

Abstract
Given the tissue-specific nature of epigenetic processes, the assessment of disease-relevant tissue is an important consideration for epigenome-wide association studies (EWAS). Little is known about whether easily accessible tissues, such as whole blood, can be used to address questions about interindividual epigenomic variation in inaccessible tissues, such as the brain. We quantified DNA methylation in matched DNA samples isolated from whole blood and 4 brain regions (prefrontal cortex, entorhinal cortex, superior temporal gyrus, and cerebellum) from 122 individuals. We explored co-variation between tissues and the extent to which methylomic variation in blood is predictive of interindividual variation identified in the brain. For the majority of DNA methylation sites, interindividual variation in whole blood is not a strong predictor of interindividual variation in the brain, although the relationship with cortical regions is stronger than with the cerebellum. Variation at a subset of probes is strongly correlated across tissues, even in instances when the actual level of DNA methylation is significantly different between them. A substantial proportion of this co-variation, however, is likely to result from genetic influences. Our data suggest that for the majority of the genome, a blood-based EWAS for disorders where brain is presumed to be the primary tissue of interest will give limited information relating to underlying pathological processes. These results do not, however, discount the utility of using a blood-based EWAS to identify biomarkers of disease phenotypes manifest in the brain. We have generated a searchable database for the interpretation of data from blood-based EWAS analyses (http://epigenetics.essex.ac.uk/bloodbrain/).
Keywords: brain, blood, cortex, cerebellum, DNA methylation, epigenetic epidemiology, Illumina 450K array
Introduction
There is increasing interest in the role of epigenetic processes in health and disease, with the primary focus of most epigenetic epidemiological studies to date being DNA methylation.1 Platforms such as the Illumina Infinium HumanMethylation450 BeadChip (450K) have enabled the economical, high-throughput profiling of methylomic variation across large numbers of samples and epigenome-wide association studies (EWAS), which aim to identify DNA methylation differences associated with environmental exposure and disease, are now underway for many types of pathology, including cancer,2-4 autoimmune disorders,5,6 psychiatric phenotypes,7 neurodevelopmental disorders,8,9 and dementia.10,11 Despite the recent successes in identifying disease-associated epigenetic variation, the design, analysis, and interpretation of EWAS requires careful attention; there are a number of critical issues that need to be considered in epigenetic epidemiology that preclude a simple re-analysis of DNA samples collected for genome-wide association studies (GWAS).9,12-14
Of particular importance is the fact that, unlike germline genetic variation, epigenetic signatures are tissue-specific; therefore, the selection of tissue type for epigenetic epidemiology is potentially critical. The ENCODE and the NIH Epigenomics Roadmap projects,15-17 for example, have recently characterized the distinct epigenetic profiles defining human cell-types, highlighting how these reflect the developmental relationships between them. It is clear that intraindividual epigenetic differences (i.e., between tissues within a single person) greatly outweigh interindividual differences within a specific tissue type.18-23 Although many clinical and epidemiological studies are examining epigenetic variation in easily accessible cells obtained from tissues, such as whole blood, the extent to which these can be used to address questions about interindividual epigenomic variation in inaccessible tissues, such as the brain, has not yet been systematically explored. Addressing this issue will be critical given the paucity of high-quality brain tissue from clinically well-phenotyped patients and controls, especially if EWAS analyses require sample sizes approaching those necessary to identify genetic associations with complex disease phenotypes. Because the brain and blood originate from different developmental cell lineages and are epigenetically distinct,23 it is clearly inappropriate to use blood as a proxy measure for actual brain DNA methylation profiles. Despite this, epidemiological studies using accessible peripheral tissues may still be informative in an epidemiological context if interindividual variation is correlated across tissues.
In this study we quantified DNA methylation using the 450K array in a collection of matched DNA samples isolated from pre-mortem whole blood and 4 post-mortem brain regions [prefrontal cortex (PFC), entorhinal cortex (EC), superior temporal gyrus (STG), and cerebellum (CER)] dissected at autopsy from 122 individuals. We describe patterns of co-variation across tissues and identify sites where estimates of DNA methylation in whole blood are predictive of interindividual variation in DNA methylation across the 4 brain regions. Our data are available in an online searchable database (http://epigenetics.essex.ac.uk/bloodbrain/) to enable the research community to explore the relationship between whole blood and brain DNA methylation patterns at specific locations across the genome.
Results and Discussion
Cortex, cerebellum, and blood are defined by very distinct profiles of DNA methylation
We used the Illumina 450K array to quantify DNA methylation in 4 dissected brain regions (PFC: n = 114, EC: n = 108, STG: n = 117 and CER: n = 112) and matched pre-mortem whole blood samples (n = 80) from an overlapping set of 122 individuals (Table S1). Following pre-processing, normalization, and stringent quality control (see Materials and Methods), principal component (PC) analysis was performed across the full dataset (comprising 531 individual DNA samples and data for a pruned set of 427,018 high-quality probes). The first PC, explaining 51.4% of the variance, clearly distinguishes between whole blood, cerebellum, and the three cortical regions (Fig. S1). A similar separation of these tissues is observed with the second PC, which explains 29.4% of the variance. This observation concurs with previous studies comparing whole blood and brain samples based on smaller samples and using alternative technologies for assessing DNA methylation.23-25 These differences between tissues are reflected in gene expression data,26,27 which confirm the cerebellum as being clearly distinguishable from cortical brain regions. The three cortical regions have strikingly similar DNA methylation profiles; however, while examination of further PCs, none of which explain more than 5% of the variance, starts to tease apart these regions, none of the top 20 PCs does this definitively (Fig. S1).
Interindividual variation and sex make a much smaller contribution to overall variation in autosomal DNA methylation than tissue differences
We used linear regression models to calculate the proportion of variance in DNA methylation explained by tissue, individual, and sex. Across autosomal probes, tissue is the strongest predictor of DNA methylation (Fig. S2), although there is a subset of probes for which individual predicts more of the variance (5.39% of all probes) than tissue type (see Table S2). Across all autosomal probes passing stringent QC (n = 416,872), tissue explains > 50% of the variance in DNA methylation at 193,333 (46.4%) sites, compared to individual differences which explain > 50% of the variance at 4,669 (1.12%) sites (Fig. 1A). Considering only autosomal DNA methylation sites characterized as being variable in whole blood (n = 185,060, see Materials and Methods), these percentages increase to 66.2% and 1.61%. As expected, sex makes a strong contribution to variation observed at probes on chromosomes X and Y (n = 10,146), explaining >50% of the variance at 5,920 (58.3%) of these positions compared to tissue which explains >50% of the variance at only 1,359 (1.34%) sites. In contrast, sex makes a very small contribution to autosomal variation, explaining >50% of the variance at only 18 (4.32 × 10−3%) of autosomal probes (Fig. S3).
Figure 1.
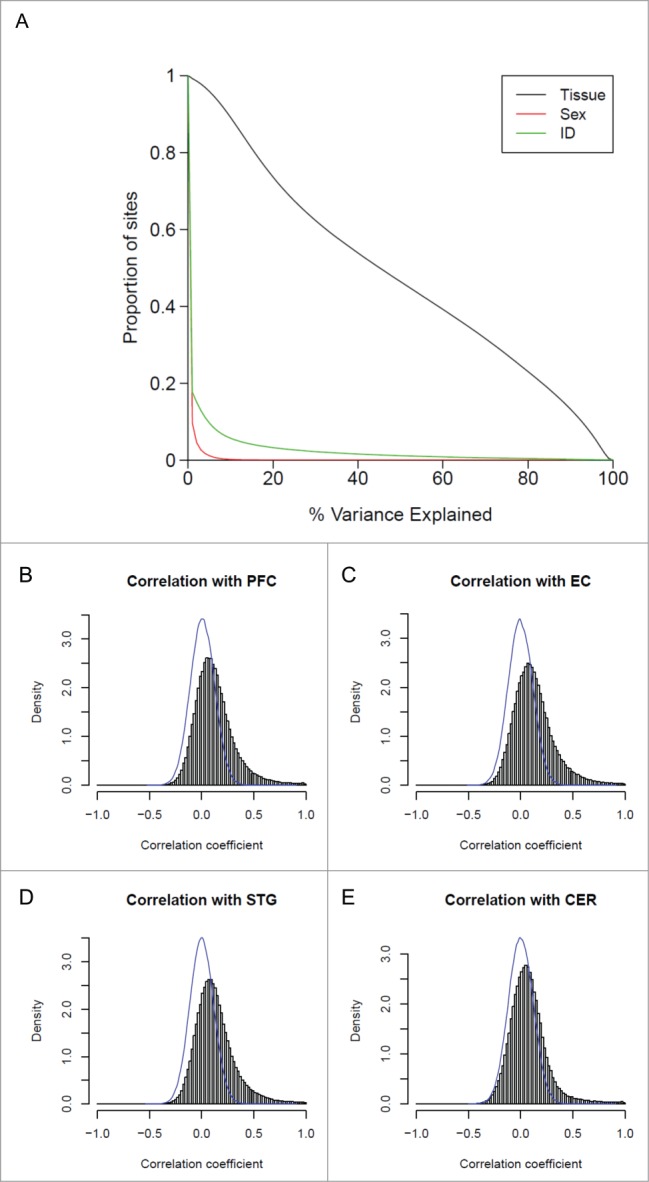
Variation in DNA methylation in whole blood is correlated with variation in the brain for a small proportion of probes. (A) The proportion of sites (y-axis) for which tissue (black), sex (red), or individual (green) explain a given percentage of DNA methylation variance (x-axis). (B) to (E) Histograms showing the distribution of correlation coefficients between DNA methylation in whole blood and the 4 brain regions (PFC, EC, STG and CER). For all 4 brain regions the distribution of correlation coefficients is significantly skewed to the right, with stronger correlations seen between whole blood and cortical regions than between whole blood and cerebellum.
Interindividual variation is correlated across tissues at a small number of sites
Although overall DNA methylation profiles are clearly distinct across different brain regions and blood, driven by highly significant mean differences in DNA methylation at multiple sites across the genome,23 we were interested in exploring the extent to which interindividual variation detected in whole blood reflects interindividual variation in the three cortical regions and cerebellum. Focusing only on probes defined as ‘blood variable’ (n = 185,060, see Materials and Methods), correlation coefficients across all individuals were calculated between DNA methylation in whole blood and each of the four brain regions. Compared to the null distribution, i.e., the scenario where there is no relationship between DNA methylation in blood and brain, established by randomly permuting samples and recalculating correlations between DNA methylation in whole blood and brain across unmatched pairs, we found a modest but highly significant positive shift in the distribution of correlations for each of the four brain regions (PFC: Wilcoxon test P < 2.2 × 10−308, EC: P < 2.2 × 10−308, STG: P < 2.2 × 10−308, CER: P < 2.2 × 10−308), with a small peak highlighting a number of probes characterized by a near perfect correlation (Fig. 1B-E). For the majority of probes, however, interindividual variation in DNA methylation in whole blood explains only a small amount of the variation seen in any of the brain regions (Fig. 2). For example, DNA methylation in whole blood is strongly correlated with levels in cerebellum (i.e., explaining >50% of the variance) for only 1.19% of “blood variable” probes, and moderately correlated (i.e., explaining >20% of the variance) with 3.68% of “blood variable” probes (see Table S3 for corresponding values with other brain regions). Of note, the extent of interindividual correlation is significantly higher (P < 1.0 × 10−308) between whole blood and each of the three cortical regions than with the cerebellum, although the proportion of correlated probes is still low (Table S3). These data concur with previous small studies correlating interindividual variation in DNA methylation between tissues. Slieker et al. compared DNA methylation profiles between blood and several internal organs and reported a comparable number of sites (5,532, 3,909, 10,905, and 2,446 sites for liver, subcutaneous fat, omentum, and skeletal muscle, respectively) with a strong relationship (r > 0.8) with variation in blood.19 Similarly, a study comparing DNA methylation in matched blood and buccal samples found only ∼3% of 998 sites were characterized by an absolute Pearson correlation > 0.5.20 Density plots of DNA methylation across the probes with the strongest positive correlations (> 0.95) between blood and brain indicate that many are characterized by a clear trimodal distribution of DNA methylation (Fig. S4), suggesting that DNA sequence variation likely mediates much of the observed cross-tissue similarities via processes such as allele-specific DNA methylation.28 Many DNA methylation quantitative trait loci (mQTL) have consistent effects across tissues29,30; Figure S5 shows a couple of examples where the correlated DNA methylation profiles across tissues are likely to result from mQTLs, as the distribution of DNA methylation levels cluster into distinct groups reflecting genotype, with consistent effects across tissues.
Figure 3.
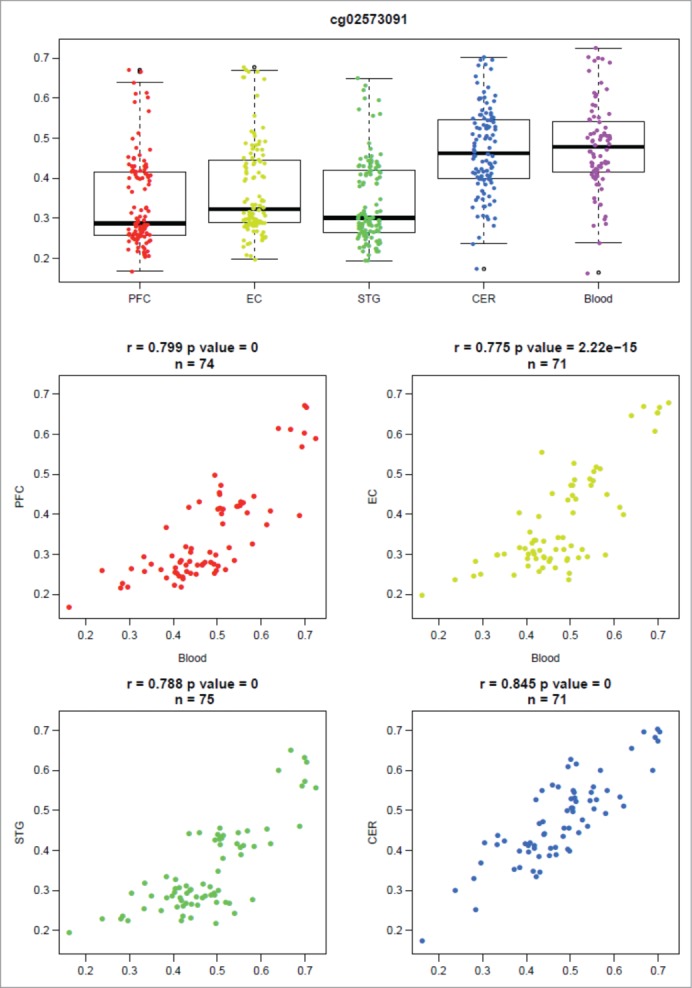
DNA methylation in whole blood significantly co-varies with that in the brain at some genomic loci. An example output of our online database (http://epigenetics.essex.ac.uk/bloodbrain/) for blood-brain correla-tions at cg26039926. Shown is a boxplot of the distribution of DNA methylation values across all individuals split by tissue and four scatterplots demonstrating the relationship between DNA methylation in whole blood and four brain regions (PFC, EC, STG, CER). At this probe there is a highly significant correlation between individual variation in whole blood and that observed in all four brain regions.
Figure 2.
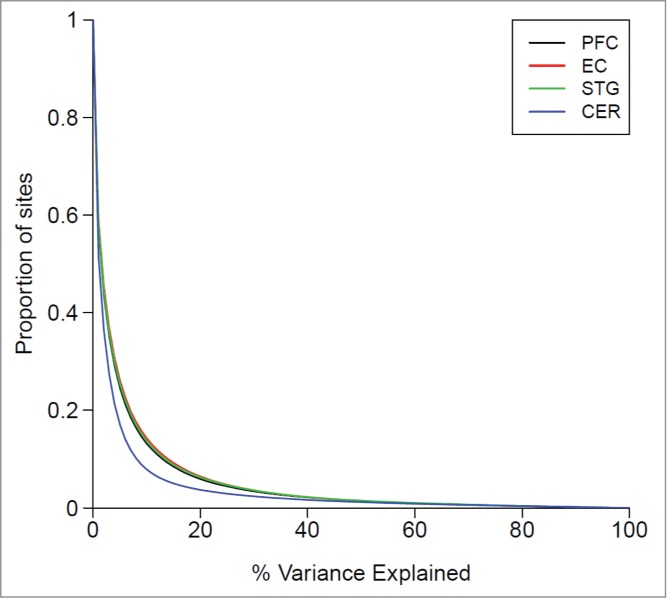
Variation in DNA methylation in whole blood as a predictor of variation in the brain. Shown is the proportion of sites (y-axis) for which variation in blood explains a certain of percentage of DNA methylation variance (x-axis) in the PFC (black), EC (red), STG (green), and CER (blue) from the same individuals.
The extent to which interindividual variation is correlated between whole blood and brain differs by brain region at some loci
Although genome-wide patterns of DNA methylation clearly distinguish between tissues, the overall extent to which interindividual variation in whole blood is correlated with that in the brain is similar across the 4 brain regions (Figs. 3 and S6), with a highly significant correlation of probe-wise correlations. Of note, the three cortical regions are more similar to each other in this regard than the cerebellum, indicating that there is a subset of probes where variation in whole blood predicts variation in the cortex and not the cerebellum, and vice versa (for example see Fig. S7). This suggests that the extent of co-variation between pairs of tissues can differ depending upon the tissues in question, and establishing a correlation between any two tissues does not imply a correlation between all tissues. Of additional interest are sites where there is a significant, but negative, correlation between blood and brain (see Fig. S8 for specific examples), a phenomenon that has been reported previously for certain loci.25 This phenomenon is relatively rare with between 2 and 5 DNA methylation sites strongly negatively correlated (explain > 50% of the variance) between blood and cortex, and most notable between DNA methylation in whole blood and cerebellum with 19 DNA methylation sites classed as strongly negatively correlated (Table S4).
Sites at which interindividual variation in DNA methylation is highly correlated between whole blood and brain are enriched in CpG-rich promoter regions
Sites at which DNA methylation is strongly correlated between whole blood and brain (r2 > 0.5) are not equally distributed across the genome. Of note, we find a significant over-representation at loci in the vicinity of transcription start sites (PFC: P = 1.34 × 10−22, EC: P = 5.15 × 10−21, STG: P = 1.06 × 10−18, CER: P = 1.34 × 10−22), 1st exon (PFC: P = 2.48 x 10−175, EC: P = 8.78 ×10−174, STG: P = 6.72 ×10−171, CER: P = 3.50 × 10−172) and 5'UTR (PFC: P = 2.07 × 10−119, EC: P = 5.04 × 10−120, STG: P = 2.98 × 10−119, CER: P = 2.28 × 10−118) and a depletion in the gene body (PFC: P = 6.15 × 10−94, EC: P = 4.43 ×10−97, STG: P = 2.56 × 10−87, CER: P = 1.85 × 10−88), 3'UTR (PFC: P = 2.92 × 10−24, EC: P = 1.06 × 10−23, STG: P = 2.02 × 10−21, CER: P = 4.74 × 10−23) and intergenic regions (PFC: P = 1.10 × 10−72, EC: P = 2.24 × 10−65, STG: P = 3.51 × 10−71, CER: P = 1.57 × 10−86) (Fig. 4 and Table S5). In addition there is enrichment in CpG islands (PFC: P < 2.2 × 10−308, EC: P < 2.2 × 10−308, STG: P < 2.2 × 10−308, CER: P < 2.2 × 10−308) and depletion in open sea (PFC: P = 8.16 × 10−262, EC: P = 3.57 × 10−276, STG: P = 2.87 × 10−273, CER: P = 7.10 × 10−244) (Fig. 4 and Table S5). Although these differences across genomic regions are highly significant, they may be partly biased by the relative paucity of Illumina 450K microarray probes away from CpG-rich promoter regions.
Figure 4.
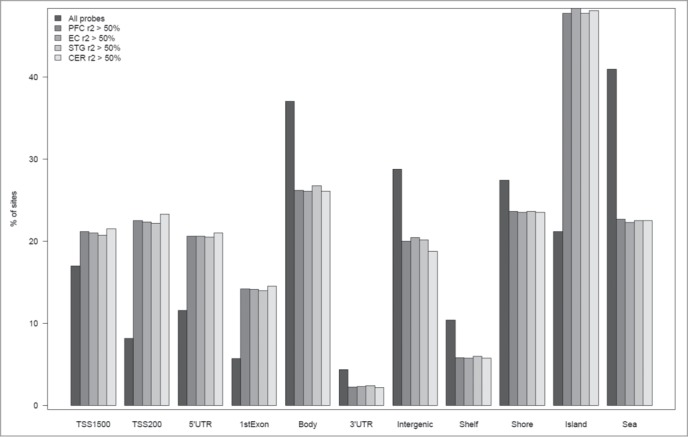
Sites at which interindividual variation correlates between whole blood and brain are enriched in specific genic features. Bar charts plotting the percentage of sites annotated to particular genic feature categories and CpG Island annotations for the full set of “blood variable” sites, in addition to the subset of sites characterized by the highest correlation (r2 > 50%) between blood and brain. Fisher's exact tests were used to test for either over or underrepresentation for each type of feature and are presented in Table S2.
Co-variation between tissues often occurs when absolute levels of DNA methylation are different
It is a common misconception that a similar average level of DNA methylation between two tissues at a given locus is sufficient to establish that one of these tissues may be used as a proxy for the other.31 In fact, for epidemiological studies that use peripheral tissues as a proxy, it is actually more important that the 2 tissues co-vary, regardless of their absolute DNA methylation levels. To demonstrate this point, Table S6 lists 887 sites characterized by similar levels of DNA methylation between tissues (paired t-test P > 0.1) but no evidence for interindividual co-variation (r2 < 0.05), with specific examples shown in Figure S9. In contrast, Table S7 and Figure S10 demonstrate sites that are characterized by highly tissue-specific levels of DNA methylation (paired t-test P < 1.00×10−5) but strong evidence for interindividual co-variation (r2 > 0.5).
Whole blood cannot be used as a proxy for DNA methylation sites that are only variable in the brain
One potential caveat to performing an EWAS of a neurological/psychiatric phenotype using a peripheral tissue as a proxy is that a proportion of sites are characterized by limited interindividual variation in whole blood but high levels of interindividual variation in the brain, and vice versa. We defined probes as having ‘low’ variation when the range of DNA methylation values across the total sample < 5% and ‘high’ variation when the range of DNA methylation in the middle 80th percentile of samples > 5%. Figure 5A shows that there are 2,505 sites characterized by high interindividual variation in whole blood but not in the cortex (STG) (see Fig. S11 and Table S8 for corresponding data for the other cortical regions) and 6,909 sites that vary in whole blood but not in the cerebellum. Whether these sites are omitted from analyses (for example, to reduce multiple testing burden) depends upon the ultimate aim of the study being undertaken; while they may not be able to inform directly about mechanistic processes in disease, they could still represent useful biomarkers. In contrast, some sites are non-variable in whole blood but vary in the brain (see Fig. 5B).
Figure 5.
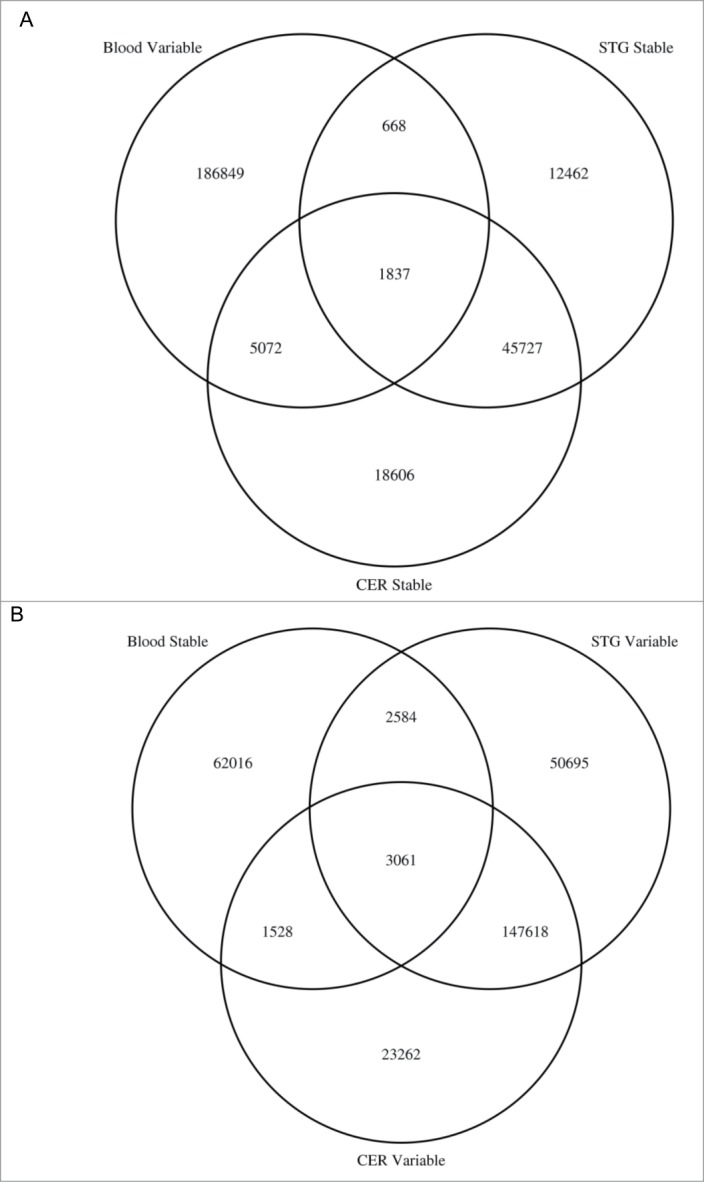
EWAS analyses of brain phenotypes using whole blood DNA may potentially miss disease associated variation and interrogate DNA methylation sites that are not actually variable in the brain. Venn diagrams showing the overlap of DNA methylation sites that are (A) variable in whole blood but not variable in the cortex (STG) or cerebellum and (B) variable in the cortex (STG) and cerebellum but not in whole blood.
Conclusion
Our data suggest that across the majority of the genome, an EWAS using whole blood for disorders where brain is the presumed to be the primary tissue of interest will give limited information relating to underlying pathological processes. However, there are a proportion of sites where interindividual variation is correlated between whole blood and brain, and these results do not discount the utility of using a blood-based EWAS to identify potential biomarkers of psychiatric disease phenotypes. We have developed a searchable online database (http://epigenetics.essex.ac.uk/bloodbrain/) to enable researchers to investigate the relationship between whole blood and brain for any probes on the Illumina 450K array to aid in the interpretation of EWAS analyses of brain disorders.
Materials and Methods
Samples
We obtained entorhinal cortex (EC), prefrontal cortex (PFC), superior temporal gyrus (STG), and cerebellum (CER) tissue from 117 individuals archived in the MRC London Neurodegenerative Disease Brain Bank (http://www.kcl.ac.uk/iop/depts/cn/research/MRC-London-Neurodegenerative-Diseases-Brain-Bank/MRC-London-Neurodegenerative-Diseases-Brain-Bank.aspx). Ethical approval for the study was provided by the NHS South East London REC 3. All samples were dissected by trained specialists, snap-frozen and stored at −80°C. Matched whole blood samples collected before death were available for 80 samples (Table S1) as part of the Alzheimer's Research UK funded study “Biomarkers of AD Neurodegeneration” with informed consent according to the Declaration of Helsinki (1991). Genomic DNA was isolated from ∼100 mg of each dissected brain region or ∼10 ml whole blood stored in EDTA collection tubes using a standard phenol-chloroform extraction method, and tested for degradation and purity before analysis. The samples used in this study included both neuropathologically unaffected controls and individuals with variable levels of neuropathology. More information about the specific samples can be found in Lunnon et al.11
Methylomic profiling
DNA (500 ng) from each sample was sodium bisulfite-treated using the Zymo EZ 96 DNA methylation kit (Zymo Research) according to the manufacturer's standard protocol. DNA methylation was quantified using the Illumina Infinium HumanMethylation450 BeadChip (Illumina) using an Illumina HiScan System (Illumina). All samples were assigned a unique code for the purpose of the experiment and grouped by tissue and randomized with respect to other variables status to avoid batch effects, and processed in batches of 4 BeadChips. Illumina Genome Studio software was used to extract the raw signal intensities of each probe (without background correction or normalization). Raw data are downloadable from GEO with accession identifier GSE59685.
Data pre-processing
All analyses were performed using R 3.0.2.32 and Bioconductor 2.13.33 Signal intensities were imported into R using the methylumi package.34 and transformed into β values. In order to confirm that each set of tissues derived from the same individual, initial quality control checks were performed using functions in the methylumi package to assess concordance between reported gender in the phenotype information and that inferred from DNA methylation sites located on the sex chromosomes. In addition, the 65 non-CpG SNP probes on the array were also used to confirm that all 4 brain regions and matched blood samples were sourced from the same individual, as their genotypes across these variants should be identical. Data was subsequently normalized in the R package wateRmelon using the dasen function as previously described.35 Prior to data analysis, we removed the 65 non-CpG SNP probes and probes characterized by either non-specific binding (n = 43,233) or containing common (minor allele frequency > 5%) SNPs within 10 bp of the CG or single base extension position (n = 15,261, identified from previously published lists).36,37 to prevent technical artifacts influencing our results. The final data set comprised 427,018 DNA methylation sites.
Data analysis
Separate linear regression models were used to calculate the proportion of variance explained (adjusted r2) by a) tissue, b) individual, and c) sex, for each DNA methylation site on the array across individuals for which data from all 5 tissues passed quality control. These linear regression models took the form

Where DNAmij is the DNA methylation value for individual j in tissue i, α is the intercept, and β the regression coefficient for each factor of interest.
A subset of “blood variable” probes was identified by calculating the DNA methylation difference between the 10th and 90th percentile across all samples, and selecting sites where this was > 5% (all chromosomes n = 194,426; autosomes n = 185,060). Sites characterized by overall differential DNA methylation between blood and each brain region were identified by a paired t-test of matched samples. Pairwise correlation coefficients were calculated between DNA methylation values from whole blood and each of the 4 brain regions across matched samples from linear regression models; the values were squared and multiplied by 100 to obtain the percentage of variance explained for each probe. Samples were permuted and correlations between DNA methylation in whole blood and brain were recalculated across unmatched pairs to establish the distribution in the scenario where there is no relationship between DNA methylation in blood and brain. The density curve of these simulated correlations was added to the histograms of the true correlation coefficients to represent the null distribution (Fig. 1 and Fig. S4). The annotation file provided by Illumina for all probes on the array was used to classify DNA methylation sites into genomic feature and CpG island feature categories; any site with no UCSC gene annotation was classed as “intergenic." Enrichment was calculated from a 2 x 2 Fisher's exact test, comparing the number of probes with blood-brain correlation r2 > 0.5 annotated to each feature category to the background of all probes.
Web Resources
A searchable database of matched blood and brain region DNA methylation data is available at http://epigenetics.essex.ac.uk/bloodbrain/. It reports the distribution of DNA methylation values in each tissue and the correlation of individual values between blood and each of the 4 brain regions for each probe on the 450K array.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the National Institute for Health (NIHR) Biomedical Research Unit in Dementia in the South London and Maudsley NHS Foundation Trust (SLaM), Brains for Dementia Research (Alzheimer Brain Bank, UK), and the donors and families who made this research possible.
Funding
Blood samples were collected as part of the Alzheimer's Research UK funded study “Biomarkers of AD Neurodegeneration." This work was funded by US National Institutes of Health grant R01 AG036039 and UK Medical Research Council grant MR/K013807/1 to J.M.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
References
- 1.Murphy TM, Mill J. Epigenetics in health and disease: heralding the EWAS era. Lancet 2014; 383:1952-4; PMID:24630775; http://dx.doi.org/ 10.1016/S0140-6736(14)60269-5 [DOI] [PubMed] [Google Scholar]
- 2.Heyn H, Carmona FJ, Gomez A, Ferreira HJ, Bell JT, Sayols S, Ward K, Stefansson OA, Moran S, Sandoval J, et al.. DNA methylation profiling in breast cancer discordant identical twins identifies DOK7 as novel epigenetic biomarker. Carcinogenesis 2013; 34:102-8; PMID:23054610; http://dx.doi.org/ 10.1093/carcin/bgs321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al.. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 2009; 41:178-86; PMID:19151715; http://dx.doi.org/ 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lange CP, Campan M, Hinoue T, Schmitz RF, van der Meulen-de Jong AE, Slingerland H, van der Meulen-de Jong AE, Slingerland H, Kok PJ, van Dijk CM, et al.. Genome-scale discovery of DNA-methylation biomarkers for blood-based detection of colorectal cancer. PLoS One 2012; 7:e50266; PMID:23209692; http://dx.doi.org/ 10.1371/journal.pone.0050266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al.. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol 2013; 31:142-7; PMID:23334450; http://dx.doi.org/ 10.1038/nbt.2487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rakyan VK, Beyan H, Down TA, Hawa MI, Maslau S, Aden D, Daunay A, Busato F, Mein CA, Manfras B, et al.. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. Plos Genet 2011; 7:e1002300; PMID:21980303; http://dx.doi.org/ 10.1371/journal.pgen.1002300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pidsley R, Viana J, Hannon E, Spiers HH, Troakes C, Al-Saraj S, Mechawar N, Turecki G, Schalkwyk LC, Bray NJ, et al.. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol 2014; 15:483; PMID:25347937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladd-Acosta C, Hansen K, Briem E, Fallin M, Kaufmann W, Feinberg A. Common DNA methylation alterations in multiple brain regions in autism. Mol Psychiatry 2014; 19:862-71; PMID:23999529; http://dx.doi.org/ 10.1038/mp.2013.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berko ER, Suzuki M, Beren F, Lemetre C, Alaimo CM, Calder RB, Ballaban-Gil K, Gounder B, Kampf K, Kirschen J, et al.. Mosaic epigenetic dysregulation of ectodermal cells in autism spectrum disorder. Plos Genet 2014; 10:e1004402; PMID:24875834; http://dx.doi.org/ 10.1371/journal.pgen.1004402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, et al.. Alzheimer's disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 2014; 17:1156-63; PMID:25129075; http://dx.doi.org/ 10.1038/nn.3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lunnon K, Smith R, Hannon E, De Jager PL, Srivastava G, Volta M, Troakes C, Al-Sarraj S, Burrage J, Macdonald R, et al.. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer's disease. Nat Neurosci 2014; 17:1164-70; PMID:25129077; http://dx.doi.org/ 10.1038/nn.3782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mill J, Heijmans BT. From promises to practical strategies in epigenetic epidemiology. Nat Rev Genet 2013; 14:585-94; PMID:23817309; http://dx.doi.org/ 10.1038/nrg3405 [DOI] [PubMed] [Google Scholar]
- 13.Relton CL, Davey Smith G. Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med 2010; 7:e1000356; PMID:21048988; http://dx.doi.org/ 10.1371/journal.pmed.1000356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 2011; 12:529-41; PMID:21747404; http://dx.doi.org/ 10.1038/nrg3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome. Nature 2012; 489:57-74; PMID:22955616; http://dx.doi.org/ 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al.. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 2011; 473:43-9; PMID:21441907; http://dx.doi.org/ 10.1038/nature09906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al.. Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317-30; PMID:25693563; http://dx.doi.org/ 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Byun HM, Siegmund KD, Pan F, Weisenberger DJ, Kanel G, Laird PW, Yang AS. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum Mol Genet 2009; 18:4808-17; PMID:19776032; http://dx.doi.org/ 10.1093/hmg/ddp445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slieker RC, Bos SD, Goeman JJ, Bovée JV, Talens RP, van der Breggen R, Suchiman HE, Lameijer EW, Putter H, van den Akker EB, et al.. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin 2013; 6:26; PMID:23919675; http://dx.doi.org/ 10.1186/1756-8935-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang R, Jones MJ, Chen E, Neumann SM, Fraser HB, Miller GE, Kobor MS. Discordance of DNA methylation variance between two accessible human tissues. Sci Rep 2015; 5:8257; PMID:25660083; http://dx.doi.org/ 10.1038/srep08257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson TM, Sharfi D, Lee M, Yrigollen CM, Naumova OY, Grigorenko EL. Comparison of whole-genome DNA methylation patterns in whole blood, saliva, and lymphoblastoid cell lines. Behav Genet 2013; 43:168-76; PMID:23269419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, et al.. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet 2006; 38:1378-85; PMID:17072317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, et al.. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol 2012; 13:R43; PMID:22703893; http://dx.doi.org/ 10.1186/gb-2012-13-6-r43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farré P, Jones MJ, Meaney MJ, Emberly E, Turecki G, Kobor MS. Concordant and discordant DNA methylation signatures of aging in human blood and brain. Epigenetics Chromatin 2015; 8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Illingworth RS, Gruenewald-Schneider U, De Sousa D, Webb S, Merusi C, Kerr AR, James KD, Smith C, Walker R, Andrews R, et al.. Inter-individual variability contrasts with regional homogeneity in the human brain DNA methylome. Nucleic Acids Res 2015; 43:732-44; PMID:25572316; http://dx.doi.org/ 10.1093/nar/gku1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.GTEx Consortium . Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348:648-60; PMID:25954001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melé M, Ferreira PG, Reverter F, DeLuca DS, Monlong J, Sammeth M, Young TR, Goldmann JM, Pervouchine DD, Sullivan TJ, et al.. Human genomics. The human transcriptome across tissues and individuals. Science 2015; 348:660-5; http://dx.doi.org/ 10.1126/science.aaa0355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schalkwyk LC, Meaburn EL, Smith R, Dempster EL, Jeffries AR, Davies MN, Plomin R, Mill J. Allelic skewing of DNA methylation is widespread across the genome. Am J Hum Genet 2010; 86:196-212; PMID:20159110; http://dx.doi.org/ 10.1016/j.ajhg.2010.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, et al.. Abundant Quantitative Trait Loci Exist for DNA Methylation and Gene Expression in Human Brain. Plos Genet 2010; 6:e1000952; http://dx.doi.org/ 10.1371/journal.pgen.1000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith AK, Kilaru V, Kocak M, Almli LM, Mercer KB, Ressler KJ, Tylavsky FA, Conneely KN. Methylation quantitative trait loci (meQTLs) are consistently detected across ancestry, developmental stage, and tissue type. Bmc Genomics 2014; 15:145; PMID:24555763; http://dx.doi.org/ 10.1186/1471-2164-15-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith AK, Kilaru V, Klengel T, Mercer KB, Bradley B, Conneely KN, Ressler KJ, Binder EB. DNA extracted from saliva for methylation studies of psychiatric traits: evidence tissue specificity and relatedness to brain. Am J Med Genet B Neuropsychiatr Genet 2015; 168B:36-44; PMID:25355443; http://dx.doi.org/ 10.1002/ajmg.b.32278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.R Development Core Team R: A Language and Environment for Statistical Computing Vienna, Austria: R Foundation for Statistical Computing, 2008. [Google Scholar]
- 33.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al.. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 2004; 5:R80; PMID:15461798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis S, Du P, Bilke S, Triche J, Bootwalla M. Methylumi: Handle Illumina Methylation Data. R Package Version 2.14.0. 2015 [Google Scholar]
- 35.Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. Bmc Genomics 2013; 14:293; PMID:23631413; http://dx.doi.org/ 10.1186/1471-2164-14-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8:203-9; PMID:23314698; http://dx.doi.org/ 10.4161/epi.23470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price ME, Cotton AM, Lam LL, Farré P, Emberly E, Brown CJ, Robinson WP, Kobor MS. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin 2013; 6:4; PMID:23452981; http://dx.doi.org/ 10.1186/1756-8935-6-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.