

Abstract
The dynamic self-organization of lipids in biological systems is a highly regulated process that enables the compartmentalization of living systems at micro- and nanoscopic scales. Consequently, quantitative methods for assaying the kinetics of supramolecular remodeling such as vesicle formation from planar lipid bilayers or multilayers are needed to understand cellular self-organization. Here, a new nanotechnology-based method for quantitative measurements of lipid–protein interactions is presented and its suitability for quantifying the membrane binding, inflation, and budding activity of the membrane-remodeling protein Sar1 is demonstrated. Lipid multilayer gratings are printed onto surfaces using nanointaglio and exposed to Sar1, resulting in the inflation of lipid multilayers into unilamellar structures, which can be observed in a label-free manner by monitoring the diffracted light. Local variations in lipid multilayer volume on the surface is used to vary substrate availability in a microarray format. A quantitative model is developed that allows quantification of binding affinity (KD) and kinetics (kon and koff). Importantly, this assay is uniquely capable of quantifying membrane remodeling. Upon Sar1-induced inflation of single bilayers from surface supported multilayers, the semicylindrical grating lines are observed to remodel into semispherical buds when a critical radius of curvature is reached.
1. Introduction
Lipid multilayer gratings are a recently invented type of label-free nanobiosensor element that transduces molecular binding events into optical signals.[1] Unlike other grating-based sensor elements,[2–4] lipid multilayer gratings can be formed from fluid lipids. This allows for nanometer scale changes in grating height and shape that lead to changes in the intensity of light diffraction. In our experiments, the lipid gratings are illuminated from an angle with white light, and the diffracted light is monitored through an optical microscope with a color camera (Figure 1). According to Bragg’s law, the wavelengths of light diffracted at different angles are primarily determined by the spacing of the lines, whereas the intensity of diffracted light is strongly dependent on the height of the grating lines.[5] Previously, lipid multilayer gratings have been used to detect vapors, streptavidin-biotin interactions, and His-tag protein-nickel binding. These binding events primarily produce decreases in diffraction intensity due to disruption of the grating structure.[1] The exception is in the binding of His-tagged proteins to nickel-chelating lipids, which slowly increased in diffraction intensity over a timescale of about an hour. We previously explained the mechanisms for changes in the diffraction signal qualitatively in terms of spreading, dewetting, and changes in lipid multilayer volume.[1] To take full advantage of lipid gratings as label-free biosensor elements, a quantitative model is needed to fit the diffraction data. We develop such a model in this paper to characterize the kinetics and thermodynamics of proteins that have innate membrane binding and remodeling activity.
Figure 1.
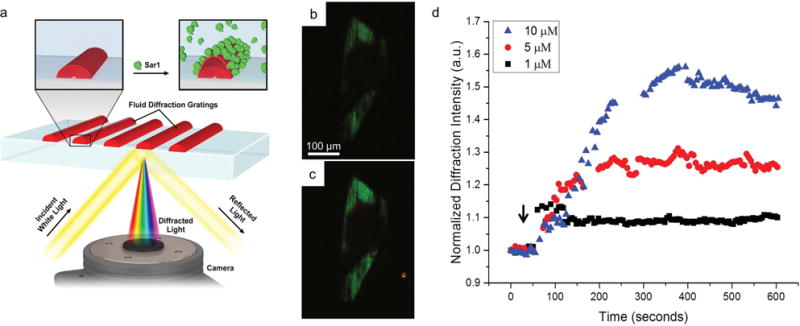
Lipid multilayer grating-based assay. a) Schematic illustration showing the optical setup and hypothesized interaction between Sar1 and the lipid multilayer gratings. b) An image of light diffracted from a lipid multilayer grating in buffer before the addition of Sar1, illuminated with white light at an angle where primarily green light is diffracted into the microscope objective. c) The same grating after 25 min of incubation with 5 × 10−6 M Sar1. The lipid gratings generally show a strong increase in diffraction intensity. The image contrast is adjusted equally for (b) and (c). d) Graph showing the dependence of the sensor response on the concentration of Sar1 applied.
Several methods have been established for measuring the membrane binding and/or remodeling activity of proteins by exposure to model membrane systems such as surface supported lipid bilayers, multilayers, or preformed vesicles in solution.[6–19] These systems have been characterized by gel electrophoresis,[6] surface plasmon resonance (SPR) spectroscopy,[7] quartz crystal microbalance (QCM),[8] fluorescence microscopy,[9–12] and transmission electron microscopy.[13–16] For example, the equilibrium dissociation constant (KD) of the membrane binding protein used in this work, Sar1, has been measured to be 10.5 ± 3.1 × 10−6 M using fluorescence microscopy to quantify the Sar1 mediated accumulation of fluorescent nucleotides to surface-supported lipid multilayer films.[9] Arrays of liposomes on agarose surfaces have recently been used to screen for interactions of fluorescently tagged proteins with hundreds of lipid formulations simultaneously.[17] Surface-based single vesicle arrays have demonstrated novel insights into membrane curvature dependent lipid–protein interactions using surface characterization techniques, principally by fluorescent microscopy.[20] Surface plasmon resonance (SPR) spectroscopy[7] and monitoring a quartz crystal microbalance (QCM)[8] are established label-free techniques for quantifying membrane binding kinetics to a surface supported lipid bilayer.[18,19]
We selected the protein Sar1 as a model system since it is a well-studied membrane binding protein with an in vivo function related to membrane remodeling. In vivo, Sar1 is one part of the coat protein complex II (COPII) proteins that facilitate transport of cargo from the endoplasmic reticulum (ER) to the Golgi apparatus by coating the transport vesicles.[21–23] Sar1 is involved in the initial conversion of semi-planar endoplasmic reticulum bilayers into small spherical vesicles, and from in vitro measurements on model membrane systems, it is known that Sar1 alone can induce membrane curvature and deforms vesicles into tubes of different shapes and sizes.[16,24] However, the exact mechanism is not fully understood. One proposed mechanism is that Sar1 exposes an amphipathic helix which inserts into the membrane and induces membrane curvature by selectively expanding the area of the outer leaflet of the ER.[25,26] Another mechanism based on in vitro membrane bending modulus measurements at different Sar1 concentrations indicates that Sar1 lowers membrane rigidity, lowering the energy cost of curvature generation below the thermal energy.[27] A third mechanism supported by cryo-electron microscopy data suggests that the formation of an ordered Sar1 lattice on the lipid membrane induces a crowding effect that leads to efficient membrane curvature.[24] It is likely that all three of these mechanisms are involved in Sar1-mediated membrane remodeling, and methods for quantifying remodeling kinetics are needed to unravel their roles in the nanoscale compartmentalization and dynamic self-organization of biological systems.
Laterally, structured lipid multilayers on surfaces are a new model membrane system that are compatible with surface based characterization techniques, yet also allow compartmentalization and remodeling, as well as entirely new properties due to the nano- and microstructure. For example, the nanometer-scale multilayer thicknesses and submicron-scale lateral dimensions of lipid multilayer gratings are crucial to their function, and innovative nanofabrication methods have been necessary to make them. So far, they have been fabricated by dip-pen nanolithography (DPN) and nanointaglio.[1,28,29] DPN involves dipping the tip of an atomic force microscope into an ink, allowing arbitrary patterns to be drawn with lateral resolution well below 100 nm.[30] Although most inks used for DPN have involved the formation of self-assembled monolayers on surfaces, when phospholipids are used as an ink, the lipid multilayer nanostructures can be formed with controllable thicknesses of 2–100 nm.[31–33] Nanointaglio is a printing-based method which we recently found to be suitable for lipid multilayer nanofabrication.[28,29] Intaglio is a mode of printing where the ink is transferred from the recesses of a patterned stamp, rather than from the protrusions. When this is carried out with a microstructured stamp using inks with appropriate viscosity such as phospholipids, multilayer patterns with nanometer-scale heights and submicron widths can be produced. An advantage of DPN is that arbitrary patterns can be directly written for rapid prototyping, whereas nanointaglio has the advantage of scalability. Furthermore, both DPN and nanointaglio are suitable for integrating multiple materials on the same surface, opening the possibility of massively parallel biosensor arrays.[29,34] Quantitative interpretation of the sensor data is needed in order to reach that goal. Here we demonstrate the suitability of lipid multilayer gratings for the rapid, simple, and label-free in vitro method of quantifying Sar1 binding and budding kinetics.
2. Results and Discussion
2.1. Sar1 Binding to Lipid Multilayer Gratings Can Be Detected by Diffracted Light
Upon exposure to Sar1, the diffraction intensity of lipid multilayer gratings was found to strongly and rapidly increase, reaching saturation within 1–5 min depending on the Sar1 concentration (Figure 1b,c). Previously, it was found that binding of His-tagged GFP to His-tag binding lipids resulted in increases in diffraction intensity from the lipid gratings; however, this interaction took over an hour. The rapid increase in diffraction intensity after only 5 min when Sar1 was added to the solution is new, and we interpret it as evidence of Sar1’s well documented membrane binding and remodeling activities.[24–27] The increase in diffraction intensity depends on the Sar1 concentration as seen in Figure 1d for grating areas with a selected initial starting intensity range between 400 and 449 arbitrary units (a.u.) of pixel values provided by the green RGB values from the color camera.
Lipid multilayer arrays have the unique capability for different areas of the surface to contain different lipid volumes, or multilayer heights. Analysis of the grating response on a pixel-by-pixel basis revealed that the amount of increase in lipid multilayer diffraction intensity upon addition of Sar1 depends on the initial local diffraction intensity (Figure 2). Previous experiments have shown the diffraction intensity of lipid multilayer gratings are directly proportional to the multilayer height of the grating, and so we explain the different initial intensities as local variations in grating height.[1] The pixels were grouped according to their initial intensities before addition of Sar1. A pixel map was generated to show the initial diffraction intensity (Figure 2a) and the intensity after 5 min of Sar1 incubation (Figure 2b). The lipid grating response was plotted as a function of time in Figure 2c. All pixels with initial intensities from 400 to 510 a.u. were identified in separate 10 a.u. bins and the time evolution of each group’s intensities is shown. Most striking is that at intensities of 450 a.u. and higher, an increase in diffraction intensity was followed by a decrease, suggesting dewetting or disruption of the lipid grating lines that can cause a decrease in diffraction intensity. A similar effect was observed previously for the binding of streptavidin to biotinylated gratings.[1] An increase in diffraction intensity observed upon Sar1 binding suggests an increase in grating volume or height, as previously observed for the binding of His-tagged proteins to gratings containing nickel-chelating lipids.[1] Sar1 binds to membranes by inserting its amphipathic α-helix into the outer leaflet of a membrane bilayer, so the expansion of the bilayer and resulting increase in grating height is an expected outcome for this protein.
Figure 2.
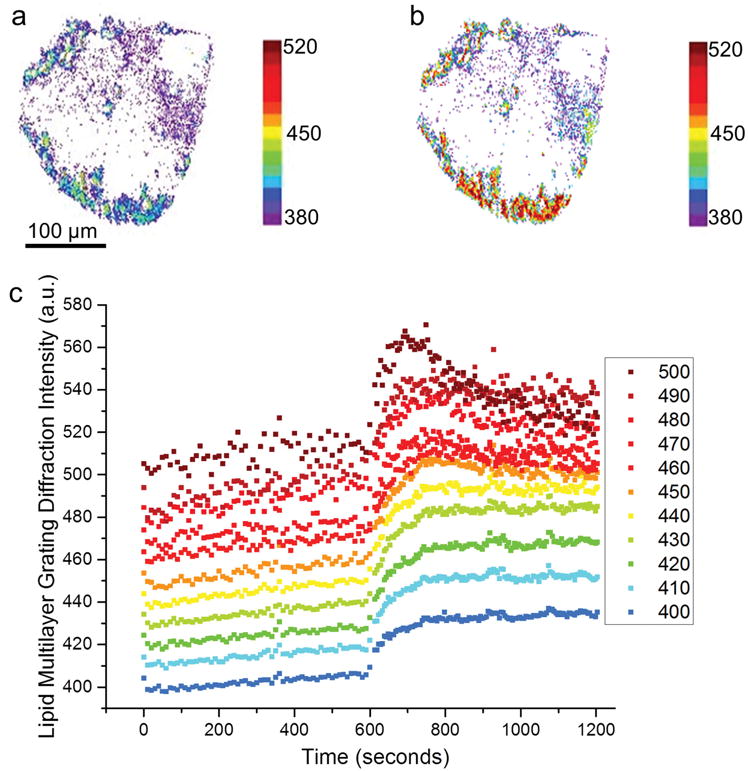
Lipid multilayer grating response depends on the initial diffraction intensity of the gratings, which has been previously shown to be directly proportional to grating height.[1] a) A map of the diffraction intensity of a lipid multilayer grating in solution before addition of Sar1. b) A map of the diffraction intensity of the same lipid multilayer grating after incubation with 5 × 10−6 M Sar1 for 5 min. c) Plots of the diffraction intensity response after application of 5 μM Sar1. The graph shows ten ranges of starting pixel intensities, indicating relative initial height of the gratings before addition of 5 × 10−6 M Sar1 at time 600 s. All analyzed pixels increased in intensity immediately upon exposure to Sar1, but only the pixels with brighter starting values eventually showed a decrease in intensity.
2.2. Sar1 Restructures Surface Supported Lipid Multilayer Patterns
To confirm the mechanisms by which Sar1 increases lipid multilayer grating diffraction intensity, we used fluorescently labeled lipid multilayer structures and monitored their optical response upon exposure to Sar1 (Figure 3). In the case of Sar1 incubation with lipid multilayer dots (of composition DOPC:DOPS:cholesterol 55:35:10 by mol%) printed onto a polylysine coated glass slide (Figure 3a), the surface-supported multilayers protruded from the dot structures as tubes (Movie S1, Supporting Information) and ultimately formed giant unilamellar vesicles (GUVs) (Figure 3b). After about 30 s, the lipid multilayer dot structures began to show deformation in their structure and by 7.5 min, almost the entire pattern had turned into GUVs with average diameters of 7.8 μm ± 1.9 μm, comparable to the 5 μm diameter and submicrometer thickness of the starting lipid multilayer spots (Figure 3b). It is likely that each spot produces more than one GUV as each multilayer dot contains a volume to produce a much larger GUV. A simple washing of the surface dispenses the GUVs heterogeneously through solution, demonstrating nearly all of the lipid multilayer patterns were converted to vesicles by Sar1. These giant vesicles appear to be unilamellar due to their uniform fluorescence intensity. These observations are consistent with a previous report that showed that yeast Sar1 could form GUVs from a continuous lipid multilayer film.[27]
Figure 3.

Fluorescence microscopy of fluorescently labeled lipid multilayer patterns upon exposure to Sar1, indicating inflation and formation of vesicles. a) Initial immersion of lipid multilayer dots of diameter 5 μm. b) The pattern in (a) after ≈7.5 min of incubation with Sar1, indicating the conversion of the lipid multilayer patterns into giant unilamellar vesicles. The vesicles were found to have an average diameter of 7.8 μm ± 1.9 μm. c) Lipid multilayer gratings of period 700 nm under buffered solution before addition of Sar1. d) The gratings in (c) after incubation of 10 × 10−6 M Sar1 for 25 min. The lines can be seen breaking up into submicron scale droplets.
When fluorescently labeled lipid multilayer grating lines were exposed to Sar1 (Figure 3c), the lines broke into small droplets or buds (Movie S2, Supporting Information) and no GUVs were observed. An image taken after about 25 min (Figure 3d) shows the breakup of the lines into droplets, with diameters on the scale of the widths of the grating lines. Closer analysis of the data indicates that the areas that tended to break into droplets began with higher fluorescence intensity (Figure S1, Supporting Information) consistent with the hypothesis that the decrease in diffraction signal is due to a dewetting mechanism.[1] The resulting differences between Sar1 interaction with 5 μm diameter dots and 300 nm wide grating lines suggest that the curvature and volume of the resulting vesicular structures is dependent on the patterning of the lipid multilayers, including the amount of deposition and available binding sites for Sar1 attachment. The rapid inflation of the lipid multilayers into GUVs in 7.5 min is consistent with the kinetics of Sar1 binding to the lipid gratings and the rapid membrane remodeling activity of Sar1, as it typically takes around an hour to inflate GUVs from surface-supported lipid multilayers by exposing them to AC electric fields.[24,35]
2.3. Modeling Sar1 Binding Using Lipid Multilayer Gratings
Overall, the diffraction and fluorescence data in Figures 2 and 3 suggest that Sar1 is increasing the grating height by binding to the lipid multilayers and inflating them. Based on this and previous observations that lipid grating height (determined by atomic force microscopy) is proportional to diffraction intensity,[1] we developed a model for the diffraction intensity as a function of Sar1 concentration and grating height (Figure 4). Our model makes the following seven assumptions:
- Sar1 binding obeys Langmuir adsorption kinetics as a function of time (t), where τ is the time at which half of the number of Sar1 are bound, which is shown in Equation (1) as
and upon reaching equilibrium the number of Sar1 bound is also a function of the Sar1 concentration [Sar1](Equation (2))(1) (2) - The number of available lipid binding sites is proportional to the initial cross-sectional area Across of the lipid grating lines (Equation (3))
where RC is the radius of curvature and h is the initial height of the lipid multilayer gratings.(3) - The surface area, or cross-sectional arc length, of the lipid grating exposed to solution increases proportional to the number of Sar1 bound (NSar1), where S′ is described by simple geometry (Equation (4))
where w is the width of the grating line.(4) The width of the lipid grating lines is fixed and approximated as 300 nm, as verified from AFM and fluorescence microscopy measurements on the stamp and printed lipid gratings.
The cross-section of the lipid grating is approximated as a section of a circle that extends below the surface, with radius of curvature RC and curvature = 1/Rc.
Dewetting leads to a decrease in diffraction intensity.
- Diffraction intensity depends on the height of the lipid multilayer gratings and also the initial starting height (Equation (5))[1]
(5)
Figure 4.

Illustration of the relevant geometric parameters of our model, and how they change as the lipid multilayers inflate upon exposure to Sar1. Upon incubation with Sar1, the protein inserts itself into the outer lipid binding sites and increases the curvature of the outer lipid multilayer, while increasing the cross-sectional area of the lipid grating.
A and B are proportionality constants. From these basic assumptions, a formula was derived to describe diffraction intensity of lipid multilayer gratings incubated with Sar1 as a function of time. To test this model, lipid gratings were exposed to 5 × 10−6 M Sar1 (Figure 2c) and Equation (6) was fit to the data
| (6) |
where ho is the initial height of lipid gratings, A and B are constants, t is the time (s), w is the width of grating lines, k is the time at which half of Sar1 is bound for a given concentration, and KD is the Equilibrium dissociation constant.
To distinguish between background and signal from diffraction, we selected all pixels that were at least 30% brighter than the background in the green channel and grouped them by their initial intensities in 10 a.u. intervals (Figure 2c). First, B was calculated by assuming that the initial height of the lipid multilayer grating is 30 nm at the intensity 400 a.u. Then, all of the initial heights were calculated, assuming that value for B. KD and width were kept as constants. A and k are fitting parameters where A = 0.026 ± 0.005 and k varies from 30 to 50 s, both of which are reasonable values for the activity curves. Diffraction intensity as a function of height, i.e., the parameters “A” and “B” are based upon previous atomic force microscopy calibration of lipid multilayer grating intensity as a function of height.[1] A is a constant that relates binding of Sar1 to lipid multilayer gratings and describes how the height of the grating line relates to the diffraction intensity of lipid-Sar1 complex and accounts for any changes in refractive index due to Sar1 binding. Similarly, B relates the intensity of the lipid grating diffraction to the height without the presence of any protein.
2.4. Measurement of Binding Affinity and Kinetics
The average intensities versus times for each group were then fitted with the model (Equation (6)), Figure 5). Intensities with a horizontal asymptote (saturation) within 1200 s and featuring no decrease in average diffraction intensity were used to calculate an average KD (Figure 5a). Groups with initial intensities above 450 a.u., which had local diffraction intensity maxima, were also fitted with the model with a cutoff time at each local maximum; shown in the inset of Figure 6. However, these data were not used in determining KD because the equilibrium condition is not well defined. KD obtained from the fits was found to be 12.8 × 10−6 M ± 2.4 × 10−6 M. The time constant τ in the model is the inverse of the parameter kobs which is defined by the kinetics of the Sar1-lipid interactions to be (Equation (7))
| (7) |
where kon and koff are the association and dissociation rate constants, respectively.[36] The average of each τ was obtained from the modeled fits to the data in Figure 2c. For fitting to the model, the time scale was defined with t = 0 being the time at which Sar1 was added to solution (600 s in Figure 2c). The model may be improved by considering refractive index of both Sar1 and the lipids. It has been shown that adsorbed protein layers on TiO2 surfaces have an associated refractive index of ≈1.5 for proteins of size ≈25 kDa,[37] like that of Sar1. While lipid layers have been shown by ellipsometry to have a similar refractive index of 1.5, interactions between protein and lipid may cause increases in refractive index which is thought to be a result of shrinking or swelling of lipid membranes.[38] Refractive index can be addressed by relating diffraction intensity as a more complicated function of height, rather than linear.
Figure 5.
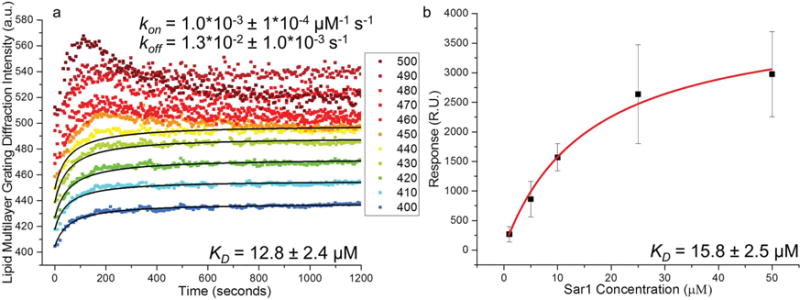
Measurement of KD and kinetics. Lipid multilayer grating calibration of Sar1 using SPR. a) Grating intensity curves as a function of time were fit by our model. The KD determined from the fit was 12.8 × 10−6 M ± 2.4 × 10−6 M, with kon = 1.0 × 10−3 ± 1 × 10−4 × 10−6 M−1 s−1 and koff = 1.3 × 10−2 ± 1 × 10−3 s−1. koff is calculated based on the experimentally determined KD and kon. b) SPR binding response assay of Sar1 to DOPC-supported lipid bilayers as a benchmark. The SPR steady state rate constant KD was found by fitting the maximum response obtained from three sets (each from a different preparation of Sar1) of SPR binding curves for different concentrations of Sar1 to a Langmuir adsorption isotherm. The KD measured by SPR was found to be 15.8 × 10−6 M ± 2.5 × 10−6 M.
Figure 6.
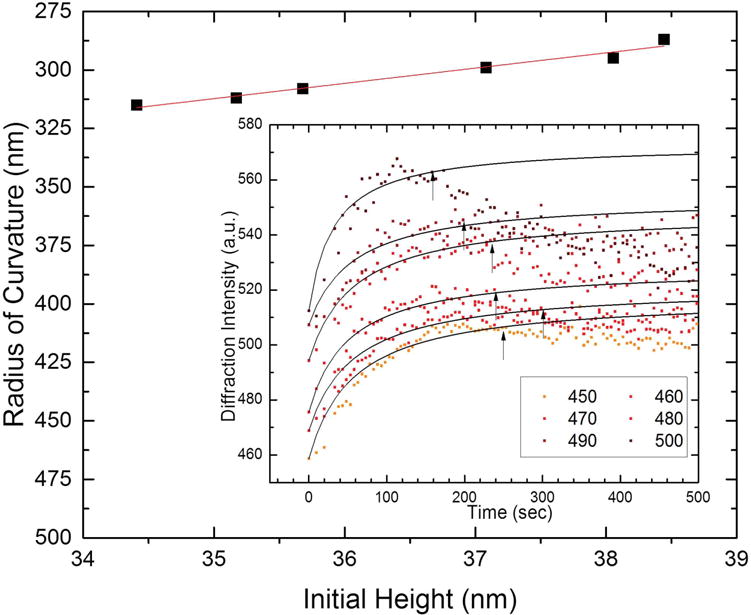
Radius of curvature at which the grating lines break into droplets, or dewet the surface for the gratings with higher initial intensity. The outer graph shows the general trend of maximum curvature as a function of initial starting heights that showed an increasing, then decreasing trend (dewetting occurs). The trend has a low slope, suggesting that curvature plays a dominant role in the dewetting. The inset shows the points that were used to calculate the maximum curvature.
As a benchmark, we used SPR to measure Sar1 binding to surface-supported lipid bilayers (Figure 5b). According to concentration dependence curves of SPR (Figure S2, Supporting Information), Langmuir adsorption isotherm fits obtained a KD of 15.8 × 10−6 M ± 2.5 × 10−6 M (Figure 5b), confirming Sar1 binding to immobilized DOPC vesicles. This value is comparable to the value of 12.8 × 10−6 M ± 2.4 × 10−6 M obtained from the lipid multilayer gratings from the same batch of Sar1. We were unable to obtain kinetic data (kon and koff) from SPR due the extreme sensitivity of SPR to changes in the refractive index of the running solution,[39] which could not be negated due to Sar1 interacting with all surfaces tested, and the lack of an established negative control for the nondiscriminate binder. Lipid multilayer gratings, however, are not as sensitive to local refractive index because the transducing mechanism is the remodeling of lipid grating lines themselves. Exchange of buffers does not significantly change the diffraction signal, as shown in a control experiment (Figure S4, Supporting Information). We were also unable to reliably carry out SPR using the negatively charged 55:35:10 DOPC:DOPS:cholesterol lipid formulation widely used for Sar1 research as a model for the endoplasmic reticulum.[22] Unlike pure DOPC nonspecific binding of the control protein BSA to the background could not be prevented, which is a requirement for reliable use of SPR to compare binding to different lipid formulations and accurate measurement of KD. We were however able to use lipid multilayer gratings to compare Sar1 binding to DOPC with binding to DOPC:DOPS:cholesterol gratings (Figure S5, Supporting Information). Lipid multilayer gratings therefore provide a promising alternative to SPR that: (a) is less sensitive to refractive index artifacts, (b) is compatible with a variety of lipid formulations, and (c) can be carried out with an easily constructed modification to a simple optical microscope.
2.5. Description of the Radius of Curvature of the Lipid Multilayer Gratings
According to assumption six of our model, we explain the decrease in diffraction intensity observed in brighter and presumably higher grating lines as the remodeling of the lipid lines into droplets. This mechanism is consistent with the fluorescence data shown in Figure 3d as well as previous observations of streptavidin binding to biotinylated lipid grating lines, where such beading up of the lipids was also observed by fluorescence microscopy.[1] Figure 6 shows the cross-sectional radius of curvature of the grating lines (defined geometrically in Figure 4) at which the grating lines remodel from a cylindrical to a spherical geometry. For the higher-intensity gratings which show the decrease in intensity, the point in time at which the grating began to decrease in intensity was identified by fitting the data to Equation (5), and systematically removing data points for longer times until the R2 value for the fit was maximized. The arrows in the inset in Figure 6b indicate the point at which R2 was maximized for each curve. Converting the data marked by arrows to curvature yielded an almost horizontal line that predicts maximum curvature of around 0.0032 (1/nm) or a radius of curvature of around 300 nm, approximately the width of the grating lines (Figure 6). The radius of curvature of the grating lines before Sar1-binding was ≈1200–1300 nm (Figure S6, Supporting Information).
The critical curvature observed may provide insights into the relationship between membrane tension and curvature.[40,41] The breakup of a liquid cylinder into droplets has been described as a Rayleigh instability where the periodicity of the droplet formation is proportional to the radius of the cylinder due to surface-tension-driven tendency toward minimization of the surface-area-to-volume ratio.[42,43] It is notable that the critical curvature observed in our experiments appears at around 300 nm, which is approximately the width of a single grating line. As the radius of curvature decreases, the Laplace contribution to the energy of the system becomes more significant. In the case of lipids, the breakup of a lipid bilayer nanocylinder into droplets has been described as a pearling instability, and can be induced by optical tweezers as well as changes in the chemical environment of the lipid nanocylinders.[44–47]
The observation of a critical radius of ≈300 nm for remodeling is similar to observation from a previous study that found that Sar1 remodels GUVs into tubules, multi-budded tubules, and at high concentrations converts the buds into vesicles.[24] That study found that Sar1 forms ordered arrays on membranes with radii greater than ≈100 nm. It was suggested that propagation of the ordered arrays could contribute to vesicle scission by forcing the nascent bud necks together. Here we propose that the Sar1 likely binds in an ordered array to the relatively flat lipid multilayers (Rc ≈ 1200–1300 nm; Figure S6 in the Supporting Information), and inflates a cylindrical bilayer until a radius-of-curvature of 300 nm or less is reached. It is then hypothesized that when Sar1 remodels the inflated bilayers into spherical buds, they can be pinched off into vesicles by the array propagation mechanism, which explains the decrease in diffraction intensity observed for the relatively thicker lipid multilayer grating lines.
Our data show that by modeling protein binding to lipid multilayer gratings, we can characterize the kinetics and thermodynamics of membrane binding and remodeling by proteins. We expect this method to be immediately applicable to a wide variety of membrane binding proteins, including other G-proteins, the large family of BAR domain proteins, lytic peptides, and cell-penetrating peptides. The technique yields a wealth of information about the characteristics of membrane binding, including determination of KD and kinetic on and off rates all in one experiment at a single protein concentration. Variations in line widths and pattern dimensions will likely provide further insights into the mechanisms of membrane remodeling. Moreover, the technique can be multiplexed by microarraying different lipids onto the stamp[29] so that experiments that test the effect of different lipid compositions on membrane binding and kinetics could be performed all at once in a high-throughput manner.
3. Conclusion
We have demonstrated that lipid multilayer gratings can be used for quantitative measurements of membrane binding protein kinetics. In the case of Sar1, a novel diffraction intensity response was observed and a quantitative model was developed to describe the inflation and dewetting of the lipid grating lines. Importantly, whereas other techniques such as SPR and QCM require the experiment to be repeated several times by varying the protein concentration, our method uses local variation in lipid multilayer height to vary the number of binding sites available, allowing quantitative data to be obtained in just a single time and cost efficient experiment. By modeling the lipid multilayer diffraction grating response, critical curvature at which an instability occurs was obtained. Furthermore, the method of nanointaglio is a scalable process that potentially allows for the rapid and quantitative screening of membrane-binding protein activity.
4. Experimental Section
Lipid Ink Preparation
Lipids used for patterning or surface plasmon resonance screening were 1,2-dioeoyl-sn-glycero-3-phosphocholine (DOPC); 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-phospho-L-serine (sodium salt) (DOPS); 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-lissamine rhodamine B sulfonyl (DOPE-RB). Formulations used were pure DOPC and 55:35:10 DOPC:DOPS:cholesterol by molar ratio. DOPS is added specifically because it is an acidic lipid. Sar1 is known to prefer negatively charged membranes[6] and this formulation was chosen as a known model for the endoplasmic reticulum.[22] Chloroform solutions of the different lipids were mixed to obtain the desired molar ratios. When making liposomal formulations, chloroform was evaporated off under a nitrogen stream, then the preparation was further dried in vacuum overnight to form a thin film of lipids on the bottom of the glass vials. Water was added to the vials containing the dried lipids to a concentration of 25 g L−1. Samples were then lightly vortexed for 10 s, then sonicated for 10 min to be used for microarraying.
Microarraying
The liposomal formulations were microarrayed from standard 384 well microtitre plates (Axygen, Inc., PMI110-07 V1., Unioncity, CA) using a BioRobotics pinspotter model BG600 (Comberton, Cambridge, England) onto a thin piece of PDMS (described as a “PDMS palette”) using a 200 μm 4 × 4 stainless steel microspot pin tool. After each lipid spot, microarray pins were washed for 2 min in acetone, 2 min in water, then dried for 30 s.
PDMS Intaglio Stamp Preparation
Polydimethylsiloxone (PDMS) (Sylgard, 184) gratings were patterned from a thermopolymeric master (EV Group, Inc) cured from a patterned silicon wafer with 700 nm pitch and 350 nm groove depth.
Lipid Multilayer Nanofabrication
Lipid multilayer gratings were fabricated by nanointaglio.[29] Dehydrated lipid was transferred onto a PDMS grating stamp (700 nm pitch, 350 nm height) from an inked PDMS palette. Excess lipid ink was removed by sacrificial printing (proofing). The PDMS grating stamp was then stamped onto PMMA-based plexiglass (HESAGlas HT, Notz Plastics) for sensing experiments. PDMS well stamps of 5 μm in diameter and approximately 1.8 μm deep were fabricated from a polystyrene pillar master. Inked PDMS well stamps were prepared the same way as grating stamps, only they were printed onto poly-lysine-coated glass slides (P35GC-1.5-14-C, MatTek Corporation) for high magnification fluorescence imaging of lipid multilayer dot structures.
Lipid Nanopattern Storage and Immersion
After nanointaglio fabrication, lipid gratings were then stored in a nitrogen glovebox (Mbraun, Inc., Model Labstar (1200/780) Stratham, NH, USA) for 24 h to remove all water from them. The nitrogen environment stabilizes the lytotropic lipid multilayer nanostructures by dehydration prior to immersion in water.[1] After immersion, the lipid multilayer gratings remain stable in buffer solution and remain physisorbed to the PMMA substrate.
Expression in Escherichia coli and Purification of Sar1
We used the constitutively active GTP-ase defective mutant H79G. H79G mutation suppresses the ability of Sar1 to hydrolyze GTP.[21,22] It has been proposed that the suppression of GTP-ase activity increases Sar1-lipid membrane binding association compared to association of wild type Sar1-lipid membrane binding.[24] Sar1 protein was expressed in BL21 E. coli cells from modified pET11d plasmid, giving the proteins an N-terminal His6 tag. BL21 cells producing the desired Sar1 protein were scraped from a frozen stock, transferred into a 25 ml LB bacterial culture medium with 100 μg mL−1 Ampicillin and cultured overnight in a shaking incubator at 37 °C. The 25 mL preculture was transferred to a 1 L LB culture with 100 μg mL−1 Ampicillin the second day and cultured for ≈4 h. The optical density was measured every 30 min using spectrophotometer. When optical density reached 1.2 the culture was induced by the addition of 1 mL of 1 molar IPTG. The induced culture was incubated at 20 °C for 5 h and the cells were collected by centrifugation at 4200 g for 15 min at 4 °C. Pellets were then resuspended in ≈30 ml lysis buffer: 300 × 10−3 M NaCl, 50 × 10−3 M HEPES, 1 × 10−3 M MgOAc, 5 × 10−3 M mercaptoethanol, and 1 protease inhibitor cocktail (PIC) tablet, pH 7.2. Cell suspension was passed through a microfluidizer for cell lysis and spun again at 25 000 rpm for 30 min. Overexpressed Sar1 was separated from the soluble bacterial proteins using FPLC from the collected supernatant. Sar1 was purified by Ni-affinity tag purification followed by anion exchange and then size-exclusion chromatography (SEC). Fractions containing Sar1 were pooled, concentrated, and stored at −80 °C until use.
Imaging Techniques
A Ti-E epifluorescence inverted microscope (Nikon Instruments, Melville, NY) fitted with a Retiga SRV (QImaging, Canada) CCD camera (1.4 MP, Peltier cooled to −45 °C) was used for diffraction imaging of the lipid nanostructures. Diffracted light was viewed through the microscope by applying an angled LED light source (Fiber lite MI-LED B1, Dolan Jenner, Inc.) at 45° by a manipulator device (M-152, Narishige, Inc.) fitted to the Ti-E inverted microscope. At an angle of 45°, the diffracted light is predominantly green, enabling the lipids to be doped with a fluorophore that emits light in the red wavelength for the purpose of rapid quality control and positioning. Diffracted light viewed in this setup produced strong green diffraction with a high signal-to-noise ratio for acquisition of changes in diffraction intensity from Sar1 interaction with the nanoscale lipid multilayer gratings. The signal from the sensor depends upon grating efficiency and wavelength does not significantly shift.
Hepes buffer (50 × 10−3 M) with 1 × 10−3 M magnesium acetate was added to the lipid multilayer gratings enclosed in a homemade PDMS barrier to concentrate the buffer onto the PMMA slide. The sensor chip was then placed onto the diffraction apparatus for light detection. Sar1 with 1 × 10−3 M GTP was added for each sensing experiment after waiting 10 min in order to establish an adequate control baseline.
Diffraction Analysis
The 16-bit sequential RGB images from the CCD camera were analyzed by using ImageJ (http://imagej.nih.gov/ij/). After removing background noise, each individual pixel from the green channel was grouped into 50 a.u. intensity bins according to its initial intensity. The average intensity of each bin had been extracted as function of time. Then, the data was exported to Origin 2015 (Originlab, Northampton, MA) for model fitting.
Modeling Software
Wolfram Mathematica (Wolfram, Champaign, IL) was used to compile and troubleshoot the modeled equation. Once Equation (6) was finalized, Origin 2015 (Originlab, Northampton, MA) was used to fit the equation to the data using the equation fitting software feature.
Surface Plasmon Resonance
Lipid inks were prepared as stated above. DOPC liposomes were then extruded to make small unilamellar vesicles using an Avanti mini extruder with a 100 nm polycarbonate filter. Surface plasmon resonance experiments were performed on a Biacore T200. The small unilamellar vesicles (SUVs) were then immobilized on an L1 sensor chip. SUVs were flowed over the L1 series S sensor chip at 5 μL min−1 for 15 min. Lipids were immobilized until saturated. Upon flowing the lipid solution over the chip, SUVs fuse to create planar bilayers on the chip surface.[48] BSA was injected for 60 s at a flow rate of 10 μL min−1 until saturated. Binding of freshly prepared 0.1 mg mL−1 BSA in LSB did not exceed 100 R.U. This indicated full coverage of the chip and its ability to be used for binding Sar1.[7] Running buffer used for SPR was 20 × 10−3 M Hepes with 1 × 10−3 M magnesium acetate, pH 7.2. Sar1 analyte was flowed over the chip surface at a flow rate of 10 uL min−1 for 10 min. The chip was washed with 3:2 50 × 10−3 M NaOH: Iso-propanol to fully regenerate the chip surface, washing away both lipid and residually bound Sar1.
Supplementary Material
Acknowledgments
The authors acknowledge the Protein Biology Laboratory, College of Medicine, FSU, for use of the SPR instrumentation. T.L., P.P., and D.V.W. are funded by the state of Florida. S.L., A.K.A., and N.V. are supported by NIH R01 GM107172. S.S. and H.H. are supported by NIH R01 GM086892. The authors thank Kevin John at the National High Magnetic Field Laboratory, Tallahassee, Florida for artwork, Arthur Omran at the Institute of Molecular Biophysics, Florida State University, Tallahassee, Florida for providing experimental resources, as well as Jen Kennedy and Amanda Stinchecum for proofreading.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Troy W. Lowry, Department of Physics, Florida State University, Tallahassee, FL 32306-4350, USA
Dr. Hanaa Hariri, Institute of Molecular Biophysics, Florida State University, Tallahassee, FL 32306-4380, USA.
Plengchart Prommapan, Department of Physics, Florida State University, Tallahassee, FL 32306-4350, USA.
Aubrey Kusi-Appiah, Department of Biological Science, Florida State University, Tallahassee, FL 32306-4370, USA.
Nicholas Vafai, Department of Biological Science, Florida State University, Tallahassee, FL 32306-4370, USA.
Prof. Ewa A. Bienkiewicz, Department of Biomedical Science, College of Medicine, Florida State University, Tallahassee, FL 32306-4300, USA
Prof. David H. Van Winkle, Department of Physics, Florida State University, Tallahassee, FL 32306-4350, USA
Prof. Scott M. Stagg, Institute of Molecular Biophysics and Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306-4380, USA
Prof. Steven Lenhert, Email: lenhert@bio.fsu.edu, Department of Biological Science and Institute of Molecular Biophysics, Florida State University, Tallahassee, FL 32306-4370, USA.
References
- 1.Lenhert S, Brinkmann F, Laue T, Walheim S, Vannahme C, Klinkhammer S, Xu M, Sekula S, Mappes T, Schimmel T, Fuchs H. Nat Nanotechnol. 2010;5:275. doi: 10.1038/nnano.2010.17. [DOI] [PubMed] [Google Scholar]
- 2.Voros J, Ramsden JJ, Csucs G, Szendro I, De Paul SM, Textor M, Spencer ND. Biomaterials. 2002;23:3699. doi: 10.1016/s0142-9612(02)00103-5. [DOI] [PubMed] [Google Scholar]
- 3.Fang Y, Ferrie AM, Fontaine NH, Mauro J, Balakrishnan J. Biophys J. 2006;91:1925. doi: 10.1529/biophysj.105.077818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rindorf L, Jensen JB, Dufva M, Pedersen LH, Hoiby PE, Bang O. Opt Express. 2006;14:8224. doi: 10.1364/oe.14.008224. [DOI] [PubMed] [Google Scholar]
- 5.Tamir T, Peng ST. Appl Phys. 1977;14:235. [Google Scholar]
- 6.Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T. Cell. 1998;93:263. doi: 10.1016/s0092-8674(00)81577-9. [DOI] [PubMed] [Google Scholar]
- 7.Schillinger AS, Grauffel C, Khan HM, Halskau O, Reuter N. Biochim Biophys Acta – Biomembranes. 2014;1838:3191. doi: 10.1016/j.bbamem.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Cho NJ, Frank CW, Kasemo B, Hook F. Nat Protoc. 2010;5:1096. doi: 10.1038/nprot.2010.65. [DOI] [PubMed] [Google Scholar]
- 9.Loftus AF, Hsieh VL, Parthasarathy R. Biochem Biophys Res Commun. 2012;426:585. doi: 10.1016/j.bbrc.2012.08.131. [DOI] [PubMed] [Google Scholar]
- 10.Forstner MB, Yee CK, Parikh AN, Groves JT. J Am Chem Soc. 2006;128:15221. doi: 10.1021/ja064093h. [DOI] [PubMed] [Google Scholar]
- 11.Tabaei SR, Rabe M, Zetterberg H, Zhdanov VP, Hook F. J Am Chem Soc. 2013;135:14151. doi: 10.1021/ja4046313. [DOI] [PubMed] [Google Scholar]
- 12.Robison AD, Huang D, Jung H, Cremer PS. Biointerphases. 2013;8:1. doi: 10.1186/1559-4106-8-1. [DOI] [PubMed] [Google Scholar]
- 13.Veesler D, Ng TS, Sendamarai AK, Eilers BJ, Lawrence CM, Lok SM, Young MJ, Johnson JE, Fu CY. Proc Natl Acad Sci USA. 2013;110:5504. doi: 10.1073/pnas.1300601110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S, Brugger B, Ringler P, Muller SA, Rammner B, Grater F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmuller H, Heuser J, Wieland F, Jahn R. Cell. 2006;127:831. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 15.Sheynis T, Friediger A, Xue WF, Hellewell AL, Tipping KW, Hewitt EW, Radford SE, Jelinek R. Biophys J. 2013;105:745. doi: 10.1016/j.bpj.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bacia K, Futai E, Prinz S, Meister A, Daum S, Glatte D, Briggs JAG, Schekman R. Sci Rep. 2011;1:17. doi: 10.1038/srep00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saliba AE, Vonkova I, Ceschia S, Findlay GM, Maeda K, Tischer C, Deghou S, van Noort V, Bork P, Pawson T, Ellenberg J, Gavin AC. Nat Methods. 2014;11:47. doi: 10.1038/nmeth.2734. [DOI] [PubMed] [Google Scholar]
- 18.Reimhult E, Zach M, Hook F, Kasemo B. Langmuir. 2006;22:3313. doi: 10.1021/la0519554. [DOI] [PubMed] [Google Scholar]
- 19.Castellana ET, Cremer PS. Surf Sci Rep. 2006;61:429. doi: 10.1016/j.surfrep.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christensen SM, Stamou DG. Sensors. 2010;10:11352. doi: 10.3390/s101211352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Russell C, Stagg SM. Traffic. 2010;11:303. doi: 10.1111/j.1600-0854.2009.01026.x. [DOI] [PubMed] [Google Scholar]
- 22.Long KR, Yamamoto Y, Baker AL, Watkins SC, Coyne CB, Conway JF, Aridor M. J Cell Biol. 2010;190:115. doi: 10.1083/jcb.201004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barlowe C, Denfert C, Schekman R. J Biol Chem. 1993;268:873. [PubMed] [Google Scholar]
- 24.Hariri H, Bhattacharya N, Johnson K, Noble AJ, Stagg SM. J Mol Biol. 2014;426:3811. doi: 10.1016/j.jmb.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee MCS, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R. Cell. 2005;122:605. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 26.Zimmerberg J, Kozlov MM. Nat Rev Mol Cell Biol. 2006;7:9. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
- 27.Settles EI, Loftus AF, McKeown AN, Parthasarathy R. Biophys J. 2010;99:1539. doi: 10.1016/j.bpj.2010.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nafday OA, Lowry TW, Lenhert S. Small. 2012;8:1021. doi: 10.1002/smll.201102096. [DOI] [PubMed] [Google Scholar]
- 29.Lowry TW, Kusi-Appiah A, Guan J, Winkle DHV, Davidson MW, Lenhert S. Adv Mater Interfaces. 2014;1:1300127. doi: 10.1002/admi.201300127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piner RD, Zhu J, Xu F, Hong SH, Mirkin CA. Science. 1999;283:661. doi: 10.1126/science.283.5402.661. [DOI] [PubMed] [Google Scholar]
- 31.Lenhert S, Sun P, Wang YH, Fuchs H, Mirkin CA. Small. 2007;3:71. doi: 10.1002/smll.200600431. [DOI] [PubMed] [Google Scholar]
- 32.Kusi-Appiah AE, Vafai N, Cranfill PJ, Davidson MW, Lenhert S. Biomaterials. 2012;33:4187. doi: 10.1016/j.biomaterials.2012.02.023. [DOI] [PubMed] [Google Scholar]
- 33.Nafday OA, Lenhert S. Nanotechnology. 2011;22:225301. doi: 10.1088/0957-4484/22/22/225301. [DOI] [PubMed] [Google Scholar]
- 34.Wang YH, Giam LR, Park M, Lenhert S, Fuchs H, Mirkin CA. Small. 2008;4:1666. doi: 10.1002/smll.200800770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang YJ, Wostein HS, Majd S. Adv Mater. 2013;25:6834. doi: 10.1002/adma.201303290. [DOI] [PubMed] [Google Scholar]
- 36.de Mol NJ, Fischer MJE. Surface Plasmon Resonance Methods and Protocols. Humana Press; New York: 2010. [Google Scholar]
- 37.Voros J. Biophys J. 2004;87:553. doi: 10.1529/biophysj.103.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cuypers PA, Corsel JW, Janssen MP, Kop JMM, Hermens WT, Hemker HC. J Biol Chem. 1983;258:2426. [PubMed] [Google Scholar]
- 39.Karlsson R, Falt A. J Immunol Methods. 1997;200:121. doi: 10.1016/s0022-1759(96)00195-0. [DOI] [PubMed] [Google Scholar]
- 40.Parikh AN, Sanii B, Smith AM, Brozell AM. Abstr Pap Am Chem Soc. 2008;236 [Google Scholar]
- 41.Shi Z, Baumgart T. Adv Colloid Interface Sci. 2014;208:76. doi: 10.1016/j.cis.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Gennes P-G, Brochard-Wyart F, Quéré D. Capillarity and Wetting Phenomena: Drops, Bubbles, Pearls, Waves. Springer Science & Business Media; New York: 2004. [Google Scholar]
- 43.Lenhert S, Gleiche M, Fuchs H, Chi LF. ChemPhysChem. 2005;6:2495. doi: 10.1002/cphc.200500276. [DOI] [PubMed] [Google Scholar]
- 44.Barziv R, Moses E. Phys Rev Lett. 1994;73:1392. doi: 10.1103/PhysRevLett.73.1392. [DOI] [PubMed] [Google Scholar]
- 45.Yu Y, Granick S. J Am Chem Soc. 2009;131:14158. doi: 10.1021/ja905900h. [DOI] [PubMed] [Google Scholar]
- 46.Sanborn J, Oglecka K, Kraut RS, Parikh AN. Faraday Discuss. 2013;161:167. doi: 10.1039/c2fd20116j. [DOI] [PubMed] [Google Scholar]
- 47.Yuan J, Hira SM, Strouse GF, Hirst LS. J Am Chem Soc. 2008;130:2067. doi: 10.1021/ja710305c. [DOI] [PubMed] [Google Scholar]
- 48.Erb EM, Chen XY, Allen S, Roberts CJ, Tendler SJB, Davies MC, Forsen S. Anal Biochem. 2000;280:29. doi: 10.1006/abio.1999.4469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.