

Abstract
Therapeutic hypothermia has emerged as a remarkably effective method of neuroprotection from ischemia and is being increasingly used in clinics. Accordingly, it is also a subject of considerable attention from a basic scientific research perspective. One of the fundamental problems, with which current studies are concerned, is the optimal method of inducing hypothermia. This review seeks to provide a broad theoretical framework for approaching this problem, and to discuss how a novel promising strategy of pharmacological modulation of the thermosensitive ion channels fits into this framework. Various physical, anatomical, physiological and molecular aspects of thermoregulation, which provide the foundation for this text, have been comprehensively reviewed and will not be discussed exhaustively here. Instead, the first part of the current review, which may be helpful for a broader readership outside of thermoregulation research, will build on this existing knowledge to outline possible opportunities and research directions aimed at controlling body temperature. The second part, aimed at a more specialist audience, will highlight the conceptual advantages and practical limitations of novel molecular agents targeting thermosensitive Transient Receptor Potential (TRP) channels in achieving this goal. Two particularly promising members of this channel family, namely TRP melastatin 8 (TRPM8) and TRP vanilloid 1 (TRPV1), will be discussed in greater detail.
Keywords: body temperature, core temperature, pharmacological hypothermia, physical cooling, therapeutic hypothermia, thermopharmacology, thermoregulation, thermosensitive ion channels, ThermoTRPs
Abbreviations: DMH, dorso-medial hypothalamus; MPA, medial preoptic area of hypothalamus; rMR, rostral medullary raphe region; ThermoTRPs, Thermosensitive Transient Receptor Potential cation channels; TRP, Transient Receptor Potential; TRPA1, Transient Receptor Potential cation channel, subfamily A, member 1; TRPM8, Transient Receptor Potential cation channel, subfamily M, member 8; TRPV1, Transient Receptor Potential cation channel, subfamily V, member 1; TRPV2, Transient Receptor Potential cation channel, subfamily V, member 2; TRPV3, Transient Receptor Potential cation channel, subfamily V, member 3; TRPV4, Transient Receptor Potential cation channel, subfamily V, member 4
Therapeutic Potential of Hypothermia
Temperature powerfully affects every biological process. Humans and many other mammals maintain their internal temperature at a stable level around 37°C,1 which appears to be optimal for their functioning under most normal conditions. However, it has been now definitively established that in some pathological states, the endogenously regulated level of internal temperature does not provide the highest chances of survival. In such cases, a more favorable outcome may be achieved by a therapeutic intervention aimed at external modulation of patient's temperature. In particular, ample basic science and clinical evidence suggests that intentional mild lowering of a patient's core temperature, also known as induced, or therapeutic hypothermia, has the potential to protect vital organs from damage, preserve their function, and improve survival during acute global ischemia and hypoxia.2-6
The therapeutic use of low temperature has been explored probably for as long as medicine exists and has demonstrated clinical benefit in a variety of contexts.7 However, only recently has this knowledge been developed into clearly defined clinical protocols of whole-body hypothermia, which have been tested in rigorous clinical trials.8,9 The first modern seminal trials of therapeutic hypothermia have demonstrated that maintaining the patient's temperature in the range of 32-34°C for 12-72 hours improved survival and neurological outcome in patients resuscitated after out-of-hospital cardiac arrest 10,11 and in neonates with perinatal asphyxial encephalopathy.12,13 Based on these positive results, recently there has been a surge of additional clinical investigations of hypothermia in traumatic brain injury,14 acute ischemic stroke,15,16 myocardial infarction,17 and other conditions.18 Many of these ongoing studies are expected to further establish therapeutic hypothermia as one of the most promising and effective new treatment methods in acute care medicine.
Unfortunately, however, some of the recently completed studies failed to demonstrate a clear benefit. Thus, while the hopes for clinical potential of the overall concept of hypothermic neuroprotection remain high, these discouraging results highlighted some of the significant challenges for the further progress of this treatment modality. Some challenges may be related to our incomplete basic understanding of the effects of low core temperature on human physiology and disease pathogenesis. More specifically, the optimal depth, duration, and timing of hypothermia have not been yet definitively established.19
In regard to the depth of hypothermia, current guidelines recommend the target core temperature of 32-34°C, based on classical animal studies 20,21 and successful human clinical trials.10,11 However, it is not clear if this temperature is the most beneficial due to paucity of studies directly comparing the relative benefit of different target core temperatures. Recently, aggressive cooling to 33°C demonstrated no additional benefit over milder cooling to 35-36°C both in a rat study 22 and in a human clinical trial in cardiac arrest patients.23 Thus, more comprehensive studies over a wide range of target temperatures are needed to determine the level that will provide the optimal balance between the protective effects of hypothermia and complications in each specific clinical context.
Of major importance is also the question of when to initiate the hypothermia in relation to the clinical event. An influential classical animal study demonstrated that delay of cooling initiation by as little as 15 min following reperfusion after cardiac arrest in dogs largely offsets the protective effect of hypothermia.24 This and other findings suggested that cooling should be initiated as soon as possible to provide the most benefit. However, 2 recent clinical trials in cardiac arrest patients have found no improvement in survival or neurological outcome with early pre-hospital cooling initiation compared with standard hospital cooling.25,26 Once again, further research is needed to understand what factors related to hypothermia timing and duration play the key role in conferring clinical benefit and to develop protocols that would take full advantage of this information.
Another major obstacle for the further development and wider adoption of therapeutic hypothermia protocols is the technical and logistical difficulty of the current methods of lowering core temperature in human patients. Most methods involve complicated equipment that is available only in a minority of treatment sites. In addition, current protocols require the use of various drugs, which often produce dangerous side effects. Moreover, even these complicated methods do not provide the level of control over the depth and rate of hypothermia required for optimal treatment effect. Therefore, there is a great need in clinical medicine for novel methods of lowering core body temperature, which will be safe, effective, easily administered and widely available.
Fortunately, the basic understanding of the molecular mechanisms of mammalian temperature regulation is rapidly advancing and may provide novel insights and potential solutions to this important problem. Before these advances are discussed, a brief overview of the conceptual approaches to lowering body temperature may be helpful.
Determinants of Body Temperature
In order to effectively manipulate the body temperature of a living organism, it is essential to understand what factors determine it. According to the fundamental principles of thermodynamics, the temperature of an organism is determined by the balance of heat generation within the organism and heat transfer between the organism and its environment (Fig. 1).27 In most experimental and clinical situations related to hypothermia, ambient temperature is lower than the core body temperature of an organism and thus the heat transfer component can more precisely be described as heat loss to the colder environment.
Figure 1.
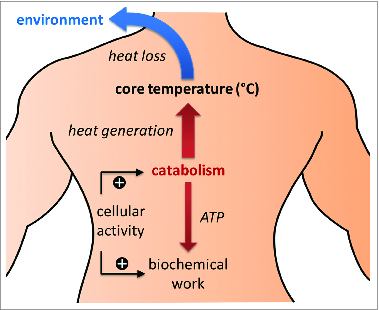
Determinants of core temperature and general approaches to its modulation. Core temperature is determined by the balance of heat generation through catabolism and heat loss to the environment (assuming most typical conditions, in which ambient temperature is lower than core temperature). Catabolism is a component of the cellular activity that provides energy for other biochemical processes. Increased cellular activity results in increased rates of catabolism and heat generation. Heat loss depends on the gradient between ambient and core temperatures. Core temperature may be lowered by decreasing heat generation through the reduction of cellular activity, and/or by increasing heat loss through the lowering of ambient temperature (i.e., application of physical cooling).
Heat generation in the body is a natural byproduct of the functional activity of cells.28 This activity encompasses catabolic, or exergonic, reactions in which complex molecules are broken down and energy is released, as well as endergonic reactions, which consume energy to perform some useful biochemical, electrochemical or mechanical work. Exergonic reactions are coupled to endergonic reactions, such that the energy liberated in the former drives the latter. However, because this coupling is less than 100% efficient, some energy escapes the transformation, is released into the surrounding body tissues in the form of heat and leads to an increase in their temperature.29 Thus, the rate of heat generation is proportional to the rates of biochemical reactions within the cell, i.e. to its activity level. Highly active cells, such as brain neurons or contracting skeletal muscle, generate more heat compared to less active cells, such as white fat or skin cells.30
Heat loss is driven by evaporation of fluids, radiation and conduction of heat to surrounding air or objects with lower temperature. The latter 2 components are heavily influenced by the gradient of temperature between the body and its environment. The conduction component of heat loss is also dependent on the thermal conductivity of the body-environment interface, which is determined by the properties of barriers, such as clothes, fur, and skin, as well as on the tone of cutaneous blood vessels.31
Altering Body Temperature
In order to modulate the internal temperature of an organism, either or both heat generation and heat loss must be altered (Fig. 1). Because heat generation is necessarily a byproduct of cellular activity, reducing heat production must involve decreasing activity. Incidentally, a decrease in activity of cells during ischemia reduces their metabolic demand and is cytoprotective by itself, independently of decrease in temperature. Thus, direct reduction in cellular activity is a complementary and parallel goal to hypothermia in achieving neuroprotection.
Figure 2.

Schematic of the thermoregulatory system and pharmacological targets for its modulation. The thermoregulatory system modifies the heat exchange between the body and the environment to maintain a particular balance level of core body temperature. The thermoregulatory system consists of thermoreceptors, afferent pathways, central thermoregulatory neurons, efferent pathways and effector organs. Strategies for controlling or blocking thermoregulatory responses may be aimed at each of these levels of organization (indicated by green captions). See text for detailed discussion. Notes: (1) thermoregulatory centers in hypothalamus are actually complex multi-neuronal circuits, but for clarity are presented here as single neurons; (2) only the most important, but not all, thermoeffector processes are shown; (3) the universal role of TRPM8 within the pathway for skin cooling is putative and has been challenged for some cold-defensive responses; (4) only 2 populations of primary sensory neurons are shown, while many others also exist.
To appreciably decrease heat generation, activity must be reduced at the level of whole organs or even of the whole organism. Incidentally, patients are typically already in the resting state, characterized by close to minimal physiological activity and near basal metabolic rate. The metabolic rate can be further reduced by general anesthesia. Thus, deeply anesthetized and mechanically ventilated humans have a metabolic rate at ∼65% of the basal level.32 However, even at this low level of heat generation and assuming typical room ambient temperatures of 22-24°C, the rate of the drop in core temperature is still suboptimal for a neuroprotective strategy.
It is tempting to speculate if even lower levels of metabolism may be achieved in humans. A promising opportunity may be related to the phenomenon of hibernation that occurs in several mammalian species.33 During hibernation, an animal is able to substantially decrease its metabolism and energy demands to survive a period of low nutrient supply. For example, black bear, a species of comparable size to humans, has 50% lower levels of metabolism in hibernation compared to pre-hibernation levels measured at the same level of core temperature.34 If a similar hypothetical state of “hibernation” could be induced in humans, the same 50% reduction in their metabolic rate and heat generation may be reasonably expected. However, this reduction would be only slightly larger than the previously mentioned reduction in metabolism of anesthetized humans (∼35%), and still would not provide the therapeutically sufficient rate of hypothermia at typical ambient temperatures. Moreover, even this seemingly marginal benefit is currently not feasible, because no strategies for inducing hibernation in humans are known. Thus, the overall strategy of decreasing heat generation through anesthesia, hibernation or other methods without an increase in heat loss doesn't appear to have the potential to achieve hypothermia within the timeframe consistent with neuroprotection.
At the same time, these methods aimed at decreasing heat generation do lower cellular activity and metabolic demand even at normal levels of core temperature, and provide cytoprotection independently from any possible temperature-lowering effect. Therefore, as mentioned earlier, they may be utilized independently or in synergy with other hypothermic strategies. Induction of the putative “hibernation” state in humans is a particularly promising and actively explored area, which is beyond the scope of the current review.
A more straightforward alternative to decreasing heat generation in achieving hypothermia is increasing heat loss. As mentioned earlier, 2 out of 3 possible mechanisms of heat loss, namely radiation and conduction, strongly depend on the gradient between core and ambient temperatures. Thus, heat loss may be greatly enhanced by lowering environmental temperature, or in other words, by application of physical cooling. This strategy has become the basis of the current clinical protocols of therapeutic hypothermia.35 A variety of practical implementations of the physical cooling strategy have been created, and new and improved methods are constantly being developed.36
Physical cooling methods can be broadly divided into surface and core cooling methods. The earliest and simplest application of surface cooling involved covering the patient's body with ice packs.10 The same principle is used in more advanced surface cooling devices of various designs, which typically utilize cold water-perfused cooling blankets and pads and include additional features such as a feedback temperature control and gradual active rewarming capabilities.37-39
Core cooling methods employ intravascular heat-exchanging catheters, which drain the heat not from the surface, but directly from the core component of the body.40,41 Intravascular devices allow higher rates of cooling, yet they have higher requirements for skilled staff and higher possibility of complications. A special case of core cooling is a rapid high-volume infusion of cold fluids.42 This strategy is often used as the very first step for a rapid induction of hypothermia, which is then maintained by other methods. Ultimately, all physical cooling methods are capable of inducing substantial heat transfer away from the body and a drop in core temperature.
However, in contrast to passive inanimate objects, complex living systems possess a critical property of homeostasis, or the ability to maintain key physiological variables at stable levels. In particular, body temperature is one of the regulated variables in homeothermic (or warm-blooded) animals, such as mammals. The structures of the body responsible for this regulation are organized into the highly complex and powerful thermoregulatory system (Fig. 2).43-46 When mammalian body temperature is challenged, the thermoregulatory system initiates a number of behavioral and physiologic responses, which modulate the thermal balance of the organism. Thus, a fully conscious mammal exposed to cold and increased heat loss responds with warm-seeking/cold-avoiding behaviors, an increase in heat generation through shivering and non-shivering thermogenesis, a decrease in heat conductivity through peripheral vasoconstriction, as well as a number of other mechanisms. As a result, the heat loss caused by cold exposure may be minimized and offset by the increased heat generation. In this case, at least initially, hypothermia does not develop.
An experimental illustration of this theoretical model may be found, for instance, in the experiment by Iampietro et al.,47 in which core temperature of nude men exposed to the cold air at 10°C and wind of 10 mph for 2 hours dropped by merely ∼0.2°C from the pre-exposure levels. Similarly, Adolph et al. reported that core temperature of nude men exposed to cold air at temperatures down to 0°C and wind speeds up to 15 mph for 4 hours did not drop below 35.8°C.48 Moreover, compensatory thermoregulatory responses prevent the clinically meaningful drop in core temperature not only during surface cooling, but also during core cooling. For instance, a study by Moore et al. demonstrated that rapid high-volume infusion of 4°C cold saline lowered core temperature of healthy awake volunteers by only 1°C and did not achieve the conventionally defined therapeutic plane of hypothermia.49
The compensatory capabilities of the body are nonetheless finite. If the cold exposure is of sufficient magnitude and duration, these capabilities may be overwhelmed by the heat loss, and core temperature will ultimately decrease.50 Thus, it is possible to achieve hypothermia even in a conscious non-medicated mammal with an intact thermoregulatory system. But along with the intended and beneficial consequence of lowered core temperature, such a forcible intervention will trigger a maximal compensatory effort by the body. This compensatory response puts a considerable load on cardiovascular, respiratory, and other homeostatic systems, and is maladaptive for a patient undergoing the hypothermic protocol. In addition, vigorous shivering, which is a major part of the compensatory cold defense response in humans, is very uncomfortable.51 This scenario demonstrates that targeting only one component of heat balance in the presence of the highly robust and redundant thermoregulatory system will result in many unintended effects and is unlikely to achieve hypothermia in a safe and specific manner. Thus, in order for the physical cooling strategy to work, it must be combined with some measures for inactivation of the thermoregulatory system.
Targets for Modulation within the Thermoregulatory System
The thermoregulatory system incorporates a series of structures,45,46 including thermoreceptors, afferent neural pathways, integrative brain centers, efferent neural pathways and effector organs such as skeletal muscle, blood vessels and brown adipose tissue, which may be amenable to inactivation or modulation (Fig. 2). Theoretically, inactivation on any of these levels has a potential to alter compensatory responses and induce tolerance to hypothermia. Accordingly, pharmacological strategies targeting various parts of the thermoregulatory system have been developed and have enabled the successful achievement of hypothermia in clinical trials. However, these strategies also have important limitations, which hinder the full therapeutic potential of induced hypothermia.
The most important targets for inactivation in the thermoregulatory system are the integrative centers in the hypothalamus. A broad suppression of the central nervous system is a natural consequence of a comatose state, which is a typical occurrence in patients resuscitated after out-of-hospital cardiac arrest.52 Further inactivation of the thermoregulatory system may be achieved by general anesthetic drugs, such as midazolam and fentanyl.10,11 Thus, patients in coma and under general anesthesia are very prone to reduction in core temperature and much of the successful experience with therapeutic hypothermia has been obtained in such patients by exposing them to the physical cooling methods discussed above. Importantly, these patients need to be mechanically ventilated and may be treated only in the most advanced acute care settings.
However, in contrast to cardiac arrest patients, victims of stroke and many other conditions typically remain in the conscious state with intact thermoregulatory responses. Induction of iatrogenic coma or general anesthesia in these patients may be logistically not feasible and provide less benefit than risk related to intubation, mechanical ventilation and adverse drug effects. Thus, many patients require hypothermia-inducing strategies, which are amenable for use outside of the intensive care setting.
One possible strategy involves the use of milder sedative drugs, such as the serotonin receptor agonist buspirone and the opioid meperidine.52 Both of these agents were shown to reduce the shivering and vasoconstriction thresholds in conscious volunteers when used individually. Furthermore, the anti-shivering effect was enhanced when the drugs were combined.53,54 Accordingly, an anti-shivering regimen based on the combination of oral buspirone and intravenous meperidine enabled the achievement of target core temperature below 34°C in an early safety and feasibility trial of hypothermia in awake stroke patients.55
However, just as general anesthetics, sedative drugs do not target thermoregulatory neurons specifically, but instead affect multiple other brain centers. When used in doses sufficient for clinically relevant hypothermia, they may produce a number of unwanted and even potentially dangerous side effects, such as sedation and respiratory depression.54,56 Impaired consciousness is especially problematic for stroke patients because it interferes with the neurological examination of their cognitive status.57 These problems of the centrally-acting drugs highlight the need for novel pharmacological strategies, which would possess greater specificity toward the thermoregulatory system and have minimal influence on other functions of the central nervous system.
Achieving this greater specificity requires the identification of distinct structures in the central nervous system, which are involved in thermoregulation. Recent functional neuroanatomical studies in rodents have greatly expanded our knowledge about the central circuitries of temperature regulation.46 According to the current model based on these studies, the integrative central thermoregulatory neurons are located in the medial preoptic area of the hypothalamus (MPA). MPA neurons are postulated to be tonically active at thermoneutral temperatures and intrinsically sensitive to local brain temperature. In addition, they are affected by skin temperature through excitatory and inhibitory inputs from peripheral thermoreceptors. On the efferent side, MPA neurons send inhibitory projections to primary effector neurons in the dorso-medial hypothalamus (DMH) and rostral medullary raphe region (rMR).
Functionally, MPA neurons are inhibited in response to skin or core cooling. Inhibition of MPA neurons disinhibits, i.e., activates, primary effector neurons in the DMH and rMR. Activation of primary effector neurons triggers cold defense responses, such as BAT thermogenesis, peripheral vascoconstriction, and shivering.
In addition to anatomical location of the relevant groups of neurons, their receptor inputs have been also identified. Pharmacological modulation of these receptor inputs allows abolishing cold defense responses at the level of central circuitries. For instance, it was shown that blocking GABA-ergic inhibitory input to the MPA neurons with the GABA antagonist bicuculline suppresses cold defense responses to skin cooling.58,59 The same effect can be achieved by pharmacological inhibition of DMH and rMR effector neurons by the GABA agonist muscimol.60-62
If these mechanisms, identified in rodents, are also relevant in humans, such interventions are predicted to effectively induce hypothermia in patients cooled by physical methods. However, specific modulation of these compactly localized neuronal populations by pharmacological modulators of the ubiquitous central nervous system receptors is possible only through spatially specific delivery of pharmacological agents. In experimental rodent studies, this is typically achieved by intra-cerebral nanoinjections of drugs to the well-defined areas of the brain. However, the clinical use of this delivery route in human patients has not been well established and at present appears impractical. Thus, the pharmacologically feasible strategy of controlling thermoregulatory system has to rely on molecular, rather than spatial, specificity of the targets.
Other common targets for inactivation within the thermoregulatory system are the effector organs. The most important cold defense response in humans is the shivering response of the skeletal muscles.63 Importantly, the shivering response is not always fully abolished even in comatose and deeply anesthetized patients, and in many cases must be managed additionally. Motor activity of the skeletal muscles, including shivering, may be effectively blocked using neuromuscular blocking pharmacological agents, or muscle paralytics.64 Accordingly, neuromuscular blockers are an essential component of most hypothermia protocols. However, effector organs, including skeletal muscles, may be involved in other critical functions of the body besides thermoregulation. In particular, muscle paralysis interferes with such a vital function of skeletal muscles as respiration. Thus, this strategy is also limited only to anesthetized and mechanically ventilated patients and is not amenable for use in other patient populations, which may benefit from hypothermic treatment.
Some effector organs may be more selectively involved in thermoregulation. In particular, brown adipose tissue has a well-established function in compensatory heat generation during cold exposure in small mammals and has been recently implicated in the same role in humans.65 At the same time, this tissue doesn't appear to be acutely involved in other vital physiological functions. Thus, its pharmacological inactivation may lead to relatively safe and specific decrease in compensatory heat generation during physical cooling. However, no such strategies in humans have been reported so far. Furthermore, because the effector systems are at least partially redundant, inactivating any one of them may not be sufficiently effective.
Targeting Thermoreceptors
Lastly, the largely untapped part of the thermoregulatory system which may be targeted in order to control thermal balance is thermoreception. Temperature in the body is sensed by central thermoreceptors in hypothalamus, spinal cord and viscera, as well as by peripheral receptors in the nerve endings of the primary sensory neurons. The central hypothalamic thermoregulatory neurons integrate all cold- and warm-sensitive peripheral and central temperature inputs, and generate an appropriate output signal determining the activity of the thermoregulatory processes. Here, an opportunity may be recognized that modulation of the neural activity of the thermoreceptors may enable deliberate control of the heat-transferring activity of the thermoeffectors.
Up to this point, it has been proposed that achieving hypothermia requires increasing heat loss by physical cooling with concomitant elimination of the compensatory responses. Accordingly, this discussion has focused on strategies for inactivation of the thermoregulatory system. However, given the capacity of the thermoregulatory system to induce net heat gain as well as net heat loss, pharmacological manipulation of thermoreceptors offers an intriguing and potentially more effective alternative for altering core temperature. In contrast to merely inactivating the thermoregulatory system, manipulating the signal from the thermoreceptors should allow shifting the thermal balance in the hypothermic direction by reducing heat retention, inhibiting thermogenesis, and facilitating active heat loss. In this scenario, the thermoregulatory system may be driving, instead of defending against, the drop in core temperature, which should result in hypothermia with less time, energy costs, and side effects. Importantly, active physical cooling thus becomes merely an auxiliary, and not a necessary measure. The fulfillment of the described strategy critically requires specific and pharmacologically accessible molecular targets within central or peripheral thermosensory neurons that would allow modulation of the activity of these neurons.
The molecular mechanisms responsible for thermosensitivity of the central neurons have not yet been clearly established. There is some evidence that this thermal sensitivity is achieved by a particular combination of common neuronal ion channels, such as hyperpolarization-activated cyclic nucleotide-gated channels (HCN), voltage-activated A-type potassium channels (Kv4.1), and tandem pore domain, or background leak potassium channels (K2P), rather than by a single and unique receptor protein.66 Because these common channels are broadly involved in nervous system functions, they have limited potential as therapeutic targets.
Until recently, the molecular markers of peripheral thermoreceptors were also unknown. In a series of major advances, several members of the Transient Receptor Potential (TRP) superfamily of non-specific cation channels were shown to be directly gated by temperature.67 These channels are collectively known as thermoTRPs and include cold-sensitive TRPM8 and TRPA1, as well as warm-sensitive TRPV1, TRPV2, TRPV3, and TRPV4.67 ThermoTRPs are expressed, with variable selectivity, in primary sensory neurons and therein fulfil one of the critical requirements for useful pharmacological targets for controlling thermoregulation. Importantly, in agreement with the model described above, modulation of at least 2 of these channels, TRPV1 and TRPM8, produced hypothermia in experimental animal studies.68-71 Next, the possible mechanism for the hypothermic action of these drugs and some practical aspects of their potential use as hypothermic agents in human patients will be discussed.
Targeting TRPM8, a molecular sensor of cold
As mentioned earlier, the activity of the thermoeffectors is controlled by an integrated signal from anatomically distinct cold- and warm-sensitive inputs. These inputs have a reciprocal influence on the thermoeffector activity, with cold inputs favoring heat gain responses, and warm inputs favoring heat loss responses. Thus, the net heat loss state of the thermoregulatory system, required for achieving hypothermia, may be induced either by increasing warm input, or by blocking the cold input. With the establishment of the cold-sensitive ion channel TRPM8 as an in vivo molecular cold sensor,72-74 its inhibition seemed likely to provide the latter opportunity, namely, to block the cold signals from reaching the thermoregulatory system and enable the drop in core temperature during exposure to subneutral ambient temperatures.
The TRPM8 channel, also known as the cold and menthol receptor, is directly gated by temperatures below ∼27°C in vitro.75-77 Within the nervous system, TRPM8 is located in a subset of primary sensory neurons of the dorsal root and trigeminal ganglia within the afferent pathway for skin cooling.78 In the context of a neuron, this ion channel converts the physical stimulus of lowered temperature into a depolarizing current, thus endowing a select population of primary neurons with cold sensitivity. Because cold-sensitive neurons transmit the signal about cold exposure to the higher-order thermoregulatory centers, TRPM8 is responsible at least for some part of this signal. Accordingly, pharmacological inhibition of TRPM8 abolishes cold-sensitivity of a subset of primary neurons and leads to a decrease in the overall cold-dependent input to the thermoregulatory system, shifting it to a less heat-generating and more heat-dissipating state.69 On the level of individual thermoeffectors, pharmacological inhibition or genetic deletion of TRPM8 decreases whole body energy production,69 thermogenesis in brown adipose tissue,69,79 peripheral vasoconstriction,69 and cold-avoidance behaviors.69,72-74 As mechanistically predicted, the net effect of these changes has been a decrease in core temperature (Fig. 3).69-71,79,80 Importantly, such a hypothermic effect has been demonstrated for at least 3 unrelated TRPM8 antagonists and in the case of one of them - in both mice and rats.69,70,80 These findings have provided an initial conceptual support for the use of pharmacological TRPM8 inhibition to induce therapeutic hypothermia.69 However, further development of this idea reveals several important limitations.
Figure 3.
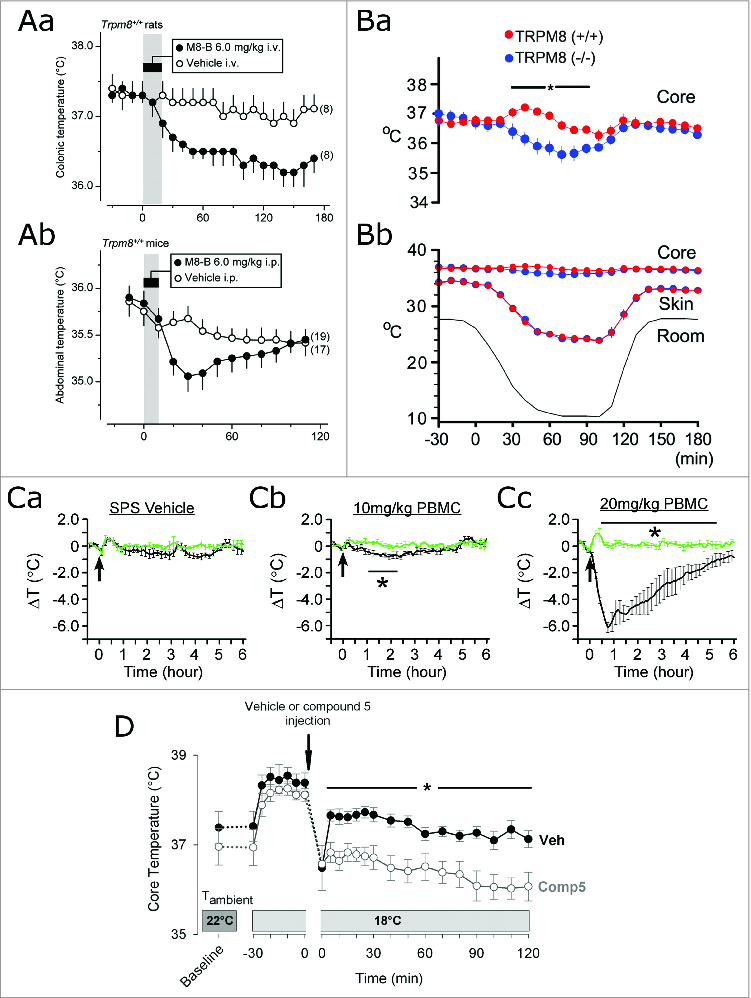
For figure legend, see page 252. Figure 3 (See previous page). The effects of the genetic deletion and pharmacological inhibition of TRPM8 channels on core body temperature. (A) Core temperature of rats after intravenous infusion (Aa) or mice after intraperitoneal infusion (Ab) of the TRPM8 antagonist M8-B (6 mg/kg) at an ambient temperature of 19°C (Aa) or 26°C (Ab). Time of infusion is indicated by a gray bar. Adapted from Almeida et al. 69 © The Society for Neuroscience/The Journal of Neuroscience. Reproduced by permission of Andrej A Romanovsky. (B) Core and skin temperatures of TRPM8+/+ and TRPM8−/− mice during exposure to a decrease in ambient temperature (indicated by the label “room” on the graph). The core temperature data from (Bb) is replotted in (Ba) with an increased scale on the Y axis. Adapted from Tajino et al. 79 © Public Library of Science. (C) Relative drop in core temperatures of TRPM8+/+ (black lines) and TRPM8−/− mice (green lines) after intraperitoneal injection (marked by an arrow) with vehicle (10% solutol/20% PEG-200/saline [SPS]) (Ca), or the TRPM8 antagonist PBMC at 10 mg/kg (Cb) or 20 mg/kg (Cc). Adapted from Knolwton et al. 70 © Public Library of Science. (D) Core temperature of mice after intraperitoneal injection of vehicle (Veh) or the TRPM8 antagonist “compound 5” 114 (20 mg/kg) held at a mildly subneutral ambient temperature (18°C). Adapted from Feketa et al. 80 © The American Physiological Society. Permission to reuse must be obtained from the rightsholders.
The first thing to consider is that TRPM8-independent pathways for cold sensing exist, and TRPM8-dependent neurons carry only some part, albeit a substantial one, of the cold signal from the skin.72-74 Even more importantly, the thermoregulatory system receives cold-generated input not only from peripheral tissues, but also from the body core, mediated at least in part by the decreased activity of the warm-sensing neurons in MPA.31 Owing to this partial redundancy, the thermoregulatory system can compensate for the failure of skin-dependent receptor input, such as that induced by TRPM8 inhibition. Based on the effects of TRPM8 antagonists on core temperature at subneutral ambient temperatures, as reported in the literature (Fig. 3),69,70,80 we propose the following model for these effects. Initially, reduced skin-dependent cold signal results in decreased heat generation and a gradual drop in core temperature. But as the core temperature drops, the core-dependent cold signal increases, compensating for the missing part of the skin-dependent signal, and drives cold defense responses. Eventually, a new steady state is reached, wherein core temperature is still defended by cold defense responses, but at a slightly lower than normal level. However, whether this lower level will be within the therapeutic range is questionable.
The magnitude of the drop in core temperature caused by TRPM8 inhibition in the described model is determined by the ambient temperature and by the relative contribution of the TRPM8-dependent part to the overall cold-induced afferent signal. There are currently no quantitative models that allow the calculation of this contribution. Based on empirical data in experimental animals (Fig. 3), only limited conclusions may be made, because there have been no studies to date that have determined the effect of the maximally effective dose of a TRPM8 antagonist within a proper dose-response experimental design. One relevant investigation reported up to a ∼6 °C drop in core temperature after a high dose of a TRPM8 antagonist.70 However, 3 other studies using different TRPM8 inhibitors demonstrated a hypothermic effect of no more than ∼2°C, even including an effect observed in mice exposed to significant ambient cooling.69,71,80 Moreover, genetic loss of TRPM8, which may be considered as a surrogate for full inhibition of TRPM8, similarly demonstrated a hypothermic effect within 1°C even at a low ambient temperature.79 It should be noted that TRPM8 germline knock-out mice may have developed some compensatory mechanisms, which would confound an analysis of TRPM8 function based on experimental findings obtained in these mice. However, the largely consistent results seen during pharmacological inhibition and genetic deletion of TRPM8 suggest only a limited hypothermic effect of even the maximal loss of TRPM8 activity. Therefore, on the whole, at this point it seems that blocking TRPM8-mediated cold afferent signal from the skin does not reduce the heat-generating mechanisms sufficiently to achieve a neuroprotective range of hypothermia. Furthermore, although this conclusion is based on rodent studies, it may be even more relevant for humans.31 The relative contribution of skin-dependent afferent signals to the thermoregulatory responses is thought to be lower in humans compared to rodents, which might make the hypothermic response to TRPM8 inhibition even less pronounced. Because there have been no studies reporting the use of TRPM8 antagonists in humans, this prediction awaits experimental confirmation.
The effects of TRPM8 antagonists also depend on ambient temperature, and thus may be increased by exposing the subject to physical cooling.69 However, as outlined above, inhibition of TRPM8 does not fully inactivate the thermoregulatory system, which still retains the ability to mount cold defense responses to prevent a further drop in core temperature. Therefore, physical cooling will still produce unwanted physiological thermoregulatory responses. Moreover, it is being increasingly recognized that individual thermoeffector mechanisms may be under the control of functionally, anatomically and molecularly distinct afferent pathways,45 and thus may be regulated by TRPM8 to a different extent. In particular, there is evidence that shivering and tachycardic responses to external cooling are not significantly controlled by TRPM8-mediated signals and are not abolished by TRPM8 inhibition.80 Thus, the use of physical cooling to enhance the hypothermic effect of TRPM8 inhibitors is expected to produce the same complications as physical cooling alone, namely, almost full activation of shivering and other cold defense responses.
Finally, pharmacological modulation of TRPM8 may be problematic because of the potential “off-target” effects. Although the field of thermoregulation is justifiably focused on TRPM8 in sensory neurons, this channel is also expressed in a number of other tissues, including prostate, liver, bladder and vascular smooth muscle, as well as in various types of tumors.81 The effects of TRPM8 modulation in these tissues are largely unknown, but may complicate its therapeutic utility.
To summarize, despite the initial encouraging prospects, both empirical data and mechanistic understanding of the functions of the TRPM8 channel in the thermoregulatory system argue against its clinical usefulness as a sole target for safe and effective hypothermia. Nevertheless, TRPM8 inhibition may not be entirely without clinical value in hypothermic protocols. Because TRPM8 antagonists seem to have at least some effects on the thermoregulatory system, their use as adjuvants may be considered and deserves further investigation.31,69 Along these lines, it was shown that TRPM8 inhibition can enhance the hypothermic effect of the TRPV1 agonist dihydrocapsaicin,71 and additional such interactions may be uncovered in future studies.
The relative lack of success of the putative universal cold sensor TRPM8 in hypothermia should not be generalized to the overall strategy of blocking cold input to the thermoregulatory system. Because this input is probably mediated by additional cold-sensitive mechanisms, identification of such mechanisms and development of the pharmacological methods for their inhibition may ultimately make this strategy more effective and promising.
Targeting TRPV1, a molecular sensor of heat
As an alternative to inhibiting cold-sensitive pathways, the net heat loss state of the thermoregulatory system may be achieved by activating the warm-sensing pathways. The conceptual validity of this idea has been confirmed in a series of studies, demonstrating that application of physical skin counter-warming techniques is able to suppress the cold defense responses to hypothermia.82-85 However, the effect of skin counter-warming alone is not sufficient to decrease core temperature into the therapeutic range. There is some evidence that a more efficient implementation of this strategy may be provided by pharmacological activation of the TRPV1 ion channel.
TRPV1, also known as the heat and capsaicin receptor, is another temperature-sensitive member of the TRP superfamily, activated by temperatures above ∼42°C in vitro.68,86 It is mainly expressed in primary and higher-order sensory neurons 86,87 and in numerous structures in the central nervous system.88,89 Although the gene encoding this channel was identified only in 1997,86 it has been known for decades that systemic administration of the TRPV1 agonist capsaicin, the pungent ingredient of chili peppers, leads to pronounced hypothermia in numerous mammalian species.90
Despite numerous studies,68,90 the site of action and the exact mechanism responsible for the hypothermic effect of TRPV1 agonists have not been definitively established. The existing findings may be largely accounted for by a model wherein TRPV1 agonists activate the skin-warming pathway. According to this model, pharmacological TRPV1 activation provides “false,” or spurious, information about the exposure of skin to high temperatures to the central thermoregulatory neurons of the hypothalamus and effectively “tricks” them into sensing heat in the absence of a real temperature challenge. In an effort to offset the consequences of the presumed overheating, central thermoregulatory neurons would be predicted to respond to such a signal with inhibition of heat-generating thermoeffector mechanisms, like warm-seeking and non-thermoregulatory locomotion, shivering, and brown adipose tissue activity, and activation of heat-dissipating mechanisms, such as peripheral vasodilation and species-dependent saliva-spreading, panting, and sweating. Such a coordinated shift in the activity of the thermoeffectors was indeed demonstrated in experimental studies90 and may explain the generation of a substantial heat flow away from the body resulting in a rapid decline in body temperature. In addition, it has been speculated that TRPV1-containing neurons responsible for these effects are most likely anatomically located in the median preoptic nucleus of the hypothalamus, and functionally belong to the afferent pathway for skin warming.68 However, whether TRPV1 agonists actually act on the hypothalamic neurons or increase the afferent signal within the skin warming pathways has yet to be conclusively demonstrated. Until then, the proposed model remains largely speculative.
Importantly, TRPV1 activation by agonists not only abolishes compensatory responses to core cooling, which actually enables a profound decrease in core temperature, but also substantially blunts the shivering and tachycardic responses to ambient cooling.80 This finding suggests that application of external cooling on the background of pharmacological TRPV1 activation has a substantially diminished effect on the overall integrated afferent thermoregulatory drive and does not trigger a significant compensatory increase in heat generation. Therefore, the rate and depth of hypothermia, induced by TRPV1 activation, may be enhanced by increasing heat loss through ambient physical cooling.
A number of additional favorable pharmacological properties further augment the practical value of TRPV1 agonists in achieving hypothermia. Importantly, it is possible to continuously deliver the agonists via intravenous or subcutaneous infusion routes, producing a long-lasting (at least 6-12 hours) and stable decrease of core temperature with rapid onset and recovery of normal temperature after discontinuation of drug delivery.71,91,92 In addition, the hypothermic effect is sufficiently large to reach the clinically relevant range and dependent on the agonist dose, which potentially allows fine control over the depth of hypothermia.71,91 Furthermore, the hypothermic effect was reproduced in a number of species, including mice, rats, monkeys and cattle, and showed minimal desensitization.91,92 Taking advantage of these favorable properties, 2 studies have demonstrated that the TRPV1 agonists dihydrocapsaicin and rinvanil decreased infarct size and improved neurological recovery in 2 different mouse models of acute ischemic stroke (Fig. 4).92,93 These findings serve as a proof-of-principle for the use of TRP channel modulators for therapeutic hypothermia.
Figure 4.

Hypothermia induced by the TRPV1 agonist dihydrocapsaicin provides neuroprotection in a cerebral ischemia and reperfusion mouse model of acute ischemic stroke. Focal cerebral ischemia in mice was induced by distal middle cerebral artery occlusion for 1 h, followed by 24 h reperfusion. Dihydrocapsaicin at 1.25 mg·kg−1·h−1 or vehicle were infused by subcutaneously implanted osmotic pumps (∼6 μl/h) from the onset of reperfusion until 10 h after reperfusion. In the “DHC (hypothermia)” (n=8) and “Vehicle” (n=10) groups, mice were kept in a warm cage only during the first 90 min of reperfusion. In the “DHC (normothermia)” group (n=6), mice were kept in a warm cage for 10 h to avoid the temperature drop, as a control for hypothermia-independent effects of dihydrocapsaicin. (A) Core temperature of mice during the initial 4 h of reperfusion. The two dashed lines demarcate the conventional therapeutic range of hypothermia (32–34°C). (B) Representative 2,3,5-triphenyltetrazolium chloride (TTC)-stained histological brain sections, (C) fractional infarct volume, and (D) scores in a panel of behavioral tests in dihydrocapsaicin- and vehicle-treated mice at 24 h reperfusion (*P < 0.05, **P < 0.001). Adapted from Cao et al. 92 © The American Physiological Society. Permission to reuse must be obtained from the rightsholders.
At the same time, despite the significant promise of TRPV1 agonists in inducing hypothermia in animal models, some aspects of targeting TRPV1 still create major obstacles for the clinical use of these agents in humans. TRPV1 was found to be expressed and functionally involved not only in thermoregulatory pathways, but also in a plethora of other physiological functions.94 Therefore, TRPV1 activation may lead to numerous “off-target” effects. Perhaps the most salient side effect is related to TRPV1 expression in nociceptors, a subset of neurons responsible for triggering pain.95 Accordingly, TRPV1 agonists, such as capsaicin, have the potential to cause, alongside the beneficial hypothermic effect on the thermoregulatory system, a conscious experience of burning pain, such as the one caused by eating chili peppers. Indeed, in addition to the widely familiar culinary experience, burning pain was reported after topical, intradermal and intramuscular administration of capsaicin in humans.96,97
However, a number of potential solutions to this problem may be foreseen. First, the induction of pain depends on the route of agonist administration 95 and may be mitigated with systemic delivery, most often associated with the hypothermic effect. Experiments in animal models reveal some behavioral changes and signs of discomfort upon intravenous infusion of dihydrocapsaicin, but are inconclusive due to inherent difficulties of assessing pain in animals.91 Because human studies have not been attempted so far, it is still unclear whether systemic administration of TRPV1 agonists in humans induces pain to the same extent as local application does. Taking this idea further, pain- and hypothermia-triggering effects of TRPV1 agonists are likely mediated by different populations of TRPV1-expressing neurons with different anatomical localizations.68 Therefore, a method of targeting exclusively those channels involved in thermoregulation could be developed in order to bypass or minimize the activation of any pain pathways.
Finally, even if TRPV1-induced pain is unavoidable, it may be managed with analgesic drugs, such as opioids. In fact, this strategy was successfully employed in the clinically approved method of using the high-dose capsaicin patch in the treatment of neuropathic pain, which faced similar challenges.97 It should be pointed out that numerous drugs also cause problematic side effects, but still provide favorable benefit-to-risk ratio and improve patient outcomes. Overall, although considerable further study is needed to determine the feasibility of using TRPV1 agonists as hypothermic agents in human patients, their dramatic hypothermic effect makes it seem worthwhile to continue basic and translational research in this direction even in the face of these challenges.
Targeting other TRP channels
Besides TRPM8 and TRPV1, other thermoTRP channels have also been implicated in thermoregulation, which suggests that modulation of their activity may similarly allow controlling thermoeffectors and altering core temperature. Although evidence for this so far is either lacking or controversial, further research is still warranted to resolve the existing issues.
For example, TRPA1 is another cold-sensitive channel, which shares some properties with TRPM8. It is activated by temperatures below ∼17°C, is expressed in sensory neurons and is required for avoidance behaviors in response to painfully low temperatures.98,99 These findings have initially suggested that, by analogy with TRPM8, TRPA1 may be responsible for autonomic cold defense responses and that its inhibition may be utilized to enable a therapeutic drop in core temperature during physical cooling. However, although the physiological role of TRPA1 was the subject of considerable controversy in the literature, a recent comprehensive study has convincingly demonstrated that pharmacological inhibition of TRPA1 has no effect on thermoeffector activity and core temperature,100 and thus does not appear to be useful in the context of hypothermia.
Warm-sensitive thermoTRPs, in addition to TRPV1, also include closely related members TRPV2,101 TRPV3102 and TRPV4.103 These channels are activated by above-neutral temperatures with distinct thresholds (52, 33 and 27°C respectively), and are expressed in primary sensory neurons as well as in skin keratinocytes, which may signal to sensory neurons. Based on these properties, similarly to TRPV1, these warm-sensitive thermoTRPs were initially thought to be responsible for thermosensation, and thus were likely candidates for mediating afferent inputs to the thermoregulatory system. However, studies in knockout animals failed to reach definitive conclusions about their roles as thermosensors, providing both supporting and opposing evidence.104-107 Nevertheless, interpretation of these studies is complicated by limitations associated with the knock-out technology, such as an influence of genetic background and the possibility of developmental compensation. Moreover, the involvement of these channels in autonomic thermoregulatory responses, as opposed to behavioral responses, has not been thoroughly explored. Finally, even if it had been definitively established that warm-sensitive themoTRPs did not serve as thermosensors, it still would not preclude the possibility that they are wired to and affect the activity of the central thermoregulatory neurons. One way to test such a possibility involves determining the specific effects of channel modulation on core temperature. However, this is complicated by the absence of specific pharmacological agents suitable for in vivo administration.108 Presently only one such study has been reported, which found no effect of intragastric administration of the TRPV3 agonists thymol and ethyl vannilin on core temperature of mice.109 However, the use of only a single agonist dose delivered by only an intragastric route precludes making definitive conclusions based on this study. Therefore, whether modulation of other warm-sensitive TRP channels has the potential of inducing hypothermia is not fully resolved at this point and may become a focus of future investigations.
It should also be noted that while this review was focused on pharmacological modulators of the TRP channels for hypothermia induction, a number of other agents have been shown to affect body temperature and have been proposed for therapeutic hypothermia, including agonists for neurotensin, dopamine, serotonin and adenosine receptors.110-113 Without a doubt, these agents have an important place in the general framework described herein and present more opportunities for controlling thermoregulation.
Conclusions
Induced hypothermia has proven to be an exceptionally powerful means to preserve brain function in conditions of hypoxia and promises to find an increasingly wider application in clinics. However, body temperature is regulated by an incredibly complex array of physical and biological processes. Therapeutic modulation of core temperature critically relies on the ability to finely tune these processes. As the study of thermoregulation has entered the molecular era, the discovery of novel specific and pharmacologically accessible molecular targets may provide just that possibility, and therein pave the way for the promising field of “thermopharmacology,” aptly named so by one of its pioneers.31,69 ThermoTRP channels represent a considerable advance in this regard, but they have important limitations that still stand in the way of producing safe and effective therapeutic hypothermia in human patients.
The fascinating advances in science and technology allow us to transcend the natural homeostatic capabilities of the human body and induce states and conditions that offer unprecedented resistance to pathological processes. However, with great power comes great responsibility. As these powerful tools increasingly enter clinical practice, it is incumbent upon us to have a deep and comprehensive understanding of their effects on physiology and interactions with the body's natural defenses. Therefore, further research is warranted and highly needed either to circumvent the limitations of the current strategies, or to discover entirely novel approaches to therapeutic temperature modulation.
Biographies
Viktor Feketa completed an MD degree at Uzhhorod National University, Ukraine, prior to entering the PhD program in Molecular Physiology & Biophysics, Cardiovascular Sciences Track at Baylor College of Medicine, Houston, TX. He is currently a PhD candidate in the laboratory of Dr. Sean Marrelli, where he is investigating potential pharmacological strategies to induce therapeutic hypothermia.

Sean Marrelli is currently an Associate Professor in the Department of Anesthesiology at Baylor College of Medicine and is the director of the Cardiovascular Sciences Track in the Molecular Physiology & Biophysics graduate program. Research in his laboratory focuses on 2 main areas involving TRP channels: 1) the role of TRP channels in endothelial control of vasodilation and cerebral blood flow and 2) the use of TRP channel modulators to promote hypothermia and neuroprotection following stroke.

Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are disclosed.
Acknowledgments
We thank Dr. Christopher Flores for critically reading the manuscript.
Funding
The studies in Dr. Marrelli's laboratory were supported by grants from the National Institute of Neurological Disorders and Stroke and the National Heart, Lung, and Blood Institute (R01HL088435, R21NS077413, and R21NS090129 to SPM).
References
- 1.Morrison PR, Ryser FA.. Weight and Body Temperature in Mammals. Science 1952; 116:231-2; PMID:17770935; http://dx.doi.org/ 10.1126/science.116.3009.231 [DOI] [PubMed] [Google Scholar]
- 2.Holzer M, Bernard SA, Hachimi-Idrissi S, Roine RO, Sterz F, Müllner M.. Hypothermia for neuroprotection after cardiac arrest: systematic review and individual patient data meta-analysis. Crit Care Med 2005; 33:414-18; PMID:15699847; http://dx.doi.org/ 10.1097/01.CCM.0000153410.87750.53 [DOI] [PubMed] [Google Scholar]
- 3.Van der Worp HB, Sena ES, Donnan GA, Howells DW, Macleod MR.. Hypothermia in animal models of acute ischaemic stroke: a systematic review and meta-analysis. Brain 2007; 130:3063-74; PMID:17478443; http://dx.doi.org/ 10.1093/brain/awm083 [DOI] [PubMed] [Google Scholar]
- 4.Fox JL, Vu EN, Doyle-Waters M, Brubacher JR, Abu-Laban R, Hu Z.. Prophylactic hypothermia for traumatic brain injury: A quantitative systematic review. Can J Emerg Med 2010; 12:355-364 [DOI] [PubMed] [Google Scholar]
- 5.Kelly FE, Nolan JP.. The effects of mild induced hypothermia on the myocardium: A systematic review. Anaesthesia 2010; 65:505-15; PMID:20151956; http://dx.doi.org/ 10.1111/j.1365-2044.2009.06237.x [DOI] [PubMed] [Google Scholar]
- 6.Scolletta S, Taccone F, Nordberg P, Donadello K, Vincent J-L, Castren M.. Intra-arrest hypothermia during cardiac arrest: a systematic review. Crit Care 2012; 16:R41; PMID:22397519; http://dx.doi.org/ 10.1186/cc11235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang H, Olivero W, Wang D, Lanzino G.. Cold as a therapeutic agent. Acta Neurochir (Wien) 2006; 148:565-70;; PMID:16489500; http://dx.doi.org/ 10.1007/s00701-006-0747-z [DOI] [PubMed] [Google Scholar]
- 8.Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloeck WGJ, Billi J, Böttiger BW, Okada K, Reyes C, Shuster M, et al.. Therapeutic hypothermia after cardiac arrest: an advisory statement by the advanced life support task force of the International Liaison Committee on Resuscitation. Circulation 2003; 108:118-21; PMID:12847056; http://dx.doi.org/ 10.1161/01.CIR.0000079019.02601.90 [DOI] [PubMed] [Google Scholar]
- 9.2005 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care . Circulation 2005; 112:IV-1-5; PMID:16314375 [DOI] [PubMed] [Google Scholar]
- 10.Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K.. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 2002; 346:557-63; PMID:11856794; http://dx.doi.org/ 10.1056/NEJMoa003289 [DOI] [PubMed] [Google Scholar]
- 11.The hypothermia after cardiac arrest study group . Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002; 346:549-56; PMID:11856793; http://dx.doi.org/ 10.1056/NEJMoa012689 [DOI] [PubMed] [Google Scholar]
- 12.Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, Fanaroff AA, Poole WK, Wright LL, Higgins RD, et al.. Whole-body hypothermia for neonates with hypoxic-ischemic encephalopathy. N Engl J Med 2005; 353:1574-84; PMID:16221780; http://dx.doi.org/ 10.1056/NEJMcps050929 [DOI] [PubMed] [Google Scholar]
- 13.Azzopardi D V, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, et al.. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 2009; 361:1349-58; PMID:19797281; http://dx.doi.org/ 10.1056/NEJMoa0900854 [DOI] [PubMed] [Google Scholar]
- 14.Andrews PJD, Sinclair HL, Battison CG, Polderman KH, Citerio G, Mascia L, Harris BA, Murray GD, Stocchetti N, Menon DK, et al.. European society of intensive care medicine study of therapeutic hypothermia (32-35°C) for intracranial pressure reduction after traumatic brain injury (the Eurotherm3235Trial). Trials 2011; 12:8; PMID:21226939; http://dx.doi.org/ 10.1186/1745-6215-12-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lyden PD, Hemmen TM, Grotta J, Rapp K, Raman R.. Endovascular therapeutic hypothermia for acute ischemic stroke: ICTuS 2/3 protocol. Int J Stroke 2014; 9:117-25; PMID:24206528; http://dx.doi.org/ 10.1111/ijs.12151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van der Worp HB, Macleod MR, Bath PMW, Demotes J, Durand-Zaleski I, Gebhardt B, Gluud C, Kollmar R, Krieger DW, Lees KR, et al.. EuroHYP-1: European multicenter, randomized, phase III clinical trial of therapeutic hypothermia plus best medical treatment vs. best medical treatment alone for acute ischemic stroke. Int J Stroke 2014; 9:642-5; PMID:24828363; http://dx.doi.org/ 10.1111/ijs.12294 [DOI] [PubMed] [Google Scholar]
- 17.Dixon SR, Whitbourn RJ, Dae MW, Grube E, Sherman W, Schaer GL, Jenkins JS, Baim DS, Gibbons RJ, Kuntz RE, et al.. Induction of mild systemic hypothermia with endovascular cooling during primary percutaneous coronary intervention for acute myocardial infarction. J Am Coll Cardiol 2002; 40:1928-34; PMID:12475451; http://dx.doi.org/ 10.1016/S0735-1097(02)02567-6 [DOI] [PubMed] [Google Scholar]
- 18.Karnatovskaia L V, Wartenberg KE, Freeman WD.. Therapeutic hypothermia for neuroprotection: history, mechanisms, risks, and clinical applications. The Neurohospitalist 2014; 4:153-63; PMID:24982721; http://dx.doi.org/ 10.1177/1941874413519802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holzer M. Targeted temperature management for comatose survivors of cardiac arrest. N Engl J Med 2010; 363:1256-64; PMID:20860507; http://dx.doi.org/ 10.1056/NEJMct1002402 [DOI] [PubMed] [Google Scholar]
- 20.Sterz F, Safar P, Tisherman S, Radovsky A, Kuboyama K, Oku K.. Mild hypothermic cardiopulmonary resuscitation improves outcome after prolonged cardiac arrest in dogs. Crit Care Med 1991; 19:379-89; PMID:1999100; http://dx.doi.org/ 10.1097/00003246-199103000-00017 [DOI] [PubMed] [Google Scholar]
- 21.Colbourne F, Corbett D.. Delayed and prolonged post-ischemic hypothermia is neuroprotective in the gerbil. Brain Res 1994; 654:265-72; PMID:7987676; http://dx.doi.org/ 10.1016/0006-8993(94)90488-X [DOI] [PubMed] [Google Scholar]
- 22.Logue ES, McMichael MJ, Callaway CW.. Comparison of the effects of hypothermia at 33 degrees C or 35 degrees C after cardiac arrest in rats. Acad Emerg Med 2007; 14:293-300; PMID:17296802; http://dx.doi.org/ 10.1111/j.1553-2712.2007.tb02010.x [DOI] [PubMed] [Google Scholar]
- 23.Nielsen N, Wetterslev J, Cronberg T, Erlinge D, Gasche Y, Hassager C, Horn J, Hovdenes J, Kjaergaard J, Kuiper M, et al.. Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med 2013; 369:2197-206; PMID:24237006; http://dx.doi.org/ 10.1056/NEJMoa1310519 [DOI] [PubMed] [Google Scholar]
- 24.Kuboyama K, Safar P, Radovsky A, Tisherman SA, Stezoski SW, Alexander H.. Delay in cooling negates the beneficial effect of mild resuscitative cerebral hypothermia after cardiac arrest in dogs: a prospective, randomized study. Crit Care Med 1993; 21:1348-58; PMID:8370299; http://dx.doi.org/ 10.1097/00003246-199309000-00019 [DOI] [PubMed] [Google Scholar]
- 25.Bernard SA, Smith K, Cameron P, Masci K, Taylor DM, Cooper DJ, Kelly A-M, Silvester W.. Induction of therapeutic hypothermia by paramedics after resuscitation from out-of-hospital ventricular fibrillation cardiac arrest: a randomized controlled trial. Circulation 2010; 122:737-42; PMID:20679551; http://dx.doi.org/ 10.1161/CIRCULATIONAHA.109.906859 [DOI] [PubMed] [Google Scholar]
- 26.Kim F, Nichol G, Maynard C, Hallstrom A, Kudenchuk PJ, Rea T, Copass MK, Carlbom D, Deem S, Longstreth WT, et al.. Effect of prehospital induction of mild hypothermia on survival and neurological status among adults with cardiac arrest: a randomized clinical trial. JAMA 2014; 311:45-52; PMID:24240712; http://dx.doi.org/ 10.1001/jama.2013.282173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke A, Rothery P.. Scaling of body temperature in mammals and birds. Funct Ecol 2007; 22: 58–67; http://dx.doi.org/ 10.1111/j.1365-2435.2007.01341.x [DOI] [Google Scholar]
- 28.Ruben J. The evolution of endothermy in mammals and birds: from physiology to fossils. Annu Rev Physiol 1995; 57:69-95; PMID:7778882; http://dx.doi.org/ 10.1146/annurev.ph.57.030195.000441 [DOI] [PubMed] [Google Scholar]
- 29.Kleiber M. The fire of life: an introduction to animal energetics Rev. Huntington, NY: R. E. Krieger Pub. Co, 1975 [Google Scholar]
- 30.Elia M. Organ and tissue contribution to metabolic rate In: Energy metabolism: tissue determinants and cellular corollaries., edited by Kinney J, Tucker H.. New York, NY: Raven Press, 1992, p. 61-80. [Google Scholar]
- 31.Romanovsky AA. Skin temperature: its role in thermoregulation. Acta Physiol 2014; 210:498-507; http://dx.doi.org/ 10.1111/apha.12231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsukawa T, Sessler DI, Sessler AM, Schroeder M, Ozaki M, Kurz A, Cheng C.. Heat flow and distribution during induction of general anesthesia. Anesthesiology 1995; 82:662-73; PMID:7879935; http://dx.doi.org/ 10.1097/00000542-199503000-00008 [DOI] [PubMed] [Google Scholar]
- 33.Nedergaard J, Cannon B.. Mammalian hibernation. Philos Trans R Soc Lond B Biol Sci 1990; 326:669-85, discussion 685-6; PMID:1969651; http://dx.doi.org/ 10.1098/rstb.1990.0038 [DOI] [PubMed] [Google Scholar]
- 34.Tøien Ø, Blake J, Edgar DM, Grahn DA, Heller HC, Barnes BM.. Hibernation in black bears: independence of metabolic suppression from body temperature. Science 2011; 331:906-9; PMID:21330544; http://dx.doi.org/ 10.1126/science.1199435 [DOI] [PubMed] [Google Scholar]
- 35.Lay C, Badjatia N.. Therapeutic hypothermia after cardiac arrest. Curr Atheroscler Rep 2010; 12:336-42; PMID:20589482; http://dx.doi.org/ 10.1007/s11883-010-0119-2 [DOI] [PubMed] [Google Scholar]
- 36.Seder DB, Van der Kloot TE.. Methods of cooling: practical aspects of therapeutic temperature management. Crit Care Med 2009; 37:S211-22; PMID:19535949; http://dx.doi.org/ 10.1097/CCM.0b013e3181aa5bad [DOI] [PubMed] [Google Scholar]
- 37.Haugk M, Sterz F, Grassberger M, Uray T, Kliegel A, Janata A, Richling N, Herkner H, Laggner AN.. Feasibility and efficacy of a new non-invasive surface cooling device in post-resuscitation intensive care medicine. Resuscitation 2007; 75:76-81; PMID:17462808; http://dx.doi.org/ 10.1016/j.resuscitation.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 38.Uray T, Malzer R.. Out-of-hospital surface cooling to induce mild hypothermia in human cardiac arrest: a feasibility trial. Resuscitation 2008; 77:331-8; PMID:18314248; http://dx.doi.org/ 10.1016/j.resuscitation.2008.01.005 [DOI] [PubMed] [Google Scholar]
- 39.Tommasi E, Lazzeri C, Bernardo P, Sori A, Chiostri M, Gensini GF, Valente S.. Cooling techniques in mild hypothermia after cardiac arrest. J Cardiovasc Med 2014; http://dx.doi.org/ 10.2459/JCM.0000000000000130 [DOI] [PubMed] [Google Scholar]
- 40.Holzer M, Müllner M, Sterz F, Robak O, Kliegel A, Losert H, Sodeck G, Uray T, Zeiner A, Laggner AN.. Efficacy and safety of endovascular cooling after cardiac arrest: cohort study and Bayesian approach. Stroke 2006; 37:1792-7; PMID:16763179; http://dx.doi.org/ 10.1161/01.STR.0000227265.52763.16 [DOI] [PubMed] [Google Scholar]
- 41.Badjatia N. Celsius Control system. Neurocrit Care 2004; 1:201-3; PMID:16174915; http://dx.doi.org/ 10.1385/NCC:1:2:201 [DOI] [PubMed] [Google Scholar]
- 42.Bernard S, Buist M, Monteiro O, Smith K.. Induced hypothermia using large volume, ice-cold intravenous fluid in comatose survivors of out-of-hospital cardiac arrest: a preliminary report. Resuscitation 2003; 56:9-13; PMID:12505732; http://dx.doi.org/ 10.1016/S0300-9572(02)00276-9 [DOI] [PubMed] [Google Scholar]
- 43.Hardy JD. Physiology of temperature regulation. Physiol Rev 1961; 41:521-606; PMID:13711528 [DOI] [PubMed] [Google Scholar]
- 44.Hensel H. Neural processes in thermoregulation. Physiol Rev 1973; 53:948-1017; PMID:4355518 [DOI] [PubMed] [Google Scholar]
- 45.Romanovsky AA. Thermoregulation: some concepts have changed. Functional architecture of the thermoregulatory system. Am J Physiol Regul Integr Comp Physiol 2007; 292:R37-46; PMID:17008453; http://dx.doi.org/ 10.1152/ajpregu.00668.2006 [DOI] [PubMed] [Google Scholar]
- 46.Nakamura K. Central circuitries for body temperature regulation and fever. Am J Physiol Regul Integr Comp Physiol 2011; 301:R1207-28; PMID:21900642; http://dx.doi.org/ 10.1152/ajpregu.00109.2011 [DOI] [PubMed] [Google Scholar]
- 47.Iampietro PF, Bass DE, Buskirk ER.. Heat exchanges of nude men in the cold: effect of humidity, temperature and wind-speed. J Appl Physiol 1958; 12:351-6; PMID:13525291 [DOI] [PubMed] [Google Scholar]
- 48.Adolph EF, Molnar GW.. Exchanges of heat and tolerances to cold in men exposed to outdoor weather. Am J Physiol 1946; 146:507-37; PMID:20992035 [DOI] [PubMed] [Google Scholar]
- 49.Moore TM, Callaway CW, Hostler D.. Core temperature cooling in healthy volunteers after rapid intravenous infusion of cold and room temperature saline solution. Ann Emerg Med 2008; 51:153-9; PMID:18045737; http://dx.doi.org/ 10.1016/j.annemergmed.2007.07.012 [DOI] [PubMed] [Google Scholar]
- 50.Behnke AR, Yaglou CP.. Physiological responses of men to chilling in ice water and to slow and fast rewarming. J Appl Physiol 1951; 3:591-602; PMID:14824042 [DOI] [PubMed] [Google Scholar]
- 51.De Witte J, Sessler DI.. Perioperative shivering: physiology and pharmacology. Anesthesiology 2002; 96:467-84; PMID:11818783; http://dx.doi.org/ 10.1097/00000542-200202000-00036 [DOI] [PubMed] [Google Scholar]
- 52.Sessler DI. Defeating normal thermoregulatory defenses: induction of therapeutic hypothermia. Stroke 2009; 40:e614-21; PMID:19679849; http://dx.doi.org/ 10.1161/STROKEAHA.108.520858 [DOI] [PubMed] [Google Scholar]
- 53.Kurz A, Ikeda T, Sessler DI, Larson MD, Bjorksten AR, Dechert M, Christensen R.. Meperidine decreases the shivering threshold twice as much as the vasoconstriction threshold. Anesthesiology 1997; 86:1046-54; PMID:9158353; http://dx.doi.org/ 10.1097/00000542-199705000-00007 [DOI] [PubMed] [Google Scholar]
- 54.Mokhtarani M, Mahgoub AN, Morioka N, Doufas AG, Dae M, Shaughnessy TE, Bjorksten AR, Sessler DI.. Buspirone and meperidine synergistically reduce the shivering threshold. Anesth Analg 2001; 93:1233-9; PMID:11682404; http://dx.doi.org/ 10.1097/00000539-200111000-00038 [DOI] [PubMed] [Google Scholar]
- 55.Lyden PD, Allgren RL, Ng K, Akins P, Meyer B, Al-Sanani F, Lutsep H, Dobak J, Matsubara BS, Zivin J.. Intravascular Cooling in the Treatment of Stroke (ICTuS): early clinical experience. J Stroke Cerebrovasc Dis 2005; 14:107-14; PMID:17904009; http://dx.doi.org/ 10.1016/j.jstrokecerebrovasdis.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 56.Mahmood MA, Zweifler RM.. Progress in shivering control. J Neurol Sci 2007; 261:47-54; PMID:17512551; http://dx.doi.org/ 10.1016/j.jns.2007.04.038 [DOI] [PubMed] [Google Scholar]
- 57.Wu T-C, Grotta JC.. Hypothermia for acute ischaemic stroke. Lancet Neurol 2013; 12:275-84; PMID:23415567; http://dx.doi.org/ 10.1016/S1474-4422(13)70013-9 [DOI] [PubMed] [Google Scholar]
- 58.Osaka T. Thermogenesis elicited by skin cooling in anaesthetized rats: lack of contribution of the cerebral cortex. J Physiol 2004; 555:503-13; PMID:14578483; http://dx.doi.org/ 10.1113/jphysiol.2003.053215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nakamura K, Morrison SF.. Central efferent pathways mediating skin cooling-evoked sympathetic thermogenesis in brown adipose tissue. Am J Physiol Regul Integr Comp Physiol 2007; 292:R127-36; PMID:16931649; http://dx.doi.org/ 10.1152/ajpregu.00427.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ootsuka Y, Blessing WW, McAllen RM.. Inhibition of rostral medullary raphé neurons prevents cold-induced activity in sympathetic nerves to rat tail and rabbit ear arteries. Neurosci Lett 2004; 357:58-62; PMID:15036613; http://dx.doi.org/ 10.1016/j.neulet.2003.11.067 [DOI] [PubMed] [Google Scholar]
- 61.Rathner JA, Madden CJ, Morrison SF.. Central pathway for spontaneous and prostaglandin E2-evoked cutaneous vasoconstriction. Am J Physiol Regul Integr Comp Physiol 2008; 295:R343-54; PMID:18463193; http://dx.doi.org/ 10.1152/ajpregu.00115.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakamura K, Morrison SF.. Central efferent pathways for cold-defensive and febrile shivering. J Physiol 2011; 589:3641-58; PMID:21610139; http://dx.doi.org/ 10.1113/jphysiol.2011.210047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hemingway A. Shivering. Physiol Rev 1963; 43:397-422; PMID:13953667 [DOI] [PubMed] [Google Scholar]
- 64.Dupuis JY, Nathan HJ, DeLima L, Wynands JE, Russell GN, Bourke M.. Pancuronium or vecuronium for treatment of shivering after cardiac surgery. Anesth Analg 1994; 79:472-81; PMID:7915090 [DOI] [PubMed] [Google Scholar]
- 65.Lee P, Swarbrick MM, Ho KKY.. Brown adipose tissue in adult humans: a metabolic renaissance. Endocr Rev 2013; 34:413-38; PMID:23550082; http://dx.doi.org/ 10.1210/er.2012-1081 [DOI] [PubMed] [Google Scholar]
- 66.Wechselberger M, Wright CL, Bishop GA, Boulant JA.. Ionic channels and conductance-based models for hypothalamic neuronal thermosensitivity. Am J Physiol Regul Integr Comp Physiol 2006; 291:R518-29; PMID:16690776; http://dx.doi.org/ 10.1152/ajpregu.00039.2006 [DOI] [PubMed] [Google Scholar]
- 67.Dhaka A, Viswanath V, Patapoutian A.. Trp ion channels and temperature sensation. Annu Rev Neurosci 2006; 29:135-61; PMID:16776582; http://dx.doi.org/ 10.1146/annurev.neuro.29.051605.112958 [DOI] [PubMed] [Google Scholar]
- 68.Romanovsky AA, Almeida MC, Garami A, Steiner AA, Norman MH, Morrison SF, Nakamura K, Burmeister JJ, Nucci TB.. The transient receptor potential vanilloid-1 channel in thermoregulation: a thermosensor it is not. Pharmacol Rev 2009; 61:228-61; PMID:19749171; http://dx.doi.org/ 10.1124/pr.109.001263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Almeida MC, Hew-Butler T, Soriano RN, Rao S, Wang W, Wang J, Tamayo N, Oliveira DL, Nucci TB, Aryal P, et al.. Pharmacological Blockade of the Cold Receptor TRPM8 Attenuates Autonomic and Behavioral Cold Defenses and Decreases Deep Body Temperature. J Neurosci 2012; 32:2086-99; PMID:22323721; http://dx.doi.org/ 10.1523/JNEUROSCI.5606-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Knowlton WM, Daniels RL, Palkar R, McCoy DD, McKemy DD.. Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One 2011; 6:e25894; PMID:21984952; http://dx.doi.org/ 10.1371/journal.pone.0025894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feketa VV, Zhang Y, Cao Z, Balasubramanian A, Flores CM, Player MR, Marrelli SP.. Transient receptor potential melastatin 8 channel inhibition potentiates the hypothermic response to transient receptor potential vanilloid 1 activation in the conscious mouse. Crit Care Med 2014; 42:e355-63; PMID:24595220; http://dx.doi.org/ 10.1097/CCM.0000000000000229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bautista DM, Siemens J, Glazer JM, Tsuruda PR, Basbaum AI, Stucky CL, Jordt S-E, Julius D.. The menthol receptor TRPM8 is the principal detector of environmental cold. Nature 2007; 448:204-8; PMID:17538622; http://dx.doi.org/ 10.1038/nature05910 [DOI] [PubMed] [Google Scholar]
- 73.Colburn RW, Lubin M Lou, Stone DJ, Wang Y, Lawrence D, D'Andrea MR, Brandt MR, Liu Y, Flores CM, Qin N.. Attenuated Cold Sensitivity in TRPM8 Null Mice. Neuron 2007; 54:379-86; PMID:17481392; http://dx.doi.org/ 10.1016/j.neuron.2007.04.017 [DOI] [PubMed] [Google Scholar]
- 74.Dhaka A, Murray AN, Mathur J, Earley TJ, Petrus MJ, Patapoutian A.. TRPM8 is required for cold sensation in mice. Neuron 2007; 54:371-8; PMID:17481391; http://dx.doi.org/ 10.1016/j.neuron.2007.02.024 [DOI] [PubMed] [Google Scholar]
- 75.McKemy DD, Neuhausser WM, Julius D.. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature 2002; 416:52-8; PMID:11882888; http://dx.doi.org/ 10.1038/nature719 [DOI] [PubMed] [Google Scholar]
- 76.Peier AM, Moqrich A, Hergarden AC, Reeve AJ, Andersson DA, Story GM, Earley TJ, Dragoni I, McIntyre P, Bevan S, et al.. A TRP channel that senses cold stimuli and menthol. Cell 2002; 108:705-15; PMID:11893340; http://dx.doi.org/ 10.1016/S0092-8674(02)00652-9 [DOI] [PubMed] [Google Scholar]
- 77.McCoy DD, Knowlton WM, McKemy DD.. Scraping through the ice: uncovering the role of TRPM8 in cold transduction. Am J Physiol Regul Integr Comp Physiol 2011; 300:R1278-87; PMID:21411765; http://dx.doi.org/ 10.1152/ajpregu.00631.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dhaka A, Earley TJ, Watson J, Patapoutian A.. Visualizing cold spots: TRPM8-expressing sensory neurons and their projections. J Neurosci 2008; 28:566-75; PMID:18199758; http://dx.doi.org/ 10.1523/JNEUROSCI.3976-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tajino K, Hosokawa H, Maegawa S, Matsumura K, Dhaka A, Kobayashi S.. Cooling-sensitive TRPM8 is thermostat of skin temperature against cooling. PLoS One 2011; 6:e17504; PMID:21407809; http://dx.doi.org/ 10.1371/journal.pone.0017504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feketa VV, Balasubramanian A, Flores CM, Player MR, Marrelli SP.. Shivering and tachycardic responses to external cooling in mice are substantially suppressed by TRPV1 activation but not by TRPM8 inhibition. Am J Physiol Regul Integr Comp Physiol 2013; 305:R1040-50; PMID:24005250; http://dx.doi.org/ 10.1152/ajpregu.00296.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Journigan VB, Zaveri NT.. TRPM8 ion channel ligands for new therapeutic applications and as probes to study menthol pharmacology. Life Sci 2013; 92:425-37; PMID:23159643; http://dx.doi.org/ 10.1016/j.lfs.2012.10.032 [DOI] [PubMed] [Google Scholar]
- 82.Cheng C, Matsukawa T, Sessler DI, Ozaki M, Kurz A, Merrifield B, Lin H, Olofsson P.. Increasing mean skin temperature linearly reduces the core-temperature thresholds for vasoconstriction and shivering in humans. Anesthesiology 1995; 82:1160-8; PMID:7741291; http://dx.doi.org/ 10.1097/00000542-199505000-00011 [DOI] [PubMed] [Google Scholar]
- 83.Sweney MT, Sigg DC, Tahvildari S, Iaizzo PA.. Shiver suppression using focal hand warming in unanesthetized normal subjects. Anesthesiology 2001; 95:1089-95; PMID:11684976; http://dx.doi.org/ 10.1097/00000542-200111000-00011 [DOI] [PubMed] [Google Scholar]
- 84.Kimberger O, Ali SZ, Markstaller M, Zmoos S, Lauber R, Hunkeler C, Kurz A.. Meperidine and skin surface warming additively reduce the shivering threshold: a volunteer study. Crit Care 2007; 11:R29; PMID:17316456; http://dx.doi.org/ 10.1186/cc5709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Badjatia N, Strongilis E, Prescutti M, Fernandez L, Fernandez A, Buitrago M, Schmidt JM, Mayer SA.. Metabolic benefits of surface counter warming during therapeutic temperature modulation. Crit Care Med 2009; 37:1893-7; PMID:19384208; http://dx.doi.org/ 10.1097/CCM.0b013e31819fffd3 [DOI] [PubMed] [Google Scholar]
- 86.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D.. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 1997; 389:816-24; PMID:9349813; http://dx.doi.org/ 10.1038/39807 [DOI] [PubMed] [Google Scholar]
- 87.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D.. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998; 21:531-43; PMID:9768840; http://dx.doi.org/ 10.1016/S0896-6273(00)80564-4 [DOI] [PubMed] [Google Scholar]
- 88.Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O'Donnell D, Nicoll RA, Shah NM, Julius D, et al.. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci 2011; 31:5067-77; PMID:21451044; http://dx.doi.org/ 10.1523/JNEUROSCI.6451-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Martins D, Tavares I, Morgado C.. “Hotheaded:” the role OF TRPV1 in brain functions. Neuropharmacology 2014; 85:151-7; PMID:24887171; http://dx.doi.org/ 10.1016/j.neuropharm.2014.05.034 [DOI] [PubMed] [Google Scholar]
- 90.Hori T. Capsaicin and central control of thermoregulation. Pharmacol Ther 1984; 26:389-416; PMID:6085515; http://dx.doi.org/ 10.1016/0163-7258(84)90041-X [DOI] [PubMed] [Google Scholar]
- 91.Fosgerau K, Weber UJ, Gotfredsen JW, Jayatissa M, Buus C, Kristensen NB, Vestergaard M, Teschendorf P, Schneider A, Hansen P, et al.. Drug-induced mild therapeutic hypothermia obtained by administration of a transient receptor potential vanilloid type 1 agonist. BMC Cardiovasc Disord 2010; 10:51; PMID:20932337; http://dx.doi.org/ 10.1186/1471-2261-10-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cao Z, Balasubramanian A, Marrelli SP.. Pharmacologically induced hypothermia via TRPV1 channel agonism provides neuroprotection following ischemic stroke when initiated 90 min after reperfusion. Am J Physiol Regul Integr Comp Physiol 2014; 306:R149-56; PMID:24305062; http://dx.doi.org/ 10.1152/ajpregu.00329.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Muzzi M, Felici R, Cavone L, Gerace E, Minassi A, Appendino G, Moroni F, Chiarugi A.. Ischemic neuroprotection by TRPV1 receptor-induced hypothermia. J Cereb Blood Flow Metab 2012; 32:978-82; PMID:22434066; http://dx.doi.org/ 10.1038/jcbfm.2012.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.White JPM, Urban L, Nagy I.. TRPV1 function in health and disease. Curr Pharm Biotechnol 2011; 12:130-44; PMID:20932253; http://dx.doi.org/ 10.2174/138920111793937844 [DOI] [PubMed] [Google Scholar]
- 95.O'Neill J, Brock C, Olesen AE, Andresen T, Nilsson M, Dickenson AH.. Unravelling the mystery of capsaicin: a tool to understand and treat pain. Pharmacol Rev 2012; 64:939-71; http://dx.doi.org/ 10.1124/pr.112.006163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Witting N, Svensson P, Gottrup H, Arendt-Nielsen L, Jensen TS.. Intramuscular and intradermal injection of capsaicin: a comparison of local and referred pain. Pain 2000; 84:407-12; PMID:10666547; http://dx.doi.org/ 10.1016/S0304-3959(99)00231-6 [DOI] [PubMed] [Google Scholar]
- 97.Simpson DM, Brown S, Tobias J.. Controlled trial of high-concentration capsaicin patch for treatment of painful HIV neuropathy. Neurology 2008; 70:2305-13; PMID:18541884; http://dx.doi.org/ 10.1212/01.wnl.0000314647.35825.9c [DOI] [PubMed] [Google Scholar]
- 98.Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, et al.. ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 2003; 112:819-29; PMID:12654248; http://dx.doi.org/ 10.1016/S0092-8674(03)00158-2 [DOI] [PubMed] [Google Scholar]
- 99.Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B, Voets T.. TRPA1 acts as a cold sensor in vitro and in vivo. Proc Natl Acad Sci U S A 2009; 106:1273-8; PMID:19144922; http://dx.doi.org/ 10.1073/pnas.0808487106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Oliveira C, Garami A, Lehto SG, Pakai E, Tekus V, Pohoczky K, Youngblood BD, Wang W, Kort ME, Kym PR, et al.. Transient receptor potential channel ankyrin-1 is not a cold sensor for autonomic thermoregulation in rodents. J Neurosci 2014; 34:4445-52; PMID:24671991; http://dx.doi.org/ 10.1523/JNEUROSCI.5387-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perálvarez-Marín A, Doñate-Macian P, Gaudet R.. What do we know about the transient receptor potential vanilloid 2 (TRPV2) ion channel? FEBS J 2013; 280:5471-87; http://dx.doi.org/ 10.1111/febs.12302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nilius B, Bíró T.. TRPV3: a “more than skinny” channel. Exp Dermatol 2013; 22:447-52; PMID:23800054; http://dx.doi.org/ 10.1111/exd.12163 [DOI] [PubMed] [Google Scholar]
- 103.Everaerts W, Nilius B, Owsianik G.. The vanilloid transient receptor potential channel TRPV4: from structure to disease. Prog Biophys Mol Biol 2010; 103:2-17; PMID:19835908; http://dx.doi.org/ 10.1016/j.pbiomolbio.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 104.Lee H, Iida T, Mizuno A, Suzuki M, Caterina MJ.. Altered thermal selection behavior in mice lacking transient receptor potential vanilloid 4. J Neurosci 2005; 25:1304-10; PMID:15689568; http://dx.doi.org/ 10.1523/JNEUROSCI.4745.04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KSR, Andahazy M, Story GM, Patapoutian A.. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science 2005; 307:1468-72; PMID:15746429; http://dx.doi.org/ 10.1126/science.1108609 [DOI] [PubMed] [Google Scholar]
- 106.Huang SM, Li X, Yu Y, Wang J, Caterina MJ.. TRPV3 and TRPV4 ion channels are not major contributors to mouse heat sensation. Mol Pain 2011; 7:37; PMID:21586160; http://dx.doi.org/ 10.1186/1744-8069-7-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Park U, Vastani N, Guan Y, Raja SN, Koltzenburg M, Caterina MJ.. TRP vanilloid 2 knockout mice are susceptible to perinatal lethality but display normal thermal and mechanical nociception. J Neurosci 2011; 31:11425-36; PMID:21832173; http://dx.doi.org/ 10.1523/JNEUROSCI.1384-09.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vriens J, Appendino G, Nilius B.. Pharmacology of vanilloid transient receptor potential cation channels. Mol Pharmacol 2009; 75:1262-79; PMID:19297520; http://dx.doi.org/ 10.1124/mol.109.055624 [DOI] [PubMed] [Google Scholar]
- 109.Masamoto Y, Kawabata F, Fushiki T.. Intragastric Administration of TRPV1, TRPV3, TRPM8, and TRPA1 Agonists Modulates Autonomic Thermoregulation in Different Manners in Mice. Biosci Biotechnol Biochem 2009; 73:1021-7; PMID:19420725; http://dx.doi.org/ 10.1271/bbb.80796 [DOI] [PubMed] [Google Scholar]
- 110.Choi K-E, Hall CL, Sun J-M, Wei L, Mohamad O, Dix TA, Yu SP.. A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. FASEB J 2012; 26:2799-810; PMID:22459147; http://dx.doi.org/ 10.1096/fj.11-201822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schneider A, Teschendorf P, Vogel P, Russ N, Knapp J, Böttiger BW, Popp E.. Facilitation of hypothermia by quinpirole and 8-OH-DPAT in a rat model of cardiac arrest. Resuscitation 2012; 83:232-7; PMID:21803015; http://dx.doi.org/ 10.1016/j.resuscitation.2011.07.023 [DOI] [PubMed] [Google Scholar]
- 112.Tupone D, Madden CJ, Morrison SF.. Central activation of the A1 adenosine receptor (A1AR) induces a hypothermic, torpor-like state in the rat. J Neurosci 2013; 33:14512-25; PMID:24005302; http://dx.doi.org/ 10.1523/JNEUROSCI.1980-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muzzi M, Blasi F, Chiarugi A.. AMP-dependent hypothermia affords protection from ischemic brain injury. J Cereb Blood Flow Metab 2013; 33:171-4; PMID:23211965; http://dx.doi.org/ 10.1038/jcbfm.2012.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Parks DJ, Parsons WH, Colburn RW, Meegalla SK, Ballentine SK, Illig CR, Qin N, Liu Y, Hutchinson TL, Lubin M Lou, et al.. Design and Optimization of Benzimidazole-Containing Transient Receptor Potential Melastatin 8 (TRPM8) Antagonists. J Med Chem 2010; 8:233-47; http://dx.doi.org/ 10.1021/jm101075v [DOI] [PubMed] [Google Scholar]