data, controls show increased synaptic gain (diminished intrinsic self‐inhibition) in all cortical sources in the deviant condition—that is, their prediction error responses to deviant tones are processed as being unduly precise and are therefore less easily suppressed. This is also the case for all individuals with a genetic risk for psychosis at the primary sensory level, but in rIFG the opposite pattern is seen. This indicates an abnormal influence of context on prediction error responses in this group, as has been seen not only in perceptual paradigms like the MMN, but also in reward learning and causal inference paradigms [Corlett et al., 2007; Murray et al., 2008].
In computational modeling work, we have shown that a loss of precision at higher levels of a hierarchical model can explain a loss of influence of context [Adams et al., 2013]. Predictive coding simulations show that aberrant precision or gain control can reproduce classic findings in the schizophrenia literature, including a reduced MMN response [Adams et al., 2013]. NMDA‐R hypofunction could confound precision or gain control in two ways, either by directly lowering synaptic gain in superficial pyramidal cell populations, or by reducing the excitability of GABAergic interneurons, thereby impairing sustained oscillatory firing of pyramidal cells and reducing their influence on lower areas [Adams et al., 2013]. Our current results lend more support to the latter mechanism, and it would be interesting to test this hypothesis directly by using DCM to assess the relative model evidences for psychosis altering the excitability of superficial pyramidal cell versus inhibitory interneuron populations.
Importantly, our results suggest that both patients and their first degree relatives have similar alterations in the excitability of superficial pyramidal cell populations, compared to controls. This indicates that these changes are linked to genetic risk factors, and are not merely a consequence of the illness state or antipsychotic medication. This alteration in the gain of superficial pyramidal cells could therefore be a potential endophenotype for psychosis [Gottesman and Gould, 2003]. The use of endophenotypes might help clarify the functional effects of genetic risk variants identified [Bramon et al., 2014; Hall and Smoller, 2010], and further research could investigate whether deviant‐related changes in excitability can predict genotype; for example, looking at candidate genes linked to NMDA‐R function. Other studies investigating effective connectivity in psychosis have also observed abnormalities in relatives of patients, including children of probands [Diwadkar et al., 2014, 2012; Winterer et al., 2003], and a previous study by Dima et al [2013] observed associations between fMRI derived measures of effective connectivity and risk genes linked to GABAergic interneuron function in patients with bipolar disorder.
Our results also suggest that patients show a further increase in excitability in rIFG across task conditions compared to unaffected relatives. This may indicate that—at least in prefrontal cortex—there are quantitative, rather than qualitative, differences between those with and without a diagnosis of a psychotic illness but at elevated genetic risk. Alternatively, this difference could be due to the effects of antipsychotic medication, which is known to influence brain function [e.g., Joutsiniemi et al., 2001; Knott et al., 2001]. The exact effects of psychotropic drugs on effective connectivity remain unclear; however, a study investigating effective connectivity in schizophrenia found abnormalities in an unmedicated at‐risk group but not in first episode patients (prescribed antipsychotics), suggesting that medication might potentially normalise abnormalities [Schmidt et al., 2013]. Future longitudinal studies and research in unmedicated patient populations are needed to address this important issue.
A limitation of the current study is that our groups differed slightly in age and gender distributions. There is evidence for both age [Cooper et al., 2006; Cooray et al., 2014; Kiang et al., 2009; Näätänen et al., 2012] and gender [Brossi et al., 2007; Matsubayashi et al., 2008] effects on MMN responses, although a DCM study did not find significant effects of aging on intrinsic connectivity [Moran et al., 2014]. Importantly, however, we found the most significant effects when comparing those with a genetic risk for psychosis (i.e. both relatives and patients) with controls, and since these two groups did not differ in age or gender distributions, our main findings are unlikely to be influenced by such confounds.
Another potential limitation is the experimental procedure used to elicit the MMN response. Because the MMN is a preattentive response not depending on the person paying attention to the sounds, it has been suggested that using a distractor task (such as watching a silent video or reading a book) can be advantageous [Duncan et al., 2009; Lang et al., 1995]. In this study, no distractor task was administered, and participants were instructed to disregard the sounds presented to them. We can therefore not control whether participants were paying attention to the task or not. Nevertheless, this distractor‐free design has been used previously and has been shown to generate clear MMN responses [Bramon et al., 2004; Haenschel et al., 2000; Javitt et al., 1998; Juckel et al., 2007]. Furthermore, attention has been found to modulate the MMN response suggesting this ERP might not actually be independent of attention [Auksztulewicz and Friston, 2015; Sussman et al., 2013; Woldorff et al., 1991].
Our Bayesian model selection result indicates that both bilateral A1 and rIFG are important in explaining group differences in modulations of intrinsic connectivity in response to deviant tones. However, modulations of self‐inhibition in STG do not seem to be so important (and were not included in the winning model). Importantly, this does not mean that the STG makes no contribution to group differences in responses, but merely suggests that including modulations in this region did not increase the evidence for the model sufficiently to justify the increased complexity. Our results furthermore suggest that group differences are most pronounced in rIFG. This is in line with past research suggesting that psychosis is associated with abnormalities at high hierarchical levels, including the prefrontal cortex [reviewed in Adams et al., 2013; Harrison et al., 2011].
We chose to calculate condition‐specific grand average responses for each group, an approach that has been used previously [e.g., Fogelson et al., 2014]. While this produces cleaner data features by reducing noise and enhancing features that are conserved over subjects, it eliminates potentially interesting individual differences. Future work could obtain subject‐specific DCM estimates, allowing the investigation of individual differences within groups, and correlations between effective connectivity parameters and various clinical and cognitive measures, as well as with genotypes.
CONCLUSION
In summary, our main finding is that patients with psychosis as well as their unaffected first‐degree relatives show increased excitability in rIFG across task conditions, relative to controls, and crucially, a loss (reversal) of the normally increased excitability in deviant trials. Hence, our results suggest that psychosis is associated with abnormalities of the sensitivity (gain) control of superficial pyramidal cell populations, which might be influenced by NMDA‐R hypofunction in prefrontal cortex. These results are in line with theories about the neuropathology and pathophysiology of psychosis. Importantly, abnormalities in unaffected relatives of patients suggest that these alterations are linked to the aetiology of psychosis and are potential endophenotypes (markers of genetic risk) for the illness.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
We would like to thank all the patients, relatives and controls who took part in this research, as well as the clinical staff who facilitated their involvement. Funding for this study was provided by the Medical Research Council (grant G0901310), the Wellcome Trust (grants 085475/B/08/Z, 085475/Z/08/Z, and 088130/Z/09/Z), and the European Commission Framework Programme for Research and Innovation (grant 330156/CODIP), and the National Institute of Health Research UK (PDA/02/06/016). We also thank the Psychiatry Research Trust, the Schizophrenia Research Fund, and the Brain and Behavior Research Foundation for financial support. All authors declare that they have no financial interests or potential conflicts of interest.
Dimitris Pinotsis and Elvira Bramon Contributed Equally as Joint Last Authors
REFERENCES
- Abi‐Saab WM, D'Souza DC, Moghaddam B, Krystal JH (1998): The NMDA antagonist model for schizophrenia: promise and pitfalls. Pharmacopsychiatry 31 Suppl 2:104–109. [DOI] [PubMed] [Google Scholar]
- Adams RA, Stephan KE, Brown HR, Friston KJ, Frith CD, Friston KJ (2013): The computational anatomy of psychosis. Front Psychiatry 47:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Gil R, Seibyl J, Sewell RA, D'Souza DC (2011): Probing GABA receptor function in schizophrenia with iomazenil. Neuropsychopharmacol 36:677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APA (1994): Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington: American Psychiatric Association.
- Auksztulewicz R, Friston K (2015): Attentional enhancement of auditory mismatch responses: A DCM/MEG study. Cereb Cortex 25:4273–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldeweg T, Hirsch SR (2015): Mismatch negativity indexes illness‐specific impairments of cortical plasticity in schizophrenia: A comparison with bipolar disorder and Alzheimer's disease. Int J Psychophysiol 95:145–155. [DOI] [PubMed] [Google Scholar]
- Bastos AM, Usrey WM, Adams RA, Mangun GR, Fries P, Friston KJ (2012): Canonical microcircuits for predictive coding. Neuron 76:695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodatsch M, Brockhaus‐Dumke A, Klosterkötter J, Ruhrmann S (2014): Forecasting psychosis by event‐related potentials — systematic review and specific meta‐analysis. Biol Psychiatry 77:951–958. [DOI] [PubMed] [Google Scholar]
- Bramon E, Croft RJ, McDonald C, Virdi GK, Gruzelier JG, Baldeweg T, Sham PC, Frangou S, Murray RM (2004): Mismatch negativity in schizophrenia: A family study. Schizophr Res 67:1–10. [DOI] [PubMed] [Google Scholar]
- Bramon E, McDonald C, Croft RJ, Landau S, Filbey F, Gruzelier JH, Sham PC, Frangou S, Murray RM (2005): Is the P300 wave an endophenotype for schizophrenia? A meta‐analysis and a family study. Neuroimage 27:960–968. [DOI] [PubMed] [Google Scholar]
- Bramon E, Psychosis Endophenotypes International Consortium Wellcome Trust Case‐Control Consortium 2 , Pirinen M, Strange A, Lin K, Freeman C, Bellenguez C, Su Z, Band G, Pearson R, Vukcevic D, Langford C, Deloukas P, Hunt S, Gray E, Dronov S, Potter SC, Tashakkori‐Ghanbaria A, Edkins S, Bumpstead SJ, Arranz MJ, Bakker S, Bender S, Bruggeman R, Cahn W, Chandler D, Collier DA, Crespo‐Facorro B, Dazzan P, de Haan L, Di Forti M, Dragović M, Giegling I, Hall J, Iyegbe C, Jablensky A, Kahn RS, Kalaydjieva L, Kravariti E, Lawrie S, Linszen DH, Mata I, McDonald C, McIntosh A, Myin‐Germeys I, Ophoff RA, Pariante CM, Paunio T, Picchioni M, Consortium PG, Ripke S, Rujescu D, Sauer H, Shaikh M, Sussmann J, Suvisaari J, Tosato S, Toulopoulou T, Van Os J, Walshe M, Weisbrod M, Whalley H, Wiersma D, Blackwell JM, Brown MA, Casas JP, Corvin A, Duncanson A, Jankowski JAZ, Markus HS, Mathew CG, Palmer CNA, Plomin R, Rautanen A, Sawcer SJ, Trembath RC, Wood NW, Barroso I, Peltonen L, Lewis CM, Murray RM, Donnelly P, Powell J Spencer CCA (2014): A genome‐wide association analysis of a broad psychosis phenotype identifies three loci for further investigation. Biol Psychiatry 75:386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brossi AB, Borba KC, Garcia CFD Reis ACMB, Isaac MdL (2007. ): Verification of the mismatch negativity (MMN) responses in normal adult subjects. Braz J Otorhinolaryngol 73:793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper RJ, Todd J, McGill K, Michie PT (2006): Auditory sensory memory and the aging brain: A mismatch negativity study. Neurobiol Aging 27:752–762. [DOI] [PubMed] [Google Scholar]
- Cooray G, Garrido MI, Hyllienmark L, Brismar T (2014): A mechanistic model of mismatch negativity in the ageing brain. Clin Neurophysiol 125:1774–1782. [DOI] [PubMed] [Google Scholar]
- Corlett PR, Murray GK, Honey GD, Aitken MRF, Shanks DR, Robbins TW, Bullmore ET, Dickinson A, Fletcher PC (2007): Disrupted prediction‐error signal in psychosis: Evidence for an associative account of delusions. Brain 130:2387–2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corlett PR, Honey GD, Krystal JH, Fletcher PC (2011): Glutamatergic model psychoses: prediction error, learning, and inference. Neuropsychopharmacol 36:294–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley NA, Mechelli A, Fusar‐Poli P, Broome MR, Matthiasson P, Johns LC, Bramon E, Valmaggia L, Williams SCR, McGuire PK (2009): Superior temporal lobe dysfunction and frontotemporal dysconnectivity in subjects at risk of psychosis and in first‐episode psychosis. Hum Brain Mapp 30:4129–4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David OO, Kiebel SJ, Harrison LM, Mattout J, Kilner JM, Friston KJ (2006): Dynamic causal modeling of evoked responses in EEG and MEG. Neuroimage 30:1255–1272. [DOI] [PubMed] [Google Scholar]
- David O, Harrison L, Friston KJ (2005): Modelling event‐related responses in the brain. Neuroimage 25:756–770. [DOI] [PubMed] [Google Scholar]
- Deouell LY, Bentin S, Giard MH (1998): Mismatch negativity in dichotic listening: evidence for interhemispheric differences and multiple generators. Psychophysiology 35:355–365. [PubMed] [Google Scholar]
- Deserno L, Sterzer P, Wüstenberg T, Heinz A, Schlagenhauf F (2012): Reduced prefrontal‐parietal effective connectivity and working memory deficits in schizophrenia. J Neurosci 32:12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz MJ, Friston KJ, Mattingley JB, Roepstorff A, Garrido MI (2014): Effective connectivity reveals right‐hemisphere dominance in audiospatial perception: implications for models of spatial neglect. J Neurosci 34:5003–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díez Á, Suazo V, Casado P, Martín‐Loeches M, Perea MV, Molina V (2014): Frontal gamma noise power and cognitive domains in schizophrenia. Psychiatry Res Neuroimaging 221:104–113. [DOI] [PubMed] [Google Scholar]
- Dima D, Frangou S, Burge L, Braeutigam S, James aC (2012): Abnormal intrinsic and extrinsic connectivity within the magnetic mismatch negativity brain network in schizophrenia: A preliminary study. Schizophr Res 135:23–27. [DOI] [PubMed] [Google Scholar]
- Dima D, Dietrich DE, Dillo W, Emrich HM (2010): Impaired top‐down processes in schizophrenia: A DCM study of ERPs. Neuroimage 52:824–832. Special issue: Computational Models of the Brain: [DOI] [PubMed] [Google Scholar]
- Dima D, Jogia J, Collier D, Vassos E, Burdick KE, Frangou S (2013): Independent modulation of engagement and connectivity of the facial network during affect processing by CACNA1C and ANK3 risk genes for bipolar disorder. JAMA Psychiatry 70:1303–1311. [DOI] [PubMed] [Google Scholar]
- Dima D, Roiser JP, Dietrich DE, Bonnemann C, Lanfermann H, Emrich HM, Dillo W (2009): Understanding why patients with schizophrenia do not perceive the hollow‐mask illusion using dynamic causal modelling. Neuroimage 46:1180–1186. [DOI] [PubMed] [Google Scholar]
- Dittmann‐Balçar A, Jüptner M, Jentzen W, Schall U (2001): Dorsolateral prefrontal cortex activation during automatic auditory duration‐mismatch processing in humans: a positron emission tomography study. Neurosci Lett 308:119–122. [DOI] [PubMed] [Google Scholar]
- Diwadkar VA, Bakshi N, Gupta G, Pruitt P, White R, Eickhoff SB (2014): Dysfunction and dysconnection in cortical‐striatal networks during sustained attention: Genetic risk for schizophrenia or bipolar disorder and its impact on brain network function. Front Psychiatry 5:50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwadkar VA, Wadehra S, Pruitt P, Keshavan MS, Rajan U, Zajac‐Benitez C, Eickhoff SB (2012): Disordered corticolimbic interactions during affective processing in children and adolescents at risk for schizophrenia revealed by functional magnetic resonance imaging and dynamic causal modeling. Arch Gen Psychiatry 69:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CC, Barry RJ, Connolly JF, Fischer C, Michie PT, Näätänen R, Polich J, Reinvang I, Van Petten C (2009): Event‐related potentials in clinical research: Guidelines for eliciting, recording, and quantifying mismatch negativity, P300, and N400. Clin Neurophysiol 120:1883–1908. [DOI] [PubMed] [Google Scholar]
- Dutt A, Ganguly T, Shaikh M, Walshe M, Schulze K, Marshall N, Constante M, McDonald C, Murray RM, Allin MPG, Bramon E (2012): Association between hippocampal volume and P300 event related potential in psychosis: Support for the Kraepelinian divide. Neuroimage 59:997–1003. [DOI] [PubMed] [Google Scholar]
- Endicott J, Spitzer RL (1978): A diagnostic interview. The schedule for affective disorders and schizophrenia. Arch Gen Psychiatry 35:837–844. [DOI] [PubMed] [Google Scholar]
- Fastenrath M, Friston KJ, Kiebel SJ (2009): Dynamical causal modelling for M/EEG: spatial and temporal symmetry constraints. Neuroimage 44:154–163. [DOI] [PubMed] [Google Scholar]
- Feldman H, Friston KJ (2010): Attention, uncertainty, and free‐energy. Front Hum Neurosci 4:215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelson N, Litvak V, Peled A, Fernandez‐Del‐Olmo M, Friston K (2014): The functional anatomy of schizophrenia: A dynamic causal modeling study of predictive coding. Schizophr Res 158:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K, Sato H, Daw N (1990): The effect of varying stimulus intensity on NMDA‐receptor activity in cat visual cortex. J Neurophysiol 64:1413–1428. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Harrison L, Penny W (2003): Dynamic causal modelling. Neuroimage 19:1273–1302. [DOI] [PubMed] [Google Scholar]
- Friston K, Kiebel S, Garrido M, David O (2007): Dynamic causal models for EEG. In: Friston KJ, Ashburner JT, Kiebel SJ, Nichols TE, Penny WD, editors. Statistical Parametric Mapping: The Analysis of Functional Brain Images. London: Elsevier Ltd; pp 561–576. [Google Scholar]
- Friston K (2008): Hierarchical Models in the Brain. PLoS Comput Biol 4:e1000211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friston KJ (1998): The disconnection hypothesis. Schizophr Res 30:115–125. [DOI] [PubMed] [Google Scholar]
- Friston K, Penny W (2011): Post hoc Bayesian model selection. Neuroimage 56:2089–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs M, Wagner M, Kastner J (2001): Boundary element method volume conductor models for {EEG} source reconstruction. Clin Neurophysiol 112:1400–1407. [DOI] [PubMed] [Google Scholar]
- Fulham WR, Michie PT, Ward PB, Rasser PE, Todd J, Johnston PJ, Thompson PM, Schall U (2014): Mismatch negativity in recent‐onset and chronic schizophrenia: A current source density analysis. PLoS One 9:e100221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido MI, Kilner JM, Kiebel SJ, Friston KJ (2007): Evoked brain responses are generated by feedback loops. Proc Natl Acad Sci USA 104:20961–20966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido MI, Kilner JM, Kiebel SJ, Friston KJ (2009a): Dynamic causal modeling of the response to frequency deviants. J Neurophysiol 101:2620–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido MI, Friston KJ, Kiebel SJ, Stephan KE, Baldeweg T, Kilner JM (2008): The functional anatomy of the MMN: A DCM study of the roving paradigm. Neuroimage 42:936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido MI, Kilner JM, Stephan KE, Friston KJ (2009b): The mismatch negativity: A review of underlying mechanisms. Clin Neurophysiol 120:453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour G, Dix S, Fellini L, Gastambide F, Plath N, Steckler T, Talpos J, Tricklebank M (2012): NMDA receptors, cognition and schizophrenia–testing the validity of the NMDA receptor hypofunction hypothesis. Neuropharmacology 62:1401–1412. [DOI] [PubMed] [Google Scholar]
- Goff DC, Coyle JT (2001): The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158:1367–1377. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Burgos G, Lewis Da (2012): NMDA receptor hypofunction, parvalbumin‐positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull 38:950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Gould TD (2003): The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry 160:636–645. [DOI] [PubMed] [Google Scholar]
- Haenschel C, Baldeweg T, Croft RJ, Whittington M, Gruzelier J (2000): Gamma and beta frequency oscillations in response to novel auditory stimuli: A comparison of human electroencephalogram (EEG) data with in vitro models. Proc Natl Acad Sci USA 97:7645–7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall MH, Schulze K, Rijsdijk F, Picchioni M, Ettinger U, Bramon E, Freedman R, Murray RM, Sham P (2006): Heritability and reliability of P300, P50 and duration mismatch negativity. Behav Genet 36:845–857. [DOI] [PubMed] [Google Scholar]
- Hall MH Smoller JW (2010): A new role for endophenotypes in the GWAS era: Functional characterization of risk variants. Harvard Rev Psychiatry 18:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M‐H, Schulze K, Rijsdijk F, Kalidindi S, McDonald C, Bramon E, Murray RM, Sham P (2009): Are auditory P300 and duration MMN heritable and putative endophenotypes of psychotic bipolar disorder? A Maudsley Bipolar Twin and Family Study. Psychol Med 39:1277–1287. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Lewis DA, Kleinman JE (2011): Neuropathology of Schizophrenia. In: Weinberger DR, Harrison PJ, editors. Schizophrenia Third Edit. Oxford: Wiley‐Blackwell. pp 372–392. [Google Scholar]
- Hong LE, Moran LV, Du X, O'Donnell P, Summerfelt A (2012): Mismatch negativity and low frequency oscillations in schizophrenia families. Clin Neurophysiol 123:1980–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasper H (1958): Report to the committee on methods of clinical examination in electroencephalography. Electroencephalogr Clin Neurophysiol 10:371–375. [Google Scholar]
- Javitt DC, Steinschneider M, Schroeder CE, Arezzo JC (1996): Role of cortical N‐methyl‐D‐aspartate receptors in auditory sensory memory and mismatch negativity generation: implications for schizophrenia. Proc Natl Acad Sci USA 93:11962–11967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR (1991): Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148:1301–1308. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Grochowski S, Shelley A‐M, Ritter W (1998): Impaired mismatch negativity (MMN) generation in schizophrenia as a function of stimulus deviance, probability, and interstimulus/interdeviant interval. Electroencephalogr Clin Neurophysiol 108:143–153. [DOI] [PubMed] [Google Scholar]
- Jemel B, Achenbach C, Muller BW, Ropcke B, Oades RD (2002): Mismatch negativity results from bilateral asymmetric dipole sources in the frontal and temporal lobes. Brain Topogr 15:13–27. [DOI] [PubMed] [Google Scholar]
- Jessen F, Fries T, Kucharski C, Nishimura T, Hoenig K, Maier W, Falkai P, Heun R (2001): Amplitude reduction of the mismatch negativity in first‐degree relatives of patients with schizophrenia. Neurosci Lett 309:185–188. [DOI] [PubMed] [Google Scholar]
- Joutsiniemi SL, Gross A, Appelberg B (2001): Marked clozapine‐induced slowing of EEG background over frontal, central, and parietal scalp areas in schizophrenic patients. J Clin Neurophysiol 18:9–13. [DOI] [PubMed] [Google Scholar]
- Juckel G, Roser P, Nadulski T, Stadelmann AM, Gallinat J (2007): Acute effects of Delta9‐tetrahydrocannabinol and standardized cannabis extract on the auditory evoked mismatch negativity. Schizophr Res 97:109–117. [DOI] [PubMed] [Google Scholar]
- Kantrowitz JT, Javitt DC (2010): N‐methyl‐d‐aspartate (NMDA) receptor dysfunction or dysregulation: The final common pathway on the road to schizophrenia? Brain Res Bull 83:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay SR, Fiszbein A, Opler LA (1987): The Positive and Negative Syndrome Scale (PANSS) for Schizophrenia. Schizophr Bull 13:261–276. [DOI] [PubMed] [Google Scholar]
- Kiang M, Braff DL, Sprock J, Light GA (2009): The relationship between preattentive sensory processing deficits and age in schizophrenia patients. Clin Neurophysiol 120:1949–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebel SJ, David O, Friston KJ (2006): Dynamic causal modelling of evoked responses in EEG/MEG with lead field parameterization. Neuroimage 30:1273–1284. [DOI] [PubMed] [Google Scholar]
- Kiebel SJ, Garrido MI, Moran R, Chen C‐CC‐C, Friston KJ (2009): Dynamic causal modeling for EEG and MEG. Hum Brain Mapp 30:1866–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebel SJ, Garrido MI, Friston KJ (2007): Dynamic causal modelling of evoked responses: The role of intrinsic connections. Neuroimage 36:332–345. [DOI] [PubMed] [Google Scholar]
- Kim M, Kim SN, Lee S, Byun MS, Shin KS, Park HY, Jang JH, Kwon JS (2014): Impaired mismatch negativity is associated with current functional status rather than genetic vulnerability to schizophrenia. Psychiatry Res 222:100–106. [DOI] [PubMed] [Google Scholar]
- Knott V, Labelle A, Jones B, Mahoney C (2001): Quantitative EEG in schizophrenia and in response to acute and chronic clozapine treatment. Schizophr Res 50:41–53. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Charney DS (1994): Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51:199–214. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Holcomb HH, Medoff DR, Tamminga CA (1995): Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 6:869–872. [DOI] [PubMed] [Google Scholar]
- Lakhan SE, Caro M, Hadzimichalis N (2013): NMDA receptor activity in neuropsychiatric disorders. Front Psychiatry 4:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AH, Eerola O, Korpilahti P, Holopainen I, Salo S, Aaltonen O (1995): Practical issues in the clinical application of mismatch negativity. Ear Hear 16:118–130. [DOI] [PubMed] [Google Scholar]
- Levanen S, Ahonen A, Hari R, McEvoy L Sams M (1996): Deviant auditory stimuli activate human left and right auditory cortex differently. Cereb Cortex 6:288–296. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Cho RY, Carter CS, Eklund K, Forster S, Kelly MA, Montrose D (2008): Subunit‐selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am J Psychiatry 165:1585–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW (2012): Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci 35:57–67. Special Issue: Neuropsychiatric Disorders: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieder F, Stephan KE, Daunizeau J, Garrido MI, Friston KJ (2013): A Neurocomputational Model of the Mismatch Negativity. PLoS Comput Biol 9:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA (2008): Circuit‐based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 31:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litvak V, Mattout J, Kiebel S, Phillips C, Henson R, Kilner J, Barnes G, Oostenveld R, Daunizeau J, Flandin G, Penny W, Friston K ( 2011): EEG and MEG data analysis in SPM8. Comput Intell Neurosci 2011:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra AK, Pinals DA, Weingartner H, Sirocco K, Missar CD, Pickar D, Breier A (1996): NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology 14:301–307. [DOI] [PubMed] [Google Scholar]
- Matsubayashi J, Kawakubo Y, Suga M, Takei Y, Kumano S, Fukuda M, Itoh K, Yumoto M, Kasai K (2008): The influence of gender and personality traits on individual difference in auditory mismatch: A magnetoencephalographic (MMNm) study. Brain Res 1236:159–165. [DOI] [PubMed] [Google Scholar]
- Maxwell M (1992): Family Interview for Genetic Studies. Bethesda, USA: Clinical Neurogenetics Branch, Intramural Research Program, National Institute of Mental Health.
- Mechelli A, Allen P, Amaro E, Fu CHY, Williams SCR, Brammer MJ, Johns LC, McGuire PK (2007): Misattribution of speech and impaired connectivity in patients with auditory verbal hallucinations. Hum Brain Mapp 28:1213–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michie PT, Innes‐Brown H, Todd J, Jablensky AV (2002): Duration mismatch negativity in biological relatives of patients with schizophrenia spectrum disorders. Biol Psychiatry 52:749–758. [DOI] [PubMed] [Google Scholar]
- Molholm S, Martinez A, Ritter W, Javitt DC, Foxe JJ (2005): The neural circuitry of pre‐attentive auditory change‐detection: An fMRI study of pitch and duration mismatch negativity generators. Cereb Cortex 15:545–551. [DOI] [PubMed] [Google Scholar]
- Moran RJ, Symmonds M, Dolan RJ, Friston KJ (2014): The brain ages optimally to model its environment: evidence from sensory learning over the adult lifespan. PLoS Comput Biol 10:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller BW, Jüptner M, Jentzen W, Müller SP (2002): Cortical activation to auditory mismatch elicited by frequency deviant and complex novel sounds: A PET study. Neuroimage 17:231–239. [DOI] [PubMed] [Google Scholar]
- Murray GK, Corlett PR, Clark L, Pessiglione M, Blackwell AD, Honey G, Jones PB, Bullmore ET, Robbins TW, Fletcher PC (2008): Substantia nigra/ventral tegmental reward prediction error disruption in psychosis. Mol Psychiatry 13:239, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JD, Anticevic A, Gancsos M, Ichinose M, Corlett PR, Krystal JH, Wang X‐J (2014): Linking microcircuit dysfunction to cognitive impairment: effects of disinhibition associated with schizophrenia in a cortical working memory model. Cereb Cortex 24:859–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näätänen R (1992): The Mismatch Negativity (MMN) In: Näätänen R, editor. Attention and brain function. Hillsdale: Lawrence Erlbaum Associates. [Google Scholar]
- Näätänen R, Kujala T, Escera C, Baldeweg T, Kreegipuu K, Carlson S, Ponton C (2012): The mismatch negativity (MMN) – A unique window to disturbed central auditory processing in ageing and different clinical conditions. Clin Neurophysiol 123:424–458. [DOI] [PubMed] [Google Scholar]
- Nagai T, Tada M, Kirihara K, Araki T, Jinde S, Kasai K (2013): Mismatch negativity as a “translatable” brain marker toward early intervention for psychosis: A review. Front Psychiatry 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICE (2014): Psychosis and schizophrenia in adults: Treatment and management. NICE clinical guideline 178. London: National Institute for Health and Care Excellence. [PubMed] [Google Scholar]
- Olney JW, Newcomer JW Farber NB (1999): NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res 33:523–533. [DOI] [PubMed] [Google Scholar]
- Oostenveld R, Fries P, Maris E, Schoffelen J‐M (2011): FieldTrip: Open source software for advanced analysis of MEG, EEG, and invasive electrophysiological data. Intell Neurosci 2011:1:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penny WD, Stephan KE, Mechelli A, Friston KJ (2004): Comparing dynamic causal models. Neuroimage 22:1157–1172. [DOI] [PubMed] [Google Scholar]
- Phillips WA, Silverstein S (2013): The coherent organization of mental life depends on mechanisms for context‐sensitive gain‐control that are impaired in schizophrenia. Front Psychol 4:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, Ell PJ (2006): First in vivo evidence of an NMDA receptor deficit in medication‐free schizophrenic patients. Mol Psychiatry 11:118–119. [DOI] [PubMed] [Google Scholar]
- Pinotsis DA, Brunet N, Bastos A, Bosman CA, Litvak V, Fries P, Friston KJ (2014): Contrast gain control and horizontal interactions in V1: A DCM study. Neuroimage 92:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinotsis DA, Schwarzkopf DS, Litvak V, Rees G, Barnes G, Friston KJ (2013): Dynamic causal modelling of lateral interactions in the visual cortex. Neuroimage 66:563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinotsis DA, Moran RJ, Friston KJ (2012): Dynamic causal modeling with neural fields. Neuroimage 59:1261–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PKE, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SGN, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll S, a Sklar P, O'Dushlaine C, Chambert K, Bergen SE, Kähler A, Duncan L, Stahl E, Genovese G, Fernández E, Collins MO, Komiyama NH, Choudhary JS, Magnusson PKE, Banks E, Shakir K, Garimella K, Fennell T, DePristo M, Grant SGN, Haggarty SJ, Gabriel S, Scolnick EM, Lander ES, Hultman CM, Sullivan PF, McCarroll S a Sklar P (2014): A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranlund S, Nottage J, Shaikh M, Dutt A, Constante M, Walshe M, Hall M‐H, Friston K, Murray R, Bramon E (2014): Resting EEG in psychosis and at‐risk populations — A possible endophenotype? Schizophr Res 153:96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RP, Ballard DH (1999): Predictive coding in the visual cortex: a functional interpretation of some extra‐classical receptive‐field effects. Nat Neurosci 2:79–87. [DOI] [PubMed] [Google Scholar]
- Rinne T, Alho K, Ilmoniemi RJ, Virtanen J, Näätänen R (2000): Separate time behaviors of the temporal and frontal mismatch negativity sources. Neuroimage 12:14–19. [DOI] [PubMed] [Google Scholar]
- Rinne T, Degerman A, Alho K (2005): Superior temporal and inferior frontal cortices are activated by infrequent sound duration decrements: An fMRI study. Neuroimage 26:66–72. [DOI] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A Walter JTR, Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014): Biological insights from 108 schizophrenia‐associated genetic loci. Nature 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roiser JP, Wigton RL, Kilner J, Mendez MA, Hon N, Friston K, Joyce E (2013): Dysconnectivity in the frontoparietal attention network in schizophrenia. Front Psychiatry 4:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Bachmann R, Kometer M, Csomor PA, Stephan KE, Seifritz E, Vollenweider FX (2012a): Mismatch negativity encoding of prediction errors predicts S‐ketamine‐induced cognitive impairments. Neuropsychopharmacol 37:865–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Diaconescu AO, Kometer M, Friston KJ, Stephan KE, Vollenweider FX (2012b): Modeling ketamine effects on synaptic plasticity during the mismatch negativity. Cereb Cortex 23:2394–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Smieskova R, Aston J, Simon A, Allen P, Fusar‐Poli P, McGuire PK, Riecher‐Rössler A, Stephan KE, Borgwardt S (2013): Brain connectivity abnormalities predating the onset of psychosis: Correlation with the effect of medication. JAMA Psychiatry 70:903–912. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Smieskova R, Simon A, Allen P, Fusar‐Poli P, McGuire PK, Bendfeldt K, Aston J, Lang UE, Walter M, Radue E‐W, Riecher‐Rössler A, Borgwardt SJ (2014): Abnormal effective connectivity and psychopathological symptoms in the psychosis high‐risk state. J Psychiatry Neurosci 39:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönwiesner M, Novitski N, Pakarinen S, Carlson S, Tervaniemi M, Näätänen R (2007): Heschl's gyrus, posterior superior temporal gyrus, and mid‐ventrolateral prefrontal cortex have different roles in the detection of acoustic changes. J Neurophysiol 97:2075–2082. [DOI] [PubMed] [Google Scholar]
- Schulze KK, Hall MHM‐H, Mcdonald C, Marshall N, Walshe M, Murray RM, Bramon E (2008): Auditory P300 in patients with bipolar disorder and their unaffected relatives. Bipolar Disord 10:377–386. [DOI] [PubMed] [Google Scholar]
- Shaikh M, Hall MHM‐H, Schulze K, Dutt A, Li K, Williams I, Walshe M, Constante M, Broome M, Picchioni M, Toulopoulou T, Collier D, Stahl D, Rijsdijk F, Powell J, Murray RM, Arranz M, Bramon E (2013): Effect of DISC1 on the P300 waveform in psychosis. Schizophr Bull 39:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh M, Valmaggia L, Broome MR, Dutt A, Lappin J, Day F, Woolley J, Tabraham P, Walshe M, Johns L, Fusar‐Poli P, Howes O, Murray RM, McGuire P, Bramon E (2012): Reduced mismatch negativity predates the onset of psychosis. Schizophr Res 134:42–48. [DOI] [PubMed] [Google Scholar]
- Shelley AM, Ward PB, Catts SV, Michie PT, Andrews S, McConaghy N (1991): Mismatch negativity: An index of a preattentive processing deficit in schizophrenia. Biol Psychiatry 30:1059–1062. [DOI] [PubMed] [Google Scholar]
- Spencer KM, Nestor PG, Perlmutter R, Niznikiewicz MA, Klump MC, Frumin M, Shenton ME, McCarley RW (2004): Neural synchrony indexes disordered perception and cognition in schizophrenia. Proc Natl Acad Sci USA 101:17288–17293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan KE, Baldeweg T, Friston KJ (2006): Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry 59:929–939. [DOI] [PubMed] [Google Scholar]
- Sussman ES, Chen S, Sussman‐Fort J, Dinces E (2013): The five myths of MMN: Redefining how to use MMN in basic and clinical research. Brain Topogr 27:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiitinen H, Salminen NH, Palomäki KJ, Mäkinen VT, Alku P, May PJC (2006): Neuromagnetic recordings reveal the temporal dynamics of auditory spatial processing in the human cortex. Neurosci Lett 396:17–22. [DOI] [PubMed] [Google Scholar]
- Todd J, Harms L, Michie P, Schall U (2013): Mismatch negativity: Translating the potential. Front Psychiatry 4:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC (2000): Ketamine‐induced deficits in auditory and visual context‐dependent processing in healthy volunteers: Implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry 57:1139–1147. [DOI] [PubMed] [Google Scholar]
- Umbricht D, Krljesb S, Krljes S (2005): Mismatch negativity in schizophrenia: A meta‐analysis. Schizophr Res 76:1–23. [DOI] [PubMed] [Google Scholar]
- Wager TD, Keller MC, Lacey SC, Jonides J (2005): Increased sensitivity in neuroimaging analyses using robust regression. Neuroimage 26:99–113. [DOI] [PubMed] [Google Scholar]
- WHO (2008): The Global Burden of Disease: 2004 Update. Geneva: World Health Organization. [Google Scholar]
- Winterer G, Coppola R, Egan MF, Goldberg TE, Weinberger DR (2003): Functional and effective frontotemporal connectivity and genetic risk for schizophrenia. Biol Psychiatry 54:1181–1192. [DOI] [PubMed] [Google Scholar]
- Woldorff MG, Hackley SA, Hillyard SA (1991): The effects of channel‐selective attention on the mismatch negativity wave elicited by deviant tones. Psychophysiology 28:30–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information