

Abstract
The matrix (MA) domain of the HIV-1 Gag is responsible for Gag targeting to the plasma membrane where virions assemble. MA also plays a role in the incorporation of the viral envelope (Env) glycoproteins, and can influence particle infectivity post-maturation and post-entry. A highly basic region of MA targets Gag to the plasma membrane via specific binding to phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2]. This binding also triggers exposure of an amino-terminal myristate moiety, which anchors Gag to the membrane. A MA mutant deficient for PI(4,5)P2 binding, 29KE/31KE, has been shown to mislocalize within the cell, leading to particle assembly in a multivesicular body compartment and defective release of cell-free particles in HeLa and 293T cells. Despite the defect in virus production in these cells, release of the 29KE/31KE mutant is not significantly reduced in primary T cells, macrophages, and Jurkat T cells. 29KE/31KE virions also display an infectivity defect associated with impaired Env incorporation, irrespective of the producer cell line. Here we examine the properties of 29KE/31KE by analyzing compensatory mutations obtained by a viral adaptation strategy. The MA mutant 16EK restores virus release through enhanced membrane binding. 16EK also influences the infectivity defect, in combination with an additional MA mutant, 62QR. Additionally, the 29KE/31KE MA mutant displays a defect in proteolytic cleavage of the murine leukemia virus Env cytoplasmic tail in pseudotyped virions. Our findings elucidate the mechanism whereby a MA mutant defective in PI(4,5)P2 binding can be rescued and highlight the ability of MA to influence Env glycoprotein function.
Keywords: Retrovirus, Gag, A-MLV, cytoplasmic tail, pseudotyping
Graphical abstract
INTRODUCTION
Assembly of human immunodeficiency virus type 1 (HIV-1) takes place on cellular membranes, beginning with the accumulation of the Pr55Gag polyprotein (henceforth referred to as Gag) on the cytoplasmic leaflet of the plasma membrane (PM), and finishing when the assembled virion is released from the host cell surface [1]. The primary factor driving assembly of HIV-1 particles is Gag; expression of Gag in the absence of other viral proteins is sufficient to generate virus-like particles (VLPs). Gag is composed of four primary domains, each essential for assembly of an infectious virion. Matrix (MA) directs Gag trafficking to the PM, and plays a role in the incorporation of the viral envelope (Env) glycoproteins. Env is composed of the receptor binding subunit, gp120, and the transmembrane fusogenic subunit, gp41. Capsid (CA) is the domain principally responsible for mediating the Gag-Gag interactions that build the hexameric Gag lattice of the immature virion. The final step in particle assembly is maturation, during which the viral protease (PR) cleaves the immature Gag lattice into the mature protein products; MA, CA, nucleocapsid (NC) and p6. Post-maturation, CA also forms the conical viral core, composed primarily of hexamers, with twelve pentamers, five at the narrow end of the cone, and seven at the broad end [2]. NC is the primary nucleic-acid binding domain, which exhibits a preference for binding to the HIV-1 genomic RNA [3]. RNA binding also promotes long-range multimerization of Gag that is necessary for VLP assembly [4]. The final carboxy (C)-terminal portion of Gag is p6, a small domain that contains the late domains Pro-[Thr/Ser]-Ala-Pro and Tyr-Pro-Xn-Leu, which recruit TSG101 and ALIX, respectively [5]. Binding to either of these host proteins recruits endosomal sorting complex required for transport (ESCRT) components that drive the scission of the budding virus from the host cell membrane.
The MA domain comprises 131 amino acids located at the amino (N)-terminus of Gag, and bears a covalently attached N-terminal myristic acid moiety [6]. In addition to the myristic acid and N-terminal residues, MA contains both a highly basic region between residues 17 and 31, and a region between residues 80 and 90, that contribute to membrane targeting and binding. MA directs Gag to the PM by the specific binding to phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] by a cleft in the basic domain [7–9]. PI(4,5)P2 is a phospholipid found in discrete microdomains on the PM [10]. Non-specific association between the basic domain of MA and negatively charged cellular membranes is inhibited by the binding of MA to cellular RNAs [11,12]. In solution, MA exists as an equilibrium mixture of myristate-sequestered and myristate-exposed conformers, and combinations of sedimentation and NMR experiments indicate that concentration-dependent protein self-association promotes myristate exposure [13]. The binding of MA to PI(4,5)P2 also triggers the myristyl switch [8,14], during which the myristic acid moiety is exposed and inserts into the lipid bilayer, anchoring Gag to the inner leaflet of the PM. Although the MA domain is evolutionarily conserved among all retroviruses, for the purposes of VLP formation, this membrane-targeting function can be replaced by an alternative membrane-binding domain, such as those found in Src or Fyn [7,15]; however, MA has additional functions that are critical for the production of an infectious particle. Residues in the basic domain have been described that reduce infectivity post viral entry [16], and various mutations have been described in MA that can prevent the incorporation of Env into the particle [17–22]. Similarly, Gag molecules that lack MA and use Src to direct trafficking cannot efficiently incorporate the HIV-1 Env unless the Env cytoplasmic tail (CT) is removed [15]. Thus, in the context of full-length HIV-1 Env and biologically relevant cell types, MA places an essential role in virus replication.
Although the unmyristylated MA protein exists in solution as a monomer [23,24], it crystallizes as a trimer [25–27]. The residues known to influence Env incorporation include Leu12, Glu16, Leu30, Val34, Thr69 and Glu98. From the crystal structure, most of these residues are located at the tips of the MA trimer, and the Env-incorporation defects can be rescued by a compensatory mutation, 62QR, near the MA trimer interface [22]. The 62QR mutation also rescues an Env-incorporation defect imposed by a deletion in the gp41 CT [22]. Thr69 is also located near the MA trimer interface, and the Env-incorporation defect imposed by mutation of this residue to Arg was not rescued by 62QR. Taken together, these data suggest that the Env CT is positioned close to the tips of the MA trimer in the Gag lattice, and that the trimeric structure of MA plays a role in the incorporation of Env into particles [28].
Following the incorporation of Env into virions, there is evidence for MA interactions regulating the function of Env [29]. In immature HIV-1 particles, Env is able to bind CD4, but does not efficiently promote membrane fusion [30,31]. This inhibition of fusion can be alleviated by gp41 CT truncation [30,31]. MLV fusogenicity, by contrast, is activated through the maturation of the Env protein [32,33]. The MLV Env CT possesses a C-terminal R peptide, which is required for Env incorporation into particles, but inhibits fusogenicity [32]. During viral maturation the R peptide is removed by PR and the Env becomes fusogenic. When MLV Env is used to pseudotype HIV-1 particles this R peptide cleavage is performed by the HIV-1 PR; however, mutation of MA has been observed to block the processing of MLV Env, thereby preventing functional pseudotyping of HIV-1 particles by MLV Env [34]. The mechanism by which MA mutants can prevent MLV Env processing is unknown, but likely reflects changes in the MA lattice structure that in some way restrict access by PR to the MLV Env CT.
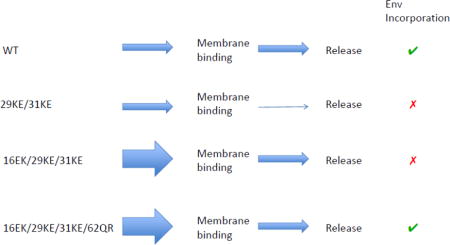
A MA mutant deficient for PI(4,5)P2 binding, 29KE/31KE, has been previously reported [8,9,35,36]. The loss of PI(4,5)P2 binding prevents efficient HIV-1 assembly and release in HeLa and 293T cells, although release in most T-cell lines, peripheral blood mononuclear cells (PBMCs) and monocyte-derived macrophages (MDMs) remains close to WT levels [35,36]. The release defect appears to be due to mislocalization of Gag and assembly of particles on intracellular membranes rather than the PM [8,9,35,36]. In addition, the 29KE/31KE mutant possesses an infectivity defect that can be alleviated by gp41 CT truncation [35]. Exploiting this observation, selection for compensatory mutations to 29KE/31KE was performed in MT4 cells, a T-cell line that is unusual in that it permits replication of CT-truncated HIV-1 [35,37]. The virus recovered was found to have acquired the second-site mutation 16EK. Combined with CT deletion, 16EK/29KE/31KE replicates in MT4 cells to WT levels, and displays approximately WT levels of virus release in HeLa and 293T cells. The 16EK mutant displays greater-than-WT virus release efficiency in HeLa and 293T [35].
In the current study, we demonstrated that the MA mutation 16EK confers enhanced membrane binding, a property that appears to alleviate the release defect of 29KE/31KE in HeLa, 293T and MT4 cells. Additionally, although the 16EK mutant itself is defective for Env incorporation [22], we found that the 16EK substitution contributes to enhancing Env incorporation, when combined with 62QR. Although the contribution of 16EK to the rescue of Env incorporation in 16EK/29KE/31KE/62QR is not understood at this time, it was found to correlate with the ability of PR to cleave the CT of A-MLV Env in pseudotyped HIV-1 particles. Gag processing is unaffected in the MA mutant particles; consequently, these data suggest the formation of an aberrant interaction between MA and the A-MLV Env CT, rather than direct effects on PR function.
RESULTS
16EK enhances 29KE/31KE release by increasing membrane binding
Our first goal was to confirm the phenotypes of the 29KE/31KE, 16EK and 16EK/29KE/31KE mutants (Fig. 1a). It has been reported previously that 29KE/31KE exhibits a virus release defect in HeLa cells [36], while 16EK is very efficiently released, and a triple mutant 16EK/29KE/31KE displays approximately WT virus release efficiency [35]. Our data confirm these observations. In addition to the improved virus release efficiency, it is apparent that 16EK displays very efficient Gag processing, as cells contain very little Pr55Gag despite high levels of intra- and extracellular CA (Fig. 1a). Gag processing occurs during maturation and is largely dependent on membrane binding and immature lattice formation. We therefore examined the membrane-binding properties of the mutants using a membrane flotation assay. WT Gag and 29KE/31KE displayed similar membrane-binding efficiency, but both 16EK and 16EK/29KE/31KE displayed markedly increased membrane binding relative to WT (Fig. 1b). Effective separation of membrane and cytosolic proteins was confirmed by western blot for GAPDH and transferrin receptor, which are cytosolic and membrane proteins, respectively (Fig. 1b).
Figure 1.
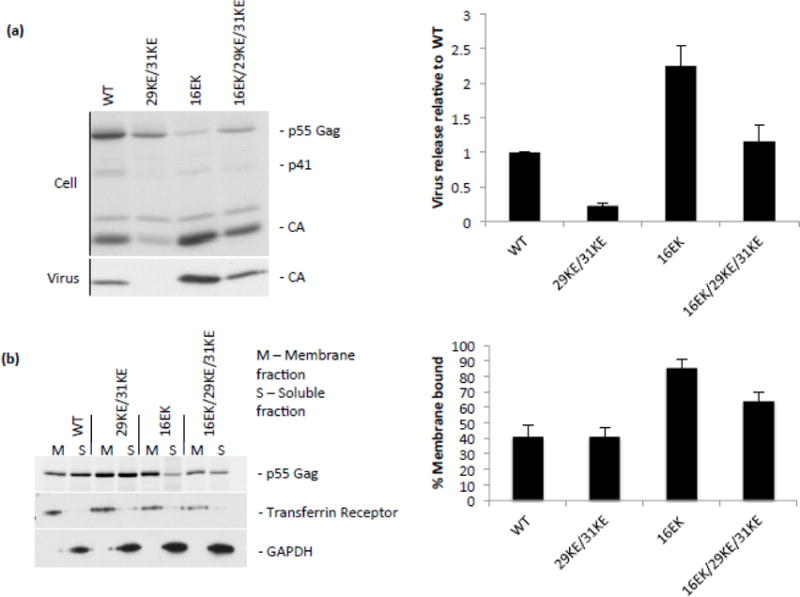
Virus release and membrane binding of 29KE/31KE and compensatory mutants. (a) HeLa cells were transfected with the HIV-1 mutants indicated, labelled with 35S Met/Cys for 3 h, then cell and virus lysates were immunoprecipitated and separated by SDS-PAGE. Samples from three independent experiments were analyzed by fluorography. Virion capsid, expressed as a percentage of total Gag, was plotted, +/− S.E.M.. (b) HeLa cells were transfected with the HIV-1 mutants indicated (in a PR– virus), then labelled with 35S Met/Cys for 20 min. Cells were harvested by scraping, lysed by sonication and lysates placed at the bottom of a sucrose gradient. Following centrifugation, membrane-associated samples were collected from the top of the gradient, and soluble samples from the bottom. Gag was immunoprecipitated from these fractions and resolved by SDS-PAGE. Samples from four independent experiments were analyzed by fluorography. Gag percentage in the membrane-associated fraction was plotted +/− S.E.M. Flotation fractions were also analyzed by western blotting for transferrin receptor and GAPDH.
To determine whether the enhanced membrane association of the 16EK mutant Gag in cells was a result of a shift in the myristyl switch equilibrium or enhanced electrostatic interactions with the PM, we synthesized recombinant WT and 16EK-mutant MA for NMR-based studies. Previous NMR studies revealed a concentration-dependent myristyl switch behavior, in which weak MA self-association (trimerization) in solution is accompanied by a shift in the myristate exposed/sequestered equilibrium toward the myristate-exposed species [13]. Concentration-dependent 1H-15N heteronuclear single quantum coherence (HSQC) NMR reveals that the 16EK mutant exhibits concentration-dependent NMR chemical shift changes similar those observed for WT (Fig. 2a and b), indicating that the enhanced membrane binding of 16EK is not due to a shift towards a myristate-exposed species.
Figure 2.
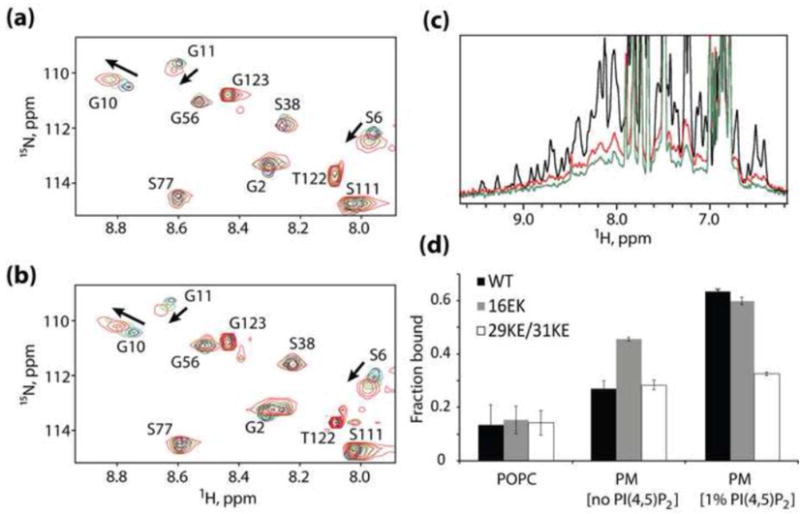
Mechanism of enhanced membrane binding by 16EK. (a,b) Concentration-dependent NMR chemical shift changes observed for residues S6, G10 and G11 in 1H-15N HSQC spectra of the wild-type (a) and 16EK (b) myrMA proteins. Spectral changes reflect a myristyl switch equilibrium switch from myristate-sequestered (low concentration) to myristate-exposed (high concentration) species [13], and indicate that the 16EK mutation does not perturb the myristyl switch. (c) One-dimensional 1H NMR spectra obtained for the myr-16EK mutant under various conditions: in the absence of liposomes (black) and in the presence of membrane mimetic liposomes that lack (red) or contain (green) PI(4,5)P2 (1%). Signal losses correlate with increasing fractions of liposome-bound protein. (d) Fractions of protein bound to POPC liposomes (left) and to membrane mimetic liposomes in the absence (center) and presence (right) of PI(4,5)P2 (1%), as determined by 1D 1H NMR. The wild type, 16EK, and 29KE/31KE proteins bind poorly to POPC liposomes but exhibit differentially enhanced binding to liposomes with PM-like compositions.
We then examined the membrane interactive properties of 16EK and WT MA using a recently developed NMR approach that involves 1H NMR-detected signal losses that accompany titrations with large unilamellar vesicles (LUVs). Studies were conducted with neutral lipid vesicles containing only 1-palmitoyl-2-oleoylphosphatidylcholine (POPC), and with liposomes containing lipid compositions designed to mimic liquid-disordered and raft-like regions of the inner leaflet of the PM [38](Mercredi, PY et al. in preparation). In these studies, binding is measured as a percentage of protein NMR signal loss that accompanies formation of the protein:liposome complex [38]. As shown in Figure 2c, WT, 16EK, and 29KE/31KE MA all exhibit poor affinity for liposomes comprising electrostatically neutral POPC lipids. However, 16EK exhibits significantly higher affinity than either WT or 29KE/31KE for PM-like liposomes that lack PI(4,5)P2 (Fig. 2d). Binding of both 16EK and WT MA to PM-like liposomes is significantly enhanced by the presence of PI(4,5)P2, whereas binding by 29KE/31KE is essentially unaffected by PI(4,5)P2 (Fig. 2d). The NMR studies collectively indicate that the 16EK mutation enhances the binding of MA to negatively charged membranes, while retaining some sensitivity to PI(4,5)P2, thereby explaining the ability of this mutation to enhance Gag membrane binding and virus production in cells. The 29KE/31KE substitutions attenuate the sensitivity of MA to PI(4,5)P2, consistent with a previous report [39].
To determine whether the high membrane binding of 16EK-containing mutants led to faster virus release kinetics, we performed a pulse-chase analysis. HeLa cells were transfected, labeled with 35S-Met/Cys, then chased with unlabeled media for up to four hours (Fig. 3a). A virus with a mutated PTAP late domain was used as a negative control. This mutant, PTAP(−) [40], is highly defective for virus release from the PM. Although the total amount of virus release was reduced by the 29KE/31KE mutations, the shape of the release curve was similar, suggesting that the virus that is released is exiting the cell over a similar time span relative to WT. By contrast, 16EK release peaks far earlier than that of WT, consistent with the highly efficient membrane binding of this mutant. The 16EK/29KE/31KE exhibited slightly slower release kinetics than WT, despite its more efficient membrane binding. It is likely that the previously reported intracellular localization of 16EK/29KE/31KE [35] offsets the more efficient membrane binding, resulting in net release kinetics that are closer to those of WT than to 29KE/31KE. In a longer pulse-chase comparing WT to 29KE/31KE, the release of 29KE/31KE remains low relative to WT even after twenty-four hours (Fig. 3b); if the residual 29KE/31KE Gag detected in cells after four hours was being released slowly, then as no newly labeled Gag is being produced, the longer chase should allow 29KE/31KE to catch up with WT. However, it appears that much of the synthesized 29KE/31KE Gag is never released, even with a long chase (Fig. 3b). This is consistent with the idea that mislocalized Gag is not released from HeLa cells even after long time periods.
Figure 3.
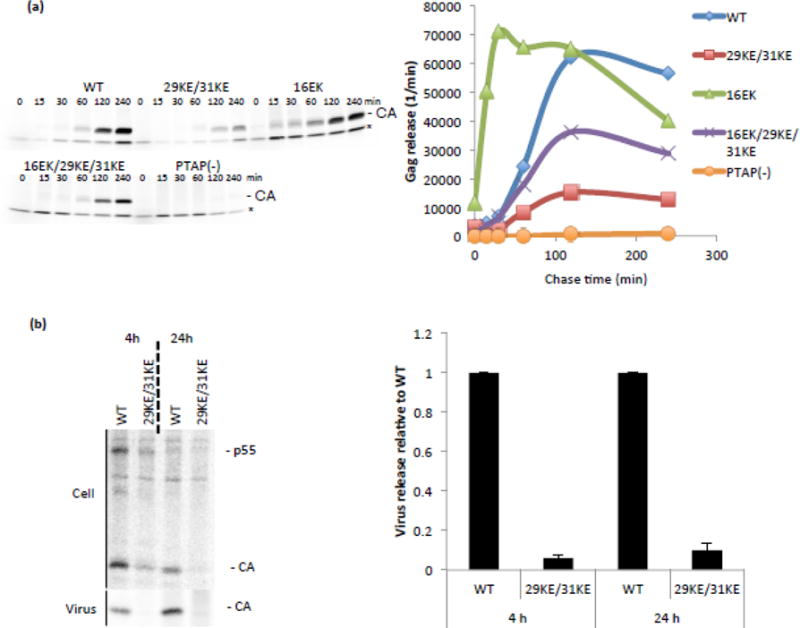
Virus release kinetics. (a) HeLa cells were transfected with the HIV-1 mutants indicated, labelled for 15 minutes with 35S Met/Cys then chased for 240 minutes. At the indicated times, media were replaced and virus harvested. Samples were separated by SDS-PAGE and analyzed by fluorography. For each time point the amount of Gag signal released per minute is plotted. Time courses were performed 4 times and data from a representative experiment is shown. A non-specific band is indicated with *. (b) HeLa cells were transfected with the HIV-1 mutants indicated, labelled with 35S Met/Cys, then cell and virus lysates were immunoprecipitated and separated by SDS-PAGE. Samples from three independent experiments were analyzed by fluorography. Virion capsid, expressed as a percentage of total Gag, was plotted relative to WT, +/− S.E.M.
In the context of HeLa cells, it appears that 16EK is able to rescue 29KE/31KE by promoting efficient membrane binding that offsets the mislocalization of Gag to intracellular membranes, a Gag localization behavior shared by 29KE/31KE and 16EK/29KE/31KE [35].
29KE/31KE is released efficiently from Jurkat but not MT4 T cells
In HeLa cells, 16EK appears to rescue 29KE/31KE by enhancing virus release; however, the replication experiments that selected 16EK as a compensatory mutation for 29KE/31KE were performed in the context of a gp41 CT-truncated virus in MT4 T cells. Based on previously reported results showing efficient release of 29KE/31KE from T cells [35], 29KE/31KE was expected to display release and infectivity levels comparable to those of WT in MT4 T cells; consequently, the nature of the 29KE/31KE defect being repaired by 16EK was unclear. We performed virus release assays with the mutants in both Jurkat and MT4 T-cell lines to verify the effect of 29KE/31KE on virus release (Fig. 4). In Jurkat T cells, all of the mutants are released efficiently; as previously reported, 29KE/31KE displayed similar release efficiency to WT [Fig. 4a and c, and [35]]. By contrast, 29KE/31KE is released less efficiently in MT4 T cells relative to the WT (Fig. 4b and c). Given these data, it is likely that 16EK was selected in MT4 cells because it increased the overall levels of virus released into the supernatant, thereby enhancing virus replication (Fig. 1 and Fig. 4b and c).
Figure 4.
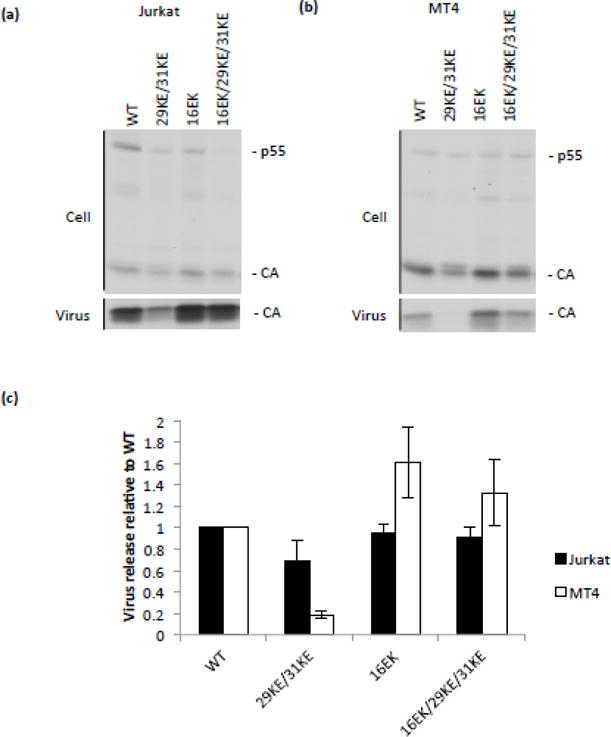
Release of 29KE/31KE from T-cell lines. VSV-G pseudotyped viruses were generated by transfecting 293T cells with the HIV-1 mutants indicated and a VSV-G expression vector. These viruses were normalized by RT assay and used to infect Jurkat and MT4 T-cell lines. 24 h postinfection cells were labelled with 35S Met/Cys for 8 or 16 h, then cell and virus lysates were immunoprecipitated and separated by SDS-PAGE. (a) Jurkat. (b) MT4. (c) Samples from three independent experiments were analyzed by fluorography. Virion capsid, expressed as a percentage of total Gag, was plotted relative to WT, +/− S.E.M.
62QR rescues Env incorporation and restores 16EK/29KE/31KE infectivity and replication
The 16EK MA mutation has previously been described as causing defective Env incorporation, and can be rescued by combination with a second-site mutation, 62QR [22]. The 62QR mutation restores 16EK infectivity in single-cycle experiments and replication in Jurkat cells; based on its distance from the 16EK, 29KE and 31KE, and proximity to the putative MA trimer interface, 62QR is thought to act via influencing the arrangement of the MA lattice (Fig. 5). As the 16EK/29KE/31KE triple mutant displays WT release efficiency but shares the defect in Env incorporation exhibited by 16EK, it is possible that the addition of 62QR to make 16EK/29KE/31KE/62QR would result in recovery of a replication-competent virus. This quadruple mutant was generated and examined for virus release, Env incorporation, infectivity and replication (Fig. 6 and Fig. 7). Although delayed relative to WT, replication of the quadruple 16EK/29KE/31KE/62QR mutant demonstrated a clear improvement in replication relative to 16EK/29KE/31KE (Fig. 6a). Virus collected from the peaks of WT and 16EK/29KE/31KE/62QR replication was used in a second passage. 16EK/29KE/31KE/62QR remained delayed relative to WT, suggesting that it did not acquire further compensatory mutations during the first passage (Fig. 6b). Sequencing of the viral DNA isolated from this 16EK/29KE/31KE/62QR-infected culture confirmed that no additional mutations in MA were present, although an S52R mutation was observed in the β-TrCP binding site in Vpu [41].
Figure 5.
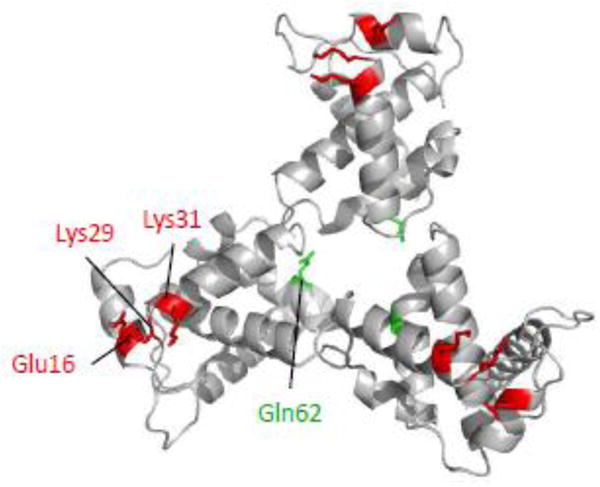
Model of the MA trimer showing residues Glu16, Lys29, Lys31 and Gln62. Model was generated in Pymol [49] based on Protein Data Bank coordinates 1HIW [25]. Gln62 (green) is found near the trimer interface, while the other residues (red) are located at the tips of the trimer.
Figure 6.
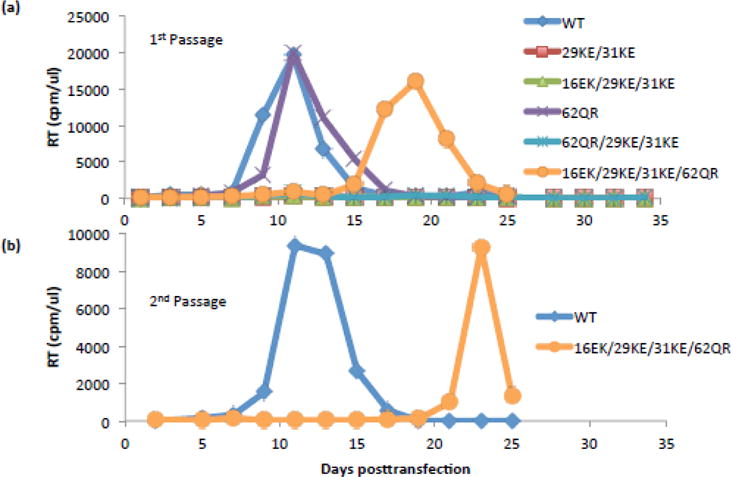
Rescue of 29KE/31KE replication in Jurkats. (a) Jurkat T cells were transfected with the HIV-1 mutants indicated. Samples were taken every two days and the cells split 1:3. Samples were assayed for RT activity. (b) Virus supernatants from the peaks of WT and 16EK/29KE/31KE/62QR were collected, normalized by RT assay and used to reinfect Jurkat T cells for a 2nd passage. Replication assays were performed three times, data from a representative experiment are shown.
Figure 7.
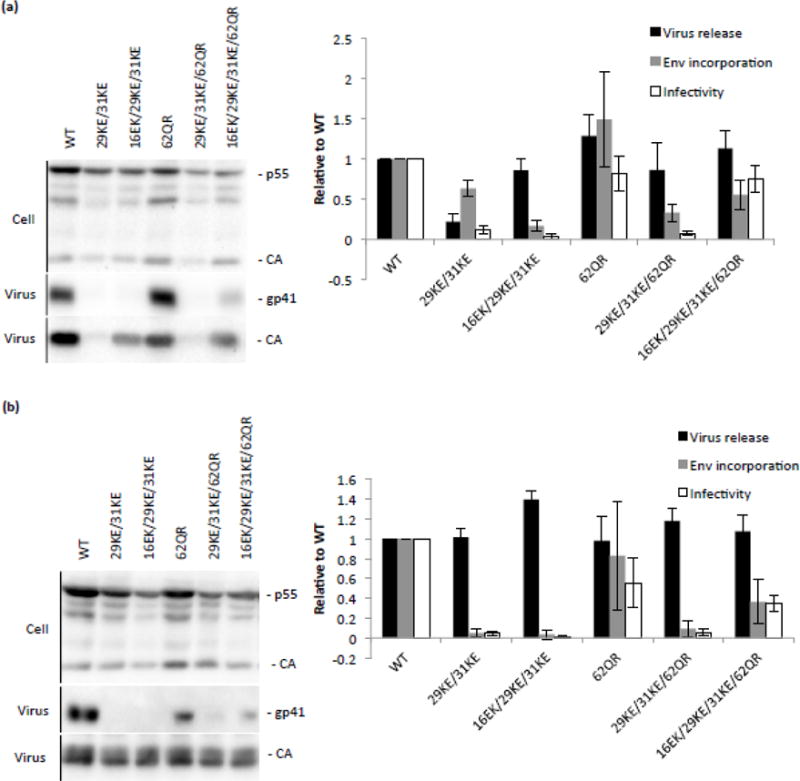
Virus release, Env incorporation and infectivity of 29KE/31KE and rescue mutations in HeLa and Jurkat cell lines. (a) HeLa cells were transfected with the HIV-1 mutants indicated. 48 h posttransfection virus and cell-associated samples were collected, separated by SDS-PAGE and analyzed by western blotting. Virus release was calculated as the amount of virion CA relative to total Gag levels, Env incorporation was expressed as amount of virion gp41 per virion CA. Virus-containing supernatants were used to infect TZM-bl cells; the resulting luciferase signal was normalized to the corresponding RT values to provide a measure of specific infectivity. Averages from four independent experiments are shown, +/− S.E.M. (b) 293T cells were transfected with the mutants indicated and a VSV-G expression vector. 48 h posttransfection supernatant was harvested and virus content determined by RT assay. These virus stocks were used to infect Jurkat T cells. 12 h postinfection the medium was replaced. After a further 48 h, virus- and cell-associated samples were harvested and analyzed as described in (a). Averages from three independent experiments are shown, +/− S.E.M.
To determine the mechanism of rescue, virus release, Env incorporation and infectivity were examined, first in virus produced from HeLa cells (Fig. 7a). In keeping with previously reported data [35,36], 29KE/31KE was defective for release and infectivity, while the addition of 16EK restored virus release but not Env incorporation. The further addition of 62QR yielded a virus that was competent for release, Env incorporation and infectivity, displaying close to WT levels in all three assays. As previously reported [22], 62QR alone performed comparably to WT. It is worth noting that the mutant 29KE/31KE/62QR was not replication competent in Jurkat cells (Fig. 6a), and was not infectious, although the addition of 62QR did appear to increase virus release efficiency (Fig. 7a).
As 29KE/31KE is inefficiently released from HeLa cells, it is difficult to accurately compare Env incorporation levels to viruses that are efficiently released because small amounts of exosome-associated Env could have a disproportionate influence on the calculation. Therefore, to examine the effect of 29KE/31KE on Env incorporation we used virus produced in Jurkat T cells (Fig. 7b); in Jurkat T cells 29KE/31KE displays virus release efficiency comparable to that of WT [Fig. 4 and [35]]. Probing for virion-associated gp41 demonstrated that 29KE/31KE is profoundly impaired for Env incorporation, consistent with the previous finding that the 30LE mutation impairs Env incorporation [18]. The addition of 62QR has been reported to rescue numerous Env-incorporation defects [22]; however, the 29KE/31KE/62QR triple mutant does not incorporate significantly more Env than does 29KE/31KE, and does not produce infectious virus (Fig. 7b). As described above for the HeLa-derived virus (Fig. 7a), the 16EK/29KE/31KE/62QR quadruple mutant displays improved levels of Env incorporation and infectivity, relative to 16EK/29KE/31KE, when produced in Jurkat T cells (Fig. 7b). While the 16EK mutation is not required to increase virus release in Jurkat cells, and is itself deficient for Env incorporation, it is required in combination with 29KE/31KE/62QR to restore Env incorporation and replication competence. These results suggest that 16EK counters some aspect of the 29KE/31KE Env incorporation defect, in addition to its effects on virus release.
The infectivity defects of 16EK and 29KE/31KE can be rescued by pseudotyping with short-tailed Env glycoproteins
It has been reported previously that Env-incorporation-defective MA mutants can be rescued by pseudotyping with CT-truncated (CTdel144) Env [18,20], as can the infectivity defect of 29KE/31KE [35]. Our data showed 29KE/31KE to be non-infectious when generated in HeLa or Jurkat cells (Fig. 7). We confirmed the infectivity defects of 29KE/31KE, 16EK and 16EK/29KE/31KE in virus produced from HeLa, Jurkat and MT4 cells (Fig. 8a and b). Rescue of 29KE/31KE by CT-truncated Env suggested that both defects related to interactions between the CT of Env and the MA domain of Gag [35]. To further substantiate this observation, Env-deficient clones of each mutant were co-expressed with a variety of short-tailed viral glycoproteins; the truncated CTdel144 HIV-1 Env, VSV-G and amphotropic MLV (A-MLV) Env (Fig. 8b). Although none of the mutants was infectious when carrying the WT HIV-1 Env protein, all were rescued by CTdel144 HIV-1 Env and VSV-G; 16EK and 16EK/29KE/31KE were also rescued by A-MLV Env (Fig. 8b). Rescue by CTdel144 and VSV-G indicates that in all three mutants the loss of infectivity with WT HIV-1 Env is associated with MA and the long HIV-1 gp41 CT.
Figure 8.
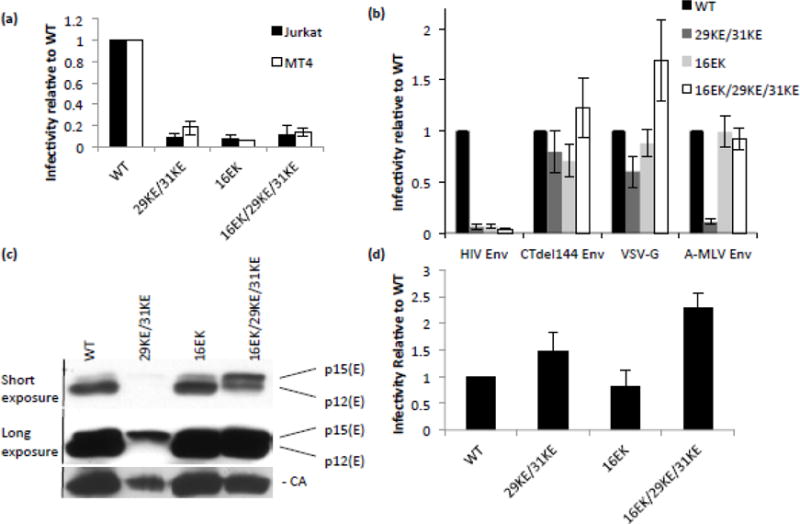
Infectivity and pseudotyping 29/31KE and 16EK. (a) Viruses pseudotyped with VSV-G were generated by transfecting 293T cells with the HIV-1 mutants indicated and a VSV-G expression vector. These viruses were normalized by RT assay and used to infect Jurkat and MT4 T-cell lines. 48 h post-infection virus was collected and RT assays and TZM-bl infections performed. Virus-containing supernatants were used to infect TZM-bl cells; the resulting luciferase signal was normalized to the corresponding RT values to provide a measure of specific infectivity. Means of three independent experiments are plotted, +/− S.E.M. (b) HeLa cells were transfected with Env-deficient clones of the HIV-1 mutants and viral glycoproteins indicated. After 24 h, virus infectivity was determined as in (a). (c) HeLa cells were transfected with Env-deficient clones of the HIV-1 mutants indicated and A-MLV Env. After 48 h, virus was pelleted by ultracentrifugation and analyzed by western blotting. (d) HeLa cells were transfected with Env-deficient clones of the HIV-1 mutants indicated and AMLV p12* Env. After 24 h, virus infectivity was determined as in (a). Means of three independent experiments are plotted +/− S.E.M.
Curiously, A-MLV Env did not rescue 29KE/31KE (Fig. 8b). Unlike CTdel144 and VSV-G, A-MLV Env undergoes a post-assembly maturation step, during which the R peptide is cleaved from p15(E) leaving the mature p12(E) (see Introduction). We previously reported a MA mutant that lacked infectivity when pseudotyped with MLV Env because it blocked cleavage of the p15(E) precursor to p12(E) [34]. A-MLV Env processing in 29KE/31KE, 16EK and 16EK/29KE/31KE was examined by western blotting of virions, revealing that in 29KE/31KE virions, p15(E) remained uncleaved, while in WT and 16EK virions the majority was cleaved to the mature p12(E) (Fig. 8c). The triple mutant 16EK/29KE/31KE displayed an intermediate level of p15(E) processing, apparently generating sufficient mature p12(E) Env to permit virus infectivity. To confirm that the infectivity defect was due to lack of processing, rather than a lack of A-MLV Env incorporation in 29KE/31KE virions, the mutants were pseudotyped with a truncated A-MLV Env protein, in which a stop codon has been added immediately prior to the R peptide-coding sequence, p12(E)* [32] (Fig. 8d). With this constitutively mature A-MLV Env, all mutants were infectious, confirming that the inability of 29KE/31KE to be pseudotyped by A-MLV Env is due to loss of A-MLV Env R-peptide processing.
DISCUSSION
The MA domain of HIV-1 Gag plays a variety of roles in regulating the formation of the virus particle; consequently a wide range of mutations in MA have been described that perturb these various functions. Mutations have been reported that reduce Gag stability, prevent Gag-membrane binding, disrupt targeting to the PM, block Env incorporation and impair infectivity post-entry [6]. The overlapping of regions conferring these phenotypes complicates mechanistic analysis of MA function. For example, mutations in the highly basic region impose defects in membrane binding, membrane targeting, Env incorporation and virus infectivity post-entry [16,36,42]. A greater understanding of how these mutations confer these distinct phenotypes will advance understanding of HIV-1 particle assembly and function.
The 29KE/31KE mutant has been described as lacking efficient particle assembly and release due to an inability to bind to PI(4,5)P2 and consequent accumulation at intracellular sites rather than the PM [35,36,39]. Although it is known that the loss of virus release can be rescued by a compensatory mutation, 16EK, the mechanism of rescue was unknown. Here we have shown that rescue is likely the result of an increase in membrane binding, irrespective of the membrane targeted, and consequent increase in virus assembly and release. Our data indicate that the increased membrane binding conferred by 16EK is a consequence of enhanced electrostatic interactions between MA and the cytoplasmic leaflet of the bilayer, resulting from a net increase in positive charge. It is conceivable that 16K could form a salt bridge with the 29E or 31E; however, our unpublished results (Mercredi et al., unpublished) indicate that, individually, 29KE and 31KE mutations have a relatively modest effect on virus assembly and release. Consistent with 16EK acting through a mechanism of enhanced virus release, 29KE/31KE release was reduced in the MT4 T-cell line in which 16EK was originally identified as a compensatory mutant. As 16EK does not alter the localization of Gag [35], it is unlikely that it corrects the PI(4,5)P2 binding defect, although this has not been formally shown. We and others have previously reported that Gag targeted to internal membranes is not released efficiently in adherent laboratory cell lines like HeLa and 293T [35,36,42–44]. It is unclear, however, why 29KE/31KE is released efficiently from Jurkat cells, PBMCs and MDMs, but not MT4, HeLa or 293T cells. 29KE/31KE virus produced from Jurkat cells reportedly contains more CD63 than does WT virus [35], suggesting that the intracellular, CD63+ site to which 29KE/31KE is targeted is the site of rapid secretion in some cell lines, but not others. The release efficiency would then depend on the rate of membrane binding and the rate at which the contents of that compartment are secreted, in addition to the kinetics of particle assembly. In HeLa cells, prolonged chases following pulse labeling indicated that the intracellularly targeted Gag is never secreted, again suggesting that the trafficking, or the identity, of these compartments is different between those cells that efficiently release 29KE/31KE and those that do not. Cells with a more active exosome secretion pathway may release this MVB-targeted mutant more efficiently than cells less active in exosome release. Further study of the cell type-dependent differences in the release of MVB-targeted Gag is likely to provide new insights into HIV-1 egress pathways.
The second defect of 29KE/31KE is that of impaired infectivity due to defective Env incorporation; this result was particularly clear when the experiment was performed using Jurkat T cells, where the release efficiency of 29KE/31KE is similar to that of WT. Addition of the 62QR mutation that has been shown to rescue many Env-incorporation-defective mutations [22], did not restore Env incorporation to a level sufficient to confer infectivity. The 16EK mutation, that was selected in MT4 T cells, blocks HIV-1 Env incorporation when present alone [22]; however, here we show that the 16EK mutation is required to restore Env incorporation and infectivity to 29KE/31KE in the context of full-length HIV-1 Env. That both 16EK and 62QR are required to complete the rescue of infectivity is seen in the lack of Env incorporation and infectivity of 29KE/31KE, 16EK/29KE/31KE and 29KE/31KE/62QR. These results suggest that the loss of function in 29KE/31KE is more profound than a simple loss of correct membrane targeting. Either the mutations themselves, or the failure to bind to PI(4,5)P2, leads to an alteration in the structure and/or function of MA. Indeed, it is known that the binding of PI(4,5)P2 and triggering of the myristyl switch are associated with structural changes at the N-terminus of MA [8,14]. Based on our previous work [22], the 62QR mutation, which is located at the putative MA trimer interace, likely enhances HIV-1 Env incorporation by making the MA lattice more accommodating to the long gp41 CT.
The A-MLV Env has a short CT and is efficiently incorporated into HIV particles, including MA mutants unable to incorporate full-length HIV-1 Env [18]. Fusogenicity of the A-MLV Env requires processing of the CT by the viral PR during particle maturation, converting the p15(E) precursor to the p12(E) mature protein. The 29KE/31KE mutant remained noninfectious when pseudotyped with A-MLV Env; the failure of A-MLV Env pseudotyping was shown to correlate with a failure to process p15(E), although the Gag precursor was cleaved normally, suggesting that the PR is functional. Addition of the 16EK mutation to 29KE/31KE restored virus infectivity, again suggesting that 16EK is correcting a defect in 29KE/31KE that goes beyond Gag mislocalization. Clearly the phenomena of HIV-1 Env incorporation and A-MLV Env p15(E) processing can be separated, although 29KE/31KE is defective for both.
The changes in MA structure imposed by the mutations described here appear to be too subtle to be readily discernible at the level of resolution provided by thin section EM. The EM morphology of the 29KE/31KE mutant is indistinguishable from that of WT [35]. While we have not examined the EM morphology of the triple mutant 29KE/31KE/62QR, both 29KE/31KE and 29KE/31KE/62QR mutant virions are fully infectious when pseudotyped with VSV-G or truncated HIV-1 Env, demonstrating that the particles are functional (Fig. 8). Indeed, we would argue that the defect in MLV Env CT cleavage imposed by the 29KE/31KE mutations provides structural insights not readily obtained by conventional ultrastructural analysis.
Collectively, the data presented here demonstrate that the MA mutant 29KE/31KE possesses a release and an Env incorporation defect. These defects are further associated with defective processing of the A-MLV Env CT by the viral PR. As such, cleavage of the MLV Env CT provides a useful probe for studying the MA lattice in virions.
MATERIALS AND METHODS
Cell lines and antibodies
HeLa and TZM-bl cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% v/v fetal bovine serum (FBS), 2 mM glutamine and antibiotics, 100 U/ml penicillin and 100 μg/ml streptomycin. TZM-bl is a HeLa-derived reporter cell line that expresses β-galactosidase and firefly luciferase following infection with HIV-1. Jurkat and MT4 CD4+ T-cells were cultured in Roswell Park Memorial Institute (RPMI) 1640 medium, supplemented with 10% v/v FBS, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin.
HIV-1 Gag was probed with patient-derived human anti-HIV-1 IgG (NIH AIDS Research and Reference Reagent Program), used at a 1:10,000 dilution. HIV-1 gp41 was probed with a human monoclonal antibody, 2F5 (NIH AIDS Research and Reference Reagent Program), at a 1:10,000 dilution. The A-MLV p15(E) Env protein was probed with a rabbit antiserum (a gift from A. Rein – National Cancer Institute-Frederick) at a 1:5,000 dilution. VSV-G was probed using P5D4 (Sigma), a mouse monclonal antibody at a 1:20,000 dilution. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was probed using 6C5, a mouse monoclonal antibody (Santa Cruz). Transferrin receptor was probed using H68.4, a mouse monoclonal antibody (Zymed).
Plasmids and cloning
To introduce point mutations into the full-length HIV-1 proviral clone pNL4-3 [45] a subclone was first generated in pBluescript (Stratagene), using the BssHII-SpeI fragment of pNL4-3. Mutations were introduced to the subclone by Quickchange mutagenesis (Stratagene), following the manufacture’s protocol. The BssHII-SpeI fragment of pNL4-3 was then recloned into pNL4-3 and mutants verified by sequencing (Macrogen). For membrane-binding assays, a PR-deficient isolate was used, NL4-3/PR- [40], mutations were introduced as described for NL4-3. For HIV-1 clones with a truncated CT, pNL4-3/CTdel144-2 was used [37]. For pseudotyping experiments, the pNL4-3/KFS clone was used, which expresses all HIV-1 proteins except Env [46], and mutations were introduced in the same manner as for pNL4-3. HIV-1 WT Env and CT-truncated Env were expressed from pIIINL4Env and pIIINL4Env/CTdel144, respectively [37]. VSV-G was expressed from pHCMVG (a gift from J. Burns – University of California, San Diego) and A-MLV Env was expressed from pSV-A-MLV (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: from N. Landau and D. Littman) [47]. An A-MLV Env mutant, lacking the R peptide, was expressed from pMLV-p12* (a gift from A. Rein – National Cancer Institute-Frederick). DNA for transfections was purified from bacterial cultures using the Plasmid Maxi Kit (Qiagen).
Virus release and infectivity
Virus release was assayed in HeLa cells, which were transfected with proviral clones using Polyjet (SignaGen Laboratories). After 24 h, media were removed from cells and replaced with labeling media (RPMI lacking methionine and cysteine, supplemented with 2% dialyzed FBS and Met/Cys 35S label). After 3 h labeling, supernatant was filtered and pelleted, then resuspended in lysis buffer (0.5% Triton X100, 300 mM NaCl, 10 mM iodoacetamide, 50 mM Tris pH 7.5, supplemented with 1} Complete Antiprotease Cocktail - Roche) and immunoprecipitated with anti-HIV-1 IgG. Cell-associated material was also lysed in lysis buffer, insoluble material removed by centrifugation, and the soluble material immunoprecipitated with anti-HIV-1 IgG. Precipitated material was separated by SDS-PAGE and quantified with a Personal Molecular Imager (Biorad). Gag release was defined as the virion-associated CA, calculated as a percentage of total intracellular p55Gag and CA, and virion-associated CA.
Infectivity of released virus was determined by collecting supernatant from HeLa cells (transfected as above), quantifying the amount of virus by measuring the RT activity, and infecting the indicator cell line TZM-bl. 24 h post-infection, luciferase expression in TZM-bl cell lysates was measured using Britelite (Perkin Elmer) and 1450 MicroBeta Jet luminometer (Perkin-Elmer). The ratio of luciferase signal to RT levels provides a measure of specific particle infectivity.
Membrane flotation
Membrane binding was determined by radiolabeling proteins, then floating cellular membranes through a sucrose gradient and comparing the amount of Gag which fractionated with the membrane-bound proteins at the top of the gradient to the amount remaining with non-membrane-bound proteins at the bottom of the gradient [48]. Briefly, HeLa cells were transfected with pNL4-3/PR- using Polyjet. After 24 h, cells were labeled with Met/Cys 35-S for 10 min then incubated for 15 min without radiolabel. Cells were then washed in phosphate buffered saline (PBS – 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4), scraped from the plate and placed in a microcentrifuge tube. Cells were lysed by sonication, nuclei and insoluble material were removed by centrifugation at 1000 × g for 5 min, and the soluble lysate was mixed with 85.5% w/v sucrose in a 4.4 ml ultracentrifuge tube. The samples were overlaid with layers of 65% and 10% w/v sucrose and centrifuged at 100,000 × g for 16 h at 4 °C. Following centrifugation the sucrose gradient was separated into the top fraction (membrane bound) and bottom fraction (non-membrane bound). These fractions were homogenized then Gag was isolated by immunoprecipitation and analyzed as described for virus release assays. The fractions were also probed by western blotting for transferrin receptor (a membrane-bound protein) and GAPDH (a cytosolic protein) to confirm separation.
Protein expression and purification
Unlabeled HIV-1 MA-WT and MA-16EK samples for NMR studies were prepared from recombinant E. coli cells as follows: cells were first grown in 2 l of LB medium at 37 °C [for myristoylated proteins, when the OD600 reached 0.25−0.3 the media was supplemented with myristic acid (10 mg/l; Sigma)] and allowed to grow until the OD600nm reached 0.6−0.7. Next, protein expression was induced with 1 mM of isopropyl-D – thiogalactoside. Cells expressing MA samples were grown at 37 °C for 4 h and lysed using a microfluidizer and the proteins were purified by cobalt affinity chromatography (Clontech) and hydrophobicity column chromatography (Butyl column; GE Healthcare)[13,14,23]. Molecular weights and efficiency of myristoylation were confirmed by electrospray ionization mass spectrometry.
Liposome preparation
Liposomes were prepared from stock solutions of 1-palmitoyl-2-oleoyl-sn-glycero-3-phospholcholine (POPC; Avanti), 1-palmitoyl-2-oleoyl-sn-glycero-3-[phosphor-L-serine] (POPS; Avanti), L-α-phosphatidylethanolamine (POPE; Avanti), L-α-phosphatidylinositol-4,5-bisphosphate ([PI(4,5)P2]; Avanti), 1-stearoyl-2-arachidonoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,5′-bisphosphate) ([PI(3,5)P2]; Avanti), and L-α-phosphatidylinositol-4-phosphate ([PI(4)P]; Avanti) lipids dissolved in chloroform and used without further purification. Lipids were dried to a thin film using a centrifugal evaporator. The dried lipids were then hydrated with liposome buffer, generating 5.76 mg/ml, (100 mM NaCl, 50 mM sodium phosphate [pH7.4], and 5 mM beta-mercaptoethanol) and vortexed until the lipid film was completely dissolved. To prepare large unilamellar vesicles by extrusion, the rehydrated lipid solution was subject to extrusion, using a mini-extruder block (Avanti), 28 times through a polycarbonate membrane with a pore size of 100 nm. Liposome samples were used within 24 h of preparation. Molar percentages of heterogeneous liposomes were prepared by decreasing the amount of POPC relative to the amount of other lipids added. Membrane-mimetic liposomes contained 5.23% POPE, 5.85% POPS, 15.69% Cholesterol, and 72.22% POPC, relative to the total lipid concentration.
NMR spectroscopy
NMR data were recorded at 35 °C on a 600 MHz Bruker Avance III spectrometer equipped with a cryogenic probe. Unlabeled protein samples contained 0.05 mM protein. For solution state NMR-detected liposome binding assays, a fraction of the LUV liposome suspensions (total, 866.4 μg lipids) was mixed with a protein sample (~150 μg) with 10% D2O and incubated at ambient temperature for a minimum of 5 min before NMR data acquisition in 3.0 mm NMR tubes. 1H-1D spectra were acquired with 512 transients, 1 sec relaxation delay, 32768 time domain points, and a spectral window of 12019 Hz. Liposome studies were performed in duplicate, shown with standard deviations. NMR data were processed using Topspin 3.2 (Bruker) and analyzed with dataChord Spectrum Analyst (One Moon Scientific), where the integrals of each spectra were obtained, using the regions 9.4-8.0 ppm, excluding resonances of the lipids and unstructured regions of the protein. Data were collected in duplicate and the fraction of protein bound was calculated as a function of the missing signal intensity at each lipid/protein molar ratio.
Virus replication
Virus replication was assayed in Jurkat cells as described [42]. Briefly, Jurkat cells were transfected with proviral clones using diethylaminoethanol (DEAE)-dextran, then cultured under standard conditions. Every 48 h samples were collected and the culture was split 1/3, then replenished with fresh media. Samples were assayed for RT activity to monitor viral replication.
Western Blotting
HeLa cells were transfected with Polyjet. Virus particles were harvested by filtering culture supernatant and pelleting by ultracentrifugation at 60 000 × g for 1 h at 4 °C. Supernatant was aspirated and virions were lysed in 2} Laemmli buffer (4% SDS, 20% glycerol, 10% β-mercaptoethanol, 0.02% bromophenol blue, 120 mM Tris pH 6.8). Cells were recovered in lysis buffer; insoluble material was removed by centrifugation at 5000 × g for 3 min at 4 °C. Protein samples were separated by SDS-PAGE, transferred to PVDF membrane by semi-dry electroblotting, then blocked in 10% skimmed milk in Tris-buffered saline (150 mM NaCl, 50 mM Tris-Cl pH 7.5) supplemented with 0.1% Tween 20 (TBST), for at least 1 h, at room temperature. Samples were probed with appropriate primary antibodies in TBST at overnight at 4 °C. After washing three times for five minutes, at room temperature, in TBST, membranes were probed with species-specific horse-radish peroxidase-conjugated secondary antibodies in TBST for 1 h, at room temperature. Following a further three washes in TBST, the membranes were incubated with enhanced chemiluminescence (ECL) substrate [22], and the luminescent signal captured using a ChemiDoc system (Biorad). Where greater sensitivity was required, membranes were developed using SuperSignal WestFemto Chemiluminescent Substrate (Thermo Scientific), following the manufacturer’s instructions.
Envelope Incorporation
Virion lysates were analyzed by western blotting for Env and CA and imaged using the ChemiDoc system. Quantification of Env and CA was performed using ImageLab software (Biorad). To calculate Env incorporation, volumes (average pixel intensity multiplied by the area covered by the band) were determined for each gp41 band and divided by the volume for the corresponding CA band.
Highlights.
How does MA interact during HIV particle assembly?
MA mutant 29KE/31KE lacks properly targeted Gag trafficking and Env incorporation.
Further defects were found in MA Env interactions, affecting HIV and MLV Env.
16EK enhances membrane binding and, with 62QR, envelope incorporation.
Acknowledgments
We thank members of the Freed laboratory and S. Welbourn for helpful discussion and critical review of the manuscript. The HIV-Ig was obtained through the NIH AIDS Research and Reference Reagent Program. Rabbit anti-MLV p15(E) serum and the A-MLV p12E* expression plasmid, were gifts from A. Rein, National Cancer Institute. pHCMVG was a gift from J. Burns, University of California, San Diego. Work in the Freed lab is supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, NIH, by the Intramural AIDS Targeted Antiviral Program, and by an Intramural AIDS Research Fellowship to PRT. Work in the Summers lab is supported by NIH/NIAID grant AI30917.
Abbreviations
- A-MLV
amphotropic murine leukemia virus
- CT
cytoplasmic tail
- Env
envelope
- MA
matrix
- PI(4,5)P2
phosphatidylinositol-4,5-bisphosphate
- VLP
virus-like particle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sundquist WI, Kräusslich H-G. HIV-1 Assembly, Budding, and Maturation. Cold Spring Harb Perspect Med. 2012;2:a006924. doi: 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ganser BK, Li S, Klishko VY, Finch JT, Sundquist WI. Assembly and analysis of conical models for the HIV-1 core. Science. 1999;283:80–3. doi: 10.1126/science.283.5398.80. [DOI] [PubMed] [Google Scholar]
- 3.Lu K, Heng X, Summers MF. Structural determinants and mechanism of HIV-1 genome packaging. J Mol Biol. 2011;410:609–33. doi: 10.1016/j.jmb.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell NM, Lever AML. HIV Gag polyprotein: processing and early viral particle assembly. Trends Microbiol. 2013;21:136–44. doi: 10.1016/j.tim.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Votteler J, Sundquist WI. Virus budding and the ESCRT pathway. Cell Host Microbe. 2013;14:232–41. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freed EO. HIV-1 gag proteins: diverse functions in the virus life cycle. Virology. 1998;251:1–15. doi: 10.1006/viro.1998.9398. [DOI] [PubMed] [Google Scholar]
- 7.Ono A, Ablan SD, Lockett SJ, Nagashima K, Freed EO. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci U S A. 2004;101:14889–94. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saad JS, Miller J, Tai J, Kim A, Ghanam RH, Summers MF. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci U S A. 2006;103:11364–9. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chukkapalli V, Ono A. Molecular determinants that regulate plasma membrane association of HIV-1 Gag. J Mol Biol. 2011;410:512–24. doi: 10.1016/j.jmb.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van den Bogaart G, Meyenberg K, Risselada HJ, Amin H, Willig KI, Hubrich BE, et al. Membrane protein sequestering by ionic protein-lipid interactions. Nature. 2011;479:552–5. doi: 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chukkapalli V, Inlora J, Todd GC, Ono A. Evidence in support of RNA-mediated inhibition of phosphatidylserine-dependent HIV-1 Gag membrane binding in cells. J Virol. 2013 doi: 10.1128/JVI.00075-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alfadhli A, McNett H, Tsagli S, Bächinger HP, Peyton DH, Barklis E. HIV-1 matrix protein binding to RNA. J Mol Biol. 2011;410:653–66. doi: 10.1016/j.jmb.2011.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang C, Loeliger E, Luncsford P, Kinde I, Beckett D, Summers MF. Entropic switch regulates myristate exposure in the HIV-1 matrix protein. Proc Natl Acad Sci U S A. 2004;101:517–22. doi: 10.1073/pnas.0305665101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saad JS, Loeliger E, Luncsford P, Liriano M, Tai J, Kim A, et al. Point mutations in the HIV-1 matrix protein turn off the myristyl switch. J Mol Biol. 2007;366:574–85. doi: 10.1016/j.jmb.2006.11.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reil H, Bukovsky AA, Gelderblom HR, Göttlinger HG. Efficient HIV-1 replication can occur in the absence of the viral matrix protein. EMBO J. 1998;17:2699–708. doi: 10.1093/emboj/17.9.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiernan RE, Ono A, Englund G, Freed EO. Role of matrix in an early postentry step in the human immunodeficiency virus type 1 life cycle. J Virol. 1998;72:4116–26. doi: 10.1128/jvi.72.5.4116-4126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brandano L, Stevenson M. A Highly Conserved Residue in the C-Terminal Helix of HIV-1 Matrix is Required for Envelope Incorporation into Virus Particles. J Virol. 2011 doi: 10.1128/JVI.06047-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freed EO, Martin MA. Virion incorporation of envelope glycoproteins with long but not short cytoplasmic tails is blocked by specific, single amino acid substitutions in the human immunodeficiency virus type 1 matrix. J Virol. 1995;69:1984–9. doi: 10.1128/jvi.69.3.1984-1989.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freed EO, Martin MA. Domains of the human immunodeficiency virus type 1 matrix and gp41 cytoplasmic tail required for envelope incorporation into virions. J Virol. 1996;70:341–51. doi: 10.1128/jvi.70.1.341-351.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mammano F, Kondo E, Sodroski J, Bukovsky A, Göttlinger HG. Rescue of human immunodeficiency virus type 1 matrix protein mutants by envelope glycoproteins with short cytoplasmic domains. J Virol. 1995;69:3824–30. doi: 10.1128/jvi.69.6.3824-3830.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono A, Huang M, Freed EO. Characterization of human immunodeficiency virus type 1 matrix revertants: effects on virus assembly, Gag processing, and Env incorporation into virions. J Virol. 1997;71:4409–18. doi: 10.1128/jvi.71.6.4409-4418.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tedbury PR, Ablan SD, Freed EO. Global rescue of defects in HIV-1 envelope glycoprotein incorporation: implications for matrix structure. PLoS Pathog. 2013;9:e1003739. doi: 10.1371/journal.ppat.1003739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Massiah MA, Starich MR, Paschall C, Summers Michael F, Christensen Allyson M, Sundquist WI. Three-dimensional structure of the human immunodeficiency virus type 1 matrix protein. J Mol Biol. 1994;244:198–223. doi: 10.1006/jmbi.1994.1719. [DOI] [PubMed] [Google Scholar]
- 24.Massiah MA, Worthylake D, Christensen AM, Sundquist WI, Hill CP, Summers MF. Comparison of the NMR and X-ray structures of the HIV-1 matrix protein: evidence for conformational changes during viral assembly. Protein Sci. 1996;5:2391–8. doi: 10.1002/pro.5560051202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill CP, Worthylake D, Bancroft DP, Christensen AM, Sundquist WI. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: implications for membrane association and assembly. Proc Natl Acad Sci U S A. 1996;93:3099–104. doi: 10.1073/pnas.93.7.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rao Z, Belyaev AS, Fry E, Roy P, Jones IM, Stuart DI. Crystal structure of SIV matrix antigen and implications for virus assembly. Nature. 1995;378:743–7. doi: 10.1038/378743a0. [DOI] [PubMed] [Google Scholar]
- 27.Alfadhli A, Barklis RL, Barklis E. HIV-1 matrix organizes as a hexamer of trimers on membranes containing phosphatidylinositol-(4,5)-bisphosphate. Virology. 2009;387:466–72. doi: 10.1016/j.virol.2009.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tedbury PR, Freed EO. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol. 2014;22:372–8. doi: 10.1016/j.tim.2014.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chojnacki J, Staudt T, Glass B, Bingen P, Engelhardt J, Anders M, et al. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338:524–8. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- 30.Murakami T, Ablan S, Freed E, Tanaka Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol. 2004;78:1026–31. doi: 10.1128/JVI.78.2.1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wyma DJ, Jiang J, Shi J, Zhou J, Lineberger JE, Miller MD, et al. Coupling of Human Immunodeficiency Virus Type 1 Fusion to Virion Maturation: a Novel Role of the gp41 Cytoplasmic Tail. J Virol. 2004;78:3429–35. doi: 10.1128/JVI.78.7.3429-3435.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rein A, Mirro J, Haynes JG, Ernst SM, Nagashima K. Function of the cytoplasmic domain of a retroviral transmembrane protein: p15E-p2E cleavage activates the membrane fusion capability of the murine leukemia virus Env protein. J Virol. 1994;68:1773–81. doi: 10.1128/jvi.68.3.1773-1781.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ragheb JA, Anderson WF. pH-independent murine leukemia virus ecotropic envelope-mediated cell fusion: implications for the role of the R peptide and p12E TM in viral entry. J Virol. 1994;68:3220–31. doi: 10.1128/jvi.68.5.3220-3231.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kiernan RE, Freed EO. Cleavage of the murine leukemia virus transmembrane env protein by human immunodeficiency virus type 1 protease: transdominant inhibition by matrix mutations. J Virol. 1998;72:9621–7. doi: 10.1128/jvi.72.12.9621-9627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Joshi A, Ablan SD, Soheilian F, Nagashima K, Freed EO. Evidence that productive human immunodeficiency virus type 1 assembly can occur in an intracellular compartment. J Virol. 2009;83:5375–87. doi: 10.1128/JVI.00109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ono A, Orenstein JM, Freed EO. Role of the Gag matrix domain in targeting human immunodeficiency virus type 1 assembly. J Virol. 2000;74:2855–66. doi: 10.1128/jvi.74.6.2855-2866.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murakami T, Freed EO. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc Natl Acad Sci U S A. 2000;97:343–8. doi: 10.1073/pnas.97.1.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ceccon A, D’Onofrio M, Zanzoni S, Longo DL, Aime S, Molinari H, et al. NMR investigation of the equilibrium partitioning of a water-soluble bile salt protein carrier to phospholipid vesicles. Proteins. 2013;81:1776–91. doi: 10.1002/prot.24329. [DOI] [PubMed] [Google Scholar]
- 39.Chukkapalli V, Oh SJ, Ono A. Opposing mechanisms involving RNA and lipids regulate HIV-1 Gag membrane binding through the highly basic region of the matrix domain. Proc Natl Acad Sci U S A. 2010;107:1600–5. doi: 10.1073/pnas.0908661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang M, Orenstein J, Martin M, Freed E. p6Gag is required for particle production from full-length human immunodeficiency virus type 1 molecular clones expressing protease. J Virol. 1995;69:6810–8. doi: 10.1128/jvi.69.11.6810-6818.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margottin F, Bour SP, Durand H, Selig L, Benichou S, Richard V, et al. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell. 1998;1:565–74. doi: 10.1016/s1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 42.Freed EO, Orenstein JM, Buckler-White AJ, Martin MA. Single amino acid changes in the human immunodeficiency virus type 1 matrix protein block virus particle production. J Virol. 1994;68:5311–20. doi: 10.1128/jvi.68.8.5311-5320.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ono A, Freed EO. Cell-type-dependent targeting of human immunodeficiency virus type 1 assembly to the plasma membrane and the multivesicular body. J Virol. 2004;78:1552–63. doi: 10.1128/JVI.78.3.1552-1563.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jouvenet N, Neil SJD, Bess C, Johnson MC, Virgen CA, Simon SM, et al. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006;4:e435. doi: 10.1371/journal.pbio.0040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–91. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freed EO, Delwart EL, Buchschacher GL, Panganiban AT. A mutation in the human immunodeficiency virus type 1 transmembrane glycoprotein gp41 dominantly interferes with fusion and infectivity. Proc Natl Acad Sci U S A. 1992;89:70–4. doi: 10.1073/pnas.89.1.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Landau NR, Page KA, Littman DR. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency virus host range. J Virol. 1991;65:162–9. doi: 10.1128/jvi.65.1.162-169.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Waheed AA, Ono A, Freed EO. Methods for the study of HIV-1 assembly. In: Prasad VR, Kalpana GV, editors. HIV Protoc. Second. Vol. 485. Totowa, NJ: Humana Press; 2009. pp. 163–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pymol. 2006 http://www.pymol.org/