

Abstract
Immortality is an essential characteristic of human carcinoma cells. We recently developed an efficient, reproducible method that immortalizes human mammary epithelial cells (HMEC) in the absence of gross genomic changes by targeting 2 critical senescence barriers. Consistent transcriptomic changes associated with immortality were identified using microarray analysis of isogenic normal finite pre-stasis, abnormal finite post-stasis, and immortal HMECs from 4 individuals. A total of 277 genes consistently changed in cells that transitioned from post-stasis to immortal. Gene ontology analysis of affected genes revealed biological processes significantly altered in the immortalization process. These immortalization-associated changes showed striking similarity to the gene expression changes seen in The Cancer Genome Atlas (TCGA) clinical breast cancer data. The most dramatic change in gene expression seen during the immortalization step was the downregulation of an unnamed, incompletely annotated transcript that we called MORT, for mortality, since its expression was closely associated with the mortal, finite lifespan phenotype. We show here that MORT (ZNF667-AS1) is expressed in all normal finite lifespan human cells examined to date and is lost in immortalized HMEC. MORT gene silencing at the mortal/immortal boundary was due to DNA hypermethylation of its CpG island promoter. This epigenetic silencing is also seen in human breast cancer cell lines and in a majority of human breast tumor tissues. The functional importance of DNA hypermethylation in MORT gene silencing is supported by the ability of 5-aza-2′-deoxycytidine to reactivate MORT expression. Analysis of TCGA data revealed deregulation of MORT expression due to DNA hypermethylation in 15 out of the 17 most common human cancers. The epigenetic silencing of MORT in a large majority of the common human cancers suggests a potential fundamental role in cellular immortalization during human carcinogenesis.
Keywords: breast cancer, DNA methylation, epigenetics, immortality, HMEC, lncRNA, lincRNA, MORT, mammary epithelia, ncRNA, ZNF667-AS1
Introduction
Immortality is an essential characteristic of human carcinoma cells. We recently reported the development of an efficient and reproducible method that immortalizes human mammary epithelial cells (HMEC) by targeting 2 critical senescence barriers.1 This immortalization method uses agents pathologically relevant to breast carcinogenesis, works with HMEC from all individuals tested, and produces transformed lines that reflect the various phenotypes seen in clinical breast cancer.1,2 The first senescence barrier, stasis (stress-associated senescence), is bypassed by disrupting the RB pathway—either by inhibiting p16 function using a p16-directed shRNA or by constitutively overexpressing cyclin D1. These post-stasis cells are then made to bypass the second proliferation barrier, replicative senescence due to critically shortened telomeres, by transduction of a dysregulated MYC. Importantly, this controlled genetic approach produces non-clonal immortalized cell lines lacking gross genomic alterations.1,2 Thus, these immortal cells carry minimal confounding passenger errors, thereby providing a unique system to identify important and still unknown participants in breast cancer immortalization.
To identify consistent transcriptomic changes associated with immortality, we used microarray analysis to profile the transcriptomes of isogenic pre-stasis, post-stasis, and immortal HMEC. A total of 277 genes consistently changed in cells that transitioned from post-stasis to immortal. Gene ontology analysis of affected genes revealed biological processes significantly targeted in the immortalization process, and when compared with The Cancer Genome Atlas (TCGA) breast cancer data, the gene changes observed in immortal HMEC showed striking similarity to the gene expression changes seen in clinical cancer. For example, the 8 genes that comprised the most significantly overrepresented gene ontology, the integrin-mediated signaling pathway, were all downregulated both during the HMEC immortalization step and in clinical human breast carcinomas. Other common gene ontologies affected during HMEC immortalization that are also seen in clinical breast cancer include biological processes associated with apoptosis and programmed cell death, cell adhesion and extracellular matrix organization, and stem cell division and protein tyrosine kinase activity.
The most dramatic change in gene expression seen during the immortalization step was the downregulation of an unnamed, incompletely annotated transcript that we called MORT, for mortality, since its expression was closely associated with the mortal, finite lifespan phenotype. MORT is a primate-specific long noncoding RNA (lncRNA) of the long intergenic noncoding RNA (lincRNA) class, has a long cellular half-life, and is enriched in the cytoplasmic fraction. The number of lncRNAs annotated in human genome is approaching 60,000,3 and while their diverse mechanistic roles have remained enigmatic and incompletely resolved,4-7 they are increasingly appreciated as regulators of normal cell physiology.8,9 lncRNAs that play fundamental roles in cell function include XIST in X chromosome inactivation,10-14 and HOTAIR in the control of the HOXD cluster.15 Correspondingly, lncRNA dysregulation has been well documented in various diseases including cancers.16-20
We show here that MORT is expressed in all normal finite lifespan human cells examined to date and is downregulated or lost in immortalized HMEC. MORT silencing during immortalization is linked to the aberrant epigenetic event of DNA hypermethylation of its CpG island promoter. This epigenetic silencing is also seen in human breast cancer cell lines and in a majority of human breast tumor tissues. The functional importance of DNA hypermethylation in MORT gene silencing is supported by the ability of 5-aza-2′-deoxycytidine (5-AdC) to reactivate MORT expression. Furthermore, analysis of TCGA data across 16 additional human cancers revealed that deregulation of MORT expression due to DNA hypermethylation is a frequent event in most common human cancers. Together, these results identify a lncRNA whose epigenetic silencing is likely involved in the immortalization of human epithelial cells and the progression of multiple human cancer types.
Results
We have recently published a 2-step procedure of HMEC immortalization using pathologically relevant agents that does not cause gross genomic changes.1 Here we characterize gene expression changes occurring during this immortalization process using HMEC from 4 different individuals (women aged 19, 21, 66, and 91 years). Figure 1 shows the analyzed samples and the steps taken to bypass the stasis and immortalization barriers. The first step is bypassing the stasis barrier by either targeting p16 using p16sh RNA or overexpression of a ccnd1/CDK2 fusion gene, thereby preventing RB-mediated stasis. These post-stasis cultures are then immortalized by transduction of MYC.
Figure 1.
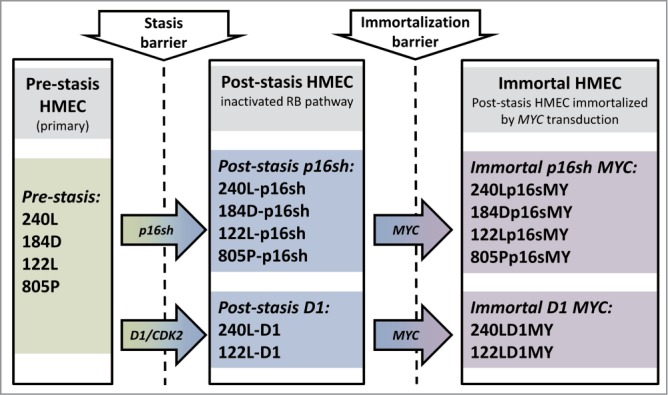
Schema of the in vitro HMEC immortalization model and of the analyzed samples. HMEC from 4 different individuals were immortalized in vitro. The stasis barrier was bypassed by either targeting p16 using p16sh RNA or overexpression of a ccnd1/CDK2 fusion gene. The immortalization barrier was bypassed by transduction of MYC.
Gene expression was analyzed using Affymetrix Human Gene 1.0 ST microarrays in all 3 types of samples: finite pre-stasis strains, finite post-stasis strains with interrupted RB pathway, and MYC-immortalized lines. Although there were 116 genes with at least 2-fold mean expression change after the first genetic modification that led to bypassing stasis (Fig. 1, Table S1), none of the changes were consistent enough across the 4 individuals to be statistically significant (Fig. 2A). However, after the second genetic modification, which led to immortalization, there were significant changes in gene expression (Fig. 2B, Table S1). A total of 277 genes were significantly changed after multiple testing P-value adjustment (adj. P-value < 0.05), and 77 of these genes were changed at least 2-fold.
Figure 2.

Gene expression changes associated with bypassing the stasis and immortalization barriers. Volcano plots of gene expression changes associated with bypassing stasis (A) and immortalization (B). Vertical dashed lines indicate 2-fold changes in expression. The horizontal dashed line indicates 0.05 adj.p-value cut-off. MORT gene (blue dot) with outstanding change in expression is labeled. (C) biological processes enriched among the genes changed during the immortalization step. The two columns on the right indicate, for each GO biological process, the number of genes that were changed in HMEC model and the proportion of these genes that are changed in TCGA cohort of 917 breast carcinomas in the same direction as in the immortalization step in the in vitro model. (D) The direction and the level of change of 8 members of integrin-mediated signaling pathway changed during immortalization in the in vitro HMEC immortalization model. The plot shows the ratio of expression change in the in vitro model in comparison to that observed in clinical TCGA breast carcinoma data.
To find relationships between gene expression and phenotypic changes, the 277 differentially expressed genes were tested for enrichment of Gene Ontology (GO) terms. The significantly overrepresented biological processes (Fig. 2C, Table S2) included positive regulation of cell death, positive regulation of apoptotic process, death, stem cell division, and integrin-mediated signaling pathway. The enrichment of these GO terms in the differentially expressed genes is consistent with the gene expression changes expected during the gain of immortality, since they include biological processes associated with continued cell proliferation, such as those associated with apoptosis and programmed cell death, cell adhesion, and extracellular matrix organization.
The integrin-mediated signaling pathway, the most overrepresented GO term, is involved in multiple processes including cell adhesion, migration, polarity, growth, and death; perturbed integrin function has long been linked to breast cancer.21-23 Eight members of the integrin-mediated signaling pathway were significantly downregulated. We determined if similar changes also occur during in vivo carcinogenesis by comparing the data from the in vitro HMEC immortalization step and TCGA data from clinical breast tumor samples (Fig. 2D). All 8 genes are also downregulated to a similar extent in clinical breast tumor tissues compared to non-tumor tissues, indicating 100% concordance between the in vitro model and clinical cancer. The data comparing the in vitro model with TCGA data for the other 16 significantly overrepresented biological processes are shown in Figure S1. Overall, there is good concordance between the in vitro model of non-malignant immortalized HMEC and the TCGA data of breast carcinomas for most of the categories (Fig. 2C), particularly considering that the in vitro model identifies only gene expression changes during immortalization, while the clinical tumor samples will have additional changes linked to malignancy. Overall, these findings indicate that the gene expression changes observed in our in vitro immortalization model are relevant for in vivo carcinogenesis and likely occur early during transformation.
Apart from the significant gene expression changes in the whole GO pathways, we identified one gene outside of GO pathways with an outstanding difference in expression between mortal vs. immortal cells. This gene, that initially had only a numeric designation LOC100128252 and no GO annotation, is completely silenced during HMEC immortalization (Fig. 2B). We named it MORT since its expression seems to be a hallmark of mortal cells. MORT was found to be a noncoding RNA and it is named ZNF667-AS1 in the current RefSeq release. To validate the microarray data we performed realtime PCR analyzis of the MORT transcript level; the microarray (Fig. 3A) and RT-PCR (Fig. 3B) data are in good concordance. We also analyzed MORT expression in 3 additional primary cell types: human mammary fibroblasts, prostate epithelial cells, and human urothelial cells, as well as in pre-stasis HMEC transduced by MYC alone. MORT is expressed in all the primary cell samples and MYC transduction alone does not cause its silencing (Fig. S2). We further expanded our analysis to 16 normal human tissues of Ilumina body map data. MORT is expressed, at variable levels, in all 16 normal human tissues tested (Fig. S3). Overall, our gene expression analysis revealed a noncoding RNA transcript that is expressed in all normal cell types examined and is the most consistently and dramatically down regulated transcript during the process of HMEC in vitro immortalization.
Figure 3.

MORT expression and DNA methylation in HMEC immortalization model. MORT expression levels in individual cell type groups of the model, determined by Affymetrix microarray (A) and RT-PCR (B). (C) MORT genomic location. The promoter part is expanded in the lower panel. The individual CpG dinucleotides in the region are displayed as vertical black bars. The region analyzed for DNA methylation by MassARRAY is indicated by red boxes. The CpGs covered by Illumina HumanMethylation450 array and used to analyze the TCGA data are indicated by red circles. (D) DNA methylation level of MORT promoter in individual cell type groups of the model. P-values indicated (paired t-test) are for contrast Immortal vs. Post-Stasis.
The MORT gene is located on chromosome 19, within the zinc finger gene cluster 19.13. Its 1.53 kb RNA consists of 2 exons of 260 and 1270 bp separated by a 16 kb intron (Fig. 3C). The MORT promoter overlaps a large CpG island (1.4 kb, 148 CpGs) that is shared with ZNF667 (head to head). ZNF667 expression is also downregulated during immortalization, although much less dramatically than MORT (Table S1), possibly due to its very low basal expression level that is about an order of magnitude lower than that of MORT. Twelve kilobases downstream of the MORT terminator there is a promoter of ZNF471. Since the MORT transcript is polyadenylated, it is a product of RNA polymerase II. Although located within a ZNF gene cluster, MORT gene does not share homology with ZNF genes. The second exon of MORT contains 2 LINE (L2 and L1MB3) and one LTR (LTR47B) repetitive elements. Based on random chance, the MORT transcript (1.5 kb) could encode a 110 aa peptide (Dinger 2008). The MORT transcript contains 5 ORFs that are larger than 50 codons; however, none of them exceeds 70 codons. BlastX searches have not produced any putative conserved protein domains in these ORFs. According to Coding-Potential Assessment Tool (CPAT),24 MORT has a negative hexamer score (−0.29) and a very low coding probability of 0.005, resulting in absence of coding label based on CPAT. Actinomycin D experiments revealed that the MORT transcript has a long half-life, comparable to the housekeeping gene GAPDH (Fig. S4). Cell fractionation experiments show enrichment of MORT in the extra-nuclear fraction, suggesting a cytoplasmic function (Fig. S5). Overall, these observations indicate that MORT is unlikely to be a protein coding RNA and likely acts as a lincRNA at a steady transcript level outside the nucleus.
Next, we searched for MORT orthologs in other species. Higher primates (chimpanzee, gorilla, and orangutan) have regions with high homology to human MORT in a homologous genomic locus, and publicly available RNA-seq data from these species indicate that it is expressed. Old world monkeys (baboon, rhesus) have a region homologous to the first exon of human MORT in the homologous genomic location; however, they do not have a region with homology to the second MORT exon at this genomic locus. Therefore, MORT seems to be an evolutionarily young lincRNA that has evolved during late primate evolution and is limited to higher primates. The emergence of a lincRNA that might have tumor suppressive activity during higher primate evolution is consistent with the long life span of higher primates and with the fact that a large fraction of noncoding RNAs found in humans are primate specific.25
Since the silencing of gene expression is frequently acompanied by DNA methylation of its promoter region, we analyzed the DNA methylation status of the MORT promoter in mortal versus immortal HMEC. The data show a consistent increase in DNA methylation of the MORT promoter in immortalized cells across the 4 individual specimens (Fig. 3D). Therefore, the lincRNA MORT seems to belong to genes that are silenced by DNA methylation of their promoters.
To find out whether MORT silencing is a more general process linked to human carcinogenesis, we first analyzed our earlier microarray expression data set from multiple breast tumors and non-tumor tissues (Figs. 4A and S6). The microarray data show deregulation of MORT transcript level across the tumor samples with a decrease in expression being the predominant trend. Similar results were found within a panel of breat tumor cell lines from the cancer cell line encyclopedia (CCLE)26 -about one half of the tumor cell lines do not express any MORT and the level in the other half is deregulated compared to non-tumor breast tissue (Figures S6 and S7 ). These data suggest that MORT silencing is a common event in human breast carcinogenesis.
Figure 4.

MORT expression and promoter methylation in breast tissue samples and in TCGA breast invasive carcinoma cohort. (A) MORT transcript level in a cohort of 27 breast carcinomas and 8 breast non-tumor samples as determined by Affymetrix microarray. MORT expression (B) and DNA methylation (C) in the large cohort of TCGA breast invasive carcinoma data. P-values are from t-test. (D) integration of DNA methylation and exression data of TCGA cohort.
We used TCGA data to further empower our analysis of MORT in breast cancer. RNA-seq data of the MORT transcript and Illumina HumanMethylation450 DNA methylation data that cover several CpGs within MORT promoter region were integrated for over 800 clinical breast tumor or non-tumor tissue samples. The integrated transcriptome and DNA methylation data show a highly significant decrease in the level of MORT expression in tumors relative to non-tumor breast tissue (Fig. 4B) that is linked to a significant DNA hypermethylation of the MORT promoter (Fig. 4C). The strong negative correlation (rho = −0.77) between DNA methylation and the expression of this lncRNA (Fig. 4D) strongly suggests that MORT is epigenetically silenced in most breast cancers.
To expand the analysis of MORT's role in human carcinogenesis beyond breast cancer, we analyzed TCGA data from 16 additional tumor types that represent the top 10 most frequent cancers found in males and females according to Cancer Facts and Figures 2015.27 MORT is deregulated in 14 out of the 16 tumor types analyzed (Table 1, Fig. S8). The majority of the tumor samples have MORT silenced in association with hypermethylation of its CpG island promoter. There is overall strong negative correlation (rho = −0.6 to −0.8, Table 1) between MORT promoter methylation and the MORT transcript level for these 14 tumor types. The two exceptions where MORT transcript level is not decreased and its promoter stays unmethylated are prostate adenocarcinomas and thyroid carcinomas. Overall, these analyses suggest that MORT is generally silenced by DNA methylation in a majority of human cancer types.
Table 1.
Correlation between MORT RNA level and MORT promoter methylation for the 17 most common cancer types. The table shows the TCGA cancer type and disease abbreviation and correlation coeficient rho between DNA methylation of MORT promoter and MORT RNA level in individual TCGA samples for particular cancer type.
| Cancer Type | rho |
|---|---|
| Acute Myeloid Leukemia [LAML] | −0.65 |
| Bladder Urothelial Carcinoma [BLCA] | −0.79 |
| Breast invasive carcinoma [BRCA] | −0.77 |
| Colon adenocarcinoma [COAD] | −0.58 |
| Head and Neck squamous cell carcinoma [HNSC] | −0.62 |
| Kidney renal clear cell carcinoma [KIRC] | −0.78 |
| Kidney renal papillary cell carcinoma [KIRP] | −0.71 |
| Liver hepatocellular carcinoma [LIHC] | −0.62 |
| Lung adenocarcinoma [LUAD] | −0.80 |
| Lung squamous cell carcinoma [LUSC] | −0.78 |
| Lymphoid Neoplasm Diffuse Large B-cell Lymphoma [DLBC] | −0.66 |
| Pancreatic adenocarcinoma [PAAD] | −0.72 |
| Prostate adenocarcinoma [PRAD] | −0.19 |
| Rectum adenocarcinoma [READ] | −0.60 |
| Skin Cutaneous Melanoma [SKCM] | −0.66 |
| Thyroid carcinoma [THCA] | −0.40 |
| Uterine Corpus Endometrial Carcinoma [UCEC] | −0.73 |
To confirm that epigenetic mechanisms are functionally involved in cancer cell specific silencing of MORT, we tested whether its silencing could be reactivated by treatment with the epigenetic modifier and DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine (5-AdC). The MORT-negative breast tumor lines MDA-MB231, MDA-MB453, and MCF7 (Figures S6 and S7) were exposed to 1 µM 5-AdC for 96 h. Each of the 3 tumor cell lines showed a significant decrease in the level of DNA methylation of the MORT promoter and reactivation of MORT expression after the 5-AdC treatment (Fig. 5). The treatment of immortalized MORT negative HMEC line 184Dp16sMY by 5-AdC also resulted in reactivation of MORT transcription (Fig. S9). These data further support that the silencing of MORT expression in cancer is due to the aberrant DNA methylation of its promoter.
Figure 5.
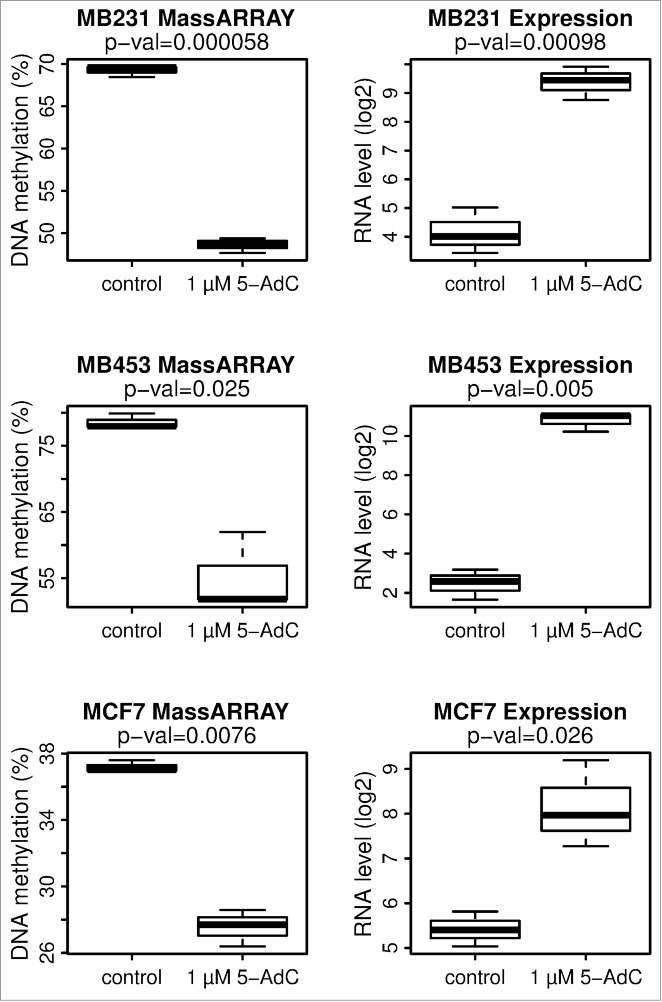
MORT is reactivated by 5-AdC treatment in 3 MORT negative breast carcinoma tumor cell lines. MDA-MB231, MDA-MB453, and MCF7 cells were grown for 96 h in the presence or absence of 1 µM 5-AdC. The experiment was repeated 3 times. DNA methylation of MORT promoter was determined by Sequenom MassARRAY and MORT transcript level by real-time PCR. P-values are from paired t-test.
Discussion
In an effort to better understand the transcriptional events associated with pathologic immortalization, we analyzed gene expression changes that arise during the controlled genetic immortalization of HMEC. This two-step model of non-clonal HMEC immortalization uses pathologically relevant genetic changes that bypass defined tumor-suppressive proliferation barriers and reproducibly create immortal, non-malignant cell lines that lack the gross genomic changes and passenger errors typically observed in tumors.1,2 We reasoned that this model system would allow for the identification of consistent gene expression changes pertinent to epithelial cell immortalization itself.
We did not observe any consistent gene expression changes among the different HMEC strains that bypassed the initial stasis proliferation barrier through p16 inhibition or cyclin D1 overexpression. Since p16 appears to be the prime mediator of cell stasis induced by sub-optimal environmental conditions, gene expression differences between the pre-stasis cells and those that were made post-stasis by transduction with p16shRNA prior to exposure to such sub-optimal environmental conditions, could well be expected to be minimal and variable among the different individual cell strains. With respect to the cyclin D1 overexpressing post-stasis cells, it is reasonable to predict that the overexpression of the ccnd1/CDK2 fusion gene resulting in hyperphosphorylated RB protein, as well as the likely phosphorylation of a number of other substrates, could lead to significant changes in gene expression; however, the D1 post-stasis group in our study was too small to have sufficient statistical power to decisively quantify these changes. Despite the minimal differences in gene expression between pre-stasis and post-stasis HMEC cultures, they display at least one significant behavioral difference—MYC can readily immortalize post-stasis HMEC, but cannot immortalize pre-stasis HMEC.1,2
In contrast to p16 shRNA-mediated bypass of cell stasis, the MYC-mediated bypass of replicative senescence and acquisition of pathologic cell immortality resulted in significant changes in expression of hundreds of genes. Evaluation of gene ontologies that were overrepresented within the differentially expressed genes include biological processes such as cell death, apoptosis, stem cell division, and regulation of protein tyrosine kinase activity; all consistent with cellular pathways one would expect to be targeted during cellular immortalization. The gene ontology term with the most significant overrepresentation was the integrin-mediated signaling pathway, which has been long associated with human breast carcinogenesis.21-23 A comparison of these immortality-linked genes with TCGA expression data revealed that a large fraction of these genes are also significantly altered in clinical breast cancer. The clinical breast tumor samples, when compared to normal breast tissues, will have, in addition to changes linked to immortality and malignancy, additional changes in cell type specific genes, due to comparing carcinomas of clonal origin to heterogeneous normal tissues containing many cell types. Despite that, for genes changed in the immortalization model, there was a good concordance between the in vitro model and clinical cancer. The fact that the most dramatic gene expression changes found in the in vitro model of HMEC immortalization are also seen in clinical breast cancer samples reinforces the relevance of the model for the study of breast carcinogenesis and suggests further that these gene expression changes seen in clinical cancer may very well occur early in transformation, prior to frank malignancy. Taken together these results illustrate the similarities between non-malignant and malignant immortal cell lines, and fundamental distinctions between non-malignant immortalized lines and normal, finite lifespan cells, lending further significance to the immortalization step in cancer progression.1,28,29
The single gene with the greatest change in expression during the immortalization step is a long intergenic noncoding RNA with unknown function. We designated this lncRNA MORT, since it is present in all mortal human cells examined and is silenced during immortalization, and might therefore be involved in preserving cellular mortality. The MORT transcript does not have a capacity to code for ORFs longer than what happens by random chance and these ORFs do not share homology with known protein domains. The MORT transcript does not have a coding label according to CPAT.24 The recent study that annotated nearly 60,000 human noncoding transcripts3 included a ZNF667-AS1 transcript variant corresponding to MORT in the noncoding category.
MORT probably arose during late primate evolution, since MORT gene orthologs are only present in the genomes of higher primates. It is possible that the increased lifespan of higher primates necessitated the rise of additional mechanisms to control undesired cellular proliferation, with MORT serving such a function. The relative level of MORT expression in normal cells is moderate to low; typical, although not exclusive, to lncRNAs.25 The second exon of the MORT transcript contains regions homologous to 3 repetitive elements, indicating that this part of the gene evolved from repetitive elements. The presence of repetitive elements within the exons and its recent appearance during evolution is also a typical feature of long noncoding RNAs.4,30,31 The noncoding nature of MORT and its exclusivity to higher primates limits the experimental models available to study MORT function, although it does not diminish its potential importance in human cellular mortality or carcinogenesis.
MORT gene silencing in transformed cells appears epigenetic in nature; we found that the loss of MORT expression was linked to aberrant DNA methylation of the MORT promoter in immortal HMEC, breast cancer cell lines, and a large fraction of the most common human cancers. Based on our studies in the HMEC system, the timing of this epigenetic dysfunction appears to be at the boundary between the mortal and immortal phenotypes. The DNA methylation level in immortalized HMEC was relatively low, although consistent between samples; much higher levels were seen in cancer cell lines or in the TCGA tumor samples. This suggests that DNA methylation is likely a consequence, or a signature, of MORT silencing, rather than its primary cause. However, DNA methylation is likely involved in the reinforcement and maintenance of MORT silencing. The ability of 5-aza-2′-deoxycytidine to robustly reactivate MORT gene expression in both immortalized HMEC and human breast cancer cell lines underscores the functional relevance of DNA methylation in this gene's silent state. Together the timing of MORT silencing during the immortalization process and its epigenetic inactivation in 15 out of the 17 most common clinical cancers suggests MORT silencing might be involved in the early, foundational stages of human carcinogenesis.
The functional role of MORT in normal and diseased cell physiology remains undetermined, with studies currently in progress. Since MORT is expressed in all finite lifespan cells and downregulated in many immortal cells, it likely plays an active role in maintaining the mortal state. Based on the circumstantial evidence to date, we hypothesize 2 complementary but distinct possible mechanisms for MORT function. It may be involved in the mechanisms that normally repress telomerase activity and telomere maintenance in finite cells, and its expression may be incompatible with an immortal state. Or MORT may be involved in mechanisms that induce cell senescence independent of telomere length, such as cell cycle components, and impose senescence under certain conditions that could otherwise lead to inappropriate cell proliferation. Our preliminary data investigating the function of MORT indicate that the MORT transcript is exported out of the nucleus and is enriched in cellular fractions containing ribosomes (unpublished data), suggesting that it might be involved in the regulation of translation of its targets. The rather long half-life of the MORT transcript indicates that the MORT RNA likely functions at steady transcript levels that do not vary during the cell cycle.
In conclusion, we have identified and characterized a lincRNA, MORT, that we found to be 1) epigenetically silenced at the immortality boundary in an experimental model of HMEC immortalization and 2) epigenetically silenced in a large majority of the most common human cancers. The temporal order of MORT loss during the in vitro arc of malignant transformation and the frequency of its aberrant silencing in clinical cancer suggests a possible important role during human carcinogenesis. Consequently, MORT may potentially represent a new target for cancer therapeutic intervention.
Materials and Methods
Cell culture
Finite and immortalized HMEC were generated and grown as previously described.1,2,32 In brief, finite lifespan HMEC from specimens 184 batch D, 240 L batch B, 122 L and 805P were obtained from reduction mammoplasty tissue or normal tissue peripheral to a tumor (805P) from women aged 21, 19, 66, and 91 years, respectively. The cells were grown in M87A medium supplemented with 0.5 ng/ml cholera toxin, and 0.1 nM oxytocin (Bachem). Retroviral transduction was performed as described.1,2 Breast tumor lines MDA-MB231, MDA-MB453, and MCF7 were grown as previously described.33,34
5-aza-2′-deoxycytidine treatment
Cells were treated with 1 µM 5-aza-2′-deoxycytidine (Sigma) for 96 h, as previously described.33
Nucleic acid isolation
Genomic DNA was isolated using the DNeasy Blood and Tissue Kit according to the manufacturer's protocol (Qiagen). Total RNA was harvested using TRIzol and purified using the miRNeasy Kit (Qiagen). The quantity of each sample was assessed using absorbance at 260 nm on the NanoDrop 1000 Spectrophotometer.
Microarray analysis
RNA labeling and hybridization to Affymetrix GeneChip® Human Gene 1.0 ST Arrays, and the microarray scanning was performed according to the manufacturer's protocols. The data were analyzed in R programming environment.35 The raw data from CEL files were normalized and summarized using package oligo.36 Differential expression was tested using the package limma.37 All p-values were adjusted to control the false discovery rate according to Benjamini and Hochberg's. Enrichment of Gene Ontology terms in differentially expressed genes was tested using package GOstats.38
The size of 2 standard deviations above the mean for random ORF was calculated using function y = 91*ln(x)−330 as described.39
DNA methylation analysis by MassARRAY
DNA methylation analysis by Sequenom MassARRAY (Sequenom) was performed as described.40 Primer sequences are listed in Table S3. Oligonucleotides used for MassARRAY analysis were ordered from Integrated DNA Technologies. Two MassARRAY amplicons covered 800 bp region including the first MORT exon and about 500 bp upstream MORT transcription start (Fig. 3C). The mean DNA methylation of all 26 informative CpG units within MassARRAY amplicons was used for data presentation and statistical analysis.
Real-time RT-PCR
The reverse transcription and real-time PCR was performed as previously described.33,41 Primers were designed for MORT, GAPDH, MYC, and XIST and used with the Human Universal Probe Library Set (Roche Diagnostics). Primer sequences are available upon request. The cycle threshold (Ct) MORT values were normalized between the samples using GAPDH Ct values. The data were then converted into a value that is in log2 scale, but increases with the expression level using formula 40-Ct. Since 40 was the total number of PCR cycles it was the lowest detectable expression level and therefore it was considered the background and set as 0 to display the data in the plots.
Online data analysis
The Illumina Human Body Map 2.0 transcriptome RNA-Seq data were downloaded from SRA (ERP000546) and aligned to hg19 human reference using tophat.42 The aligned RNA seq data for breast cancer cell lines were downloaded from Cancer Genomics Hub. MORT rpkm values were determined using samtools.43 and custom R scripts. RNASeqV2 and Illumina HumanMethylation450 DNA methylation data for tumor and normal tissue samples were downloaded from The Cancer Genome Atlas Data Portal. Mean RNA-Seq rpkm values for exons constituting the MORT RNA were plotted against the mean DNA methylation of the 7 CpGs from MORT promoter region for individual samples. The spearman correlation coefficient rho between RNA level and DNA methylation was calculated using function cor.test. For the GO categories comparison plots the raw TCGA RNA-Seq counts for individual exons were normalized using voom function44 from the package limma and the differential expression between tumor and normal samples was calculated using the limma package.
Accession Numbers
The Gene Expression Omnibus (GEO) accession number for microarray dataset is GSE72353.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Micheal P. Hailey for outstanding technical assistance. The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Funding
This work was supported by the Margaret E. and Fenton L. Maynard Endowment for Breast Cancer Research and Center grants ES006694, CA23074, CA65662, P42 ES04940, LBNL DOE: the US. Department of Energy (DE-AC02-05CH11231).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
References
- 1.Garbe JC, Vrba L, Sputova K, Fuchs L, Novak P, Brothman AR, Jackson M, Chin K, LaBarge MA, Watts G, et al.. Immortalization of normal human mammary epithelial cells in two steps by direct targeting of senescence barriers does not require gross genomic alterations. Cell Cycle 2014; 13:3423-35; PMID:25485586; http://dx.doi.org/ 10.4161/15384101.2014.954456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee JK, Garbe JC, Vrba L, Miyano M, Futscher BW, Stampfer MR, LaBarge MA. Age and the means of bypassing stasis influence the intrinsic subtype of immortalized human mammary epithelial cells. Front Cell Dev Biol 2015; 3:13; PMID:25815289; http://dx.doi.org/ 10.3389/fcell.2015.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al.. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet 2015; 47:199-208; PMID:25599403; http://dx.doi.org/ 10.1038/ng.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell 2013; 154:26-46; PMID:23827673; http://dx.doi.org/ 10.1016/j.cell.2013.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keniry A, Oxley D, Monnier P, Kyba M, Dandolo L, Smits G, Reik W. The H19 lincRNA is a developmental reservoir of miR-675 that suppresses growth and Igf1r. Nat Cell Biol 2012; 14:659-65; PMID:22684254; http://dx.doi.org/ 10.1038/ncb2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014; 157:77-94; PMID:24679528; http://dx.doi.org/ 10.1016/j.cell.2014.03.008 [DOI] [PubMed] [Google Scholar]
- 7.Brannan CI, Dees EC, Ingram RS, Tilghman SM. The product of the H19 gene may function as an RNA. Mol Cell Biol 1990; 10:28-36; PMID:1688465; http://dx.doi.org/ 10.1128/MCB.10.1.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem 2012; 81:145-66; PMID:22663078; http://dx.doi.org/ 10.1146/annurev-biochem-051410-092902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N. Requirement for Xist in X chromosome inactivation. Nature 1996; 379:131-7; PMID:8538762; http://dx.doi.org/ 10.1038/379131a0 [DOI] [PubMed] [Google Scholar]
- 10.Gendrel AV, Heard E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Annu Rev Cell Dev Biol 2014; 30:561-80; PMID:25000994; http://dx.doi.org/ 10.1146/annurev-cellbio-101512-122415 [DOI] [PubMed] [Google Scholar]
- 11.Heard E, Chaumeil J, Masui O, Okamoto I. Mammalian X-chromosome inactivation: an epigenetics paradigm. Cold Spring Harbor Symposia Quantitative Biol 2004; 69:89-102; PMID:16117637; http://dx.doi.org/ 10.1101/sqb.2004.69.89 [DOI] [PubMed] [Google Scholar]
- 12.Lee Jeannie T, Bartolomei Marisa S. X-Inactivation, Imprinting, and Long Noncoding RNAs in Health and Disease. Cell 2013; 152:1308-23; PMID:23498939; http://dx.doi.org/ 10.1016/j.cell.2013.02.016 [DOI] [PubMed] [Google Scholar]
- 13.Vincent-Salomon A, Ganem-Elbaz C, Manie E, Raynal V, Sastre-Garau X, Stoppa-Lyonnet D, Stern MH, Heard E. X inactive-specific transcript RNA coating and genetic instability of the X chromosome in BRCA1 breast tumors. Cancer Res 2007; 67:5134-40; PMID:17545591; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0465 [DOI] [PubMed] [Google Scholar]
- 14.Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, et al.. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Sci 2013; 341:1237973; PMID:23828888; http://dx.doi.org/ 10.1126/science.1237973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rinn JL, Kertesz M, Wang JK, Squazzo SL, Xu X, Brugmann SA, Goodnough LH, Helms JA, Farnham PJ, Segal E, et al.. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007; 129:1311-23; PMID:17604720; http://dx.doi.org/ 10.1016/j.cell.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet 2011; 12:861-74; PMID:22094949; http://dx.doi.org/ 10.1038/nrg3074 [DOI] [PubMed] [Google Scholar]
- 17.Ling H, Vincent K, Pichler M, Fodde R, Berindan-Neagoe I, Slack FJ, Calin GA. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015; 34(39):5003-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitra SA, Mitra AP, Triche TJ. A central role for long non-coding RNA in cancer. Front Genet 2012; 3:17; PMID:22363342; http://dx.doi.org/ 10.3389/fgene.2012.00017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al.. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010; 464:1071-6; PMID:20393566; http://dx.doi.org/ 10.1038/nature08975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et al.. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene 2003; 22:8031-41; PMID:12970751; http://dx.doi.org/ 10.1038/sj.onc.1206928 [DOI] [PubMed] [Google Scholar]
- 21.Pontier SM, Muller WJ. Integrins in mammary-stem-cell biology and breast-cancer progression–a role in cancer stem cells? J Cell Sci 2009; 122:207-14; PMID:19118213; http://dx.doi.org/ 10.1242/jcs.040394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizejewski GJ. Role of integrins in cancer: survey of expression patterns. Proc Soc Exp Biol Med 1999; 222:124-38; PMID:10564536; http://dx.doi.org/ 10.1046/j.1525-1373.1999.d01-122.x [DOI] [PubMed] [Google Scholar]
- 23.Hynes RO. Integrins: Bidirectional, Allosteric Signaling Machines. Cell 2002; 110:673-87; PMID:12297042; http://dx.doi.org/ 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Park HJ, Dasari S, Wang S, Kocher JP, Li W. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res 2013; 41:e74; PMID:23335781; http://dx.doi.org/ 10.1093/nar/gkt006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al.. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res 2012; 22:1775-89; PMID:22955988; http://dx.doi.org/ 10.1101/gr.132159.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al.. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483:603-7; PMID:22460905; http://dx.doi.org/ 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.American_Cancer_Society. Cancer Facts & Figures 2015. In: Society AC, ed. Atlanta: American Cancer Society, 2015 [Google Scholar]
- 28.Li Y, Pan J, Li JL, Lee JH, Tunkey C, Saraf K, Garbe JC, Whitley MZ, Jelinsky SA, Stampfer MR, et al.. Transcriptional changes associated with breast cancer occur as normal human mammary epithelial cells overcome senescence barriers and become immortalized. Mol Cancer 2007; 6:7; PMID:17233903; http://dx.doi.org/ 10.1186/1476-4598-6-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stampfer MR, LaBarge MA, Garbe JC. An Integrated Human Mammary Epithelial Cell Culture System for Studying Carcinogenesis and Aging In: Schatten H, ed. Cell and Molecular Biology of Breast Cancer. New York: Springer Science+Business Media, 2013:323-61 [Google Scholar]
- 30.Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep 2015; 11:1110-22; PMID:25959816; http://dx.doi.org/ 10.1016/j.celrep.2015.04.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kapusta A, Kronenberg Z, Lynch VJ, Zhuo X, Ramsay L, Bourque G, Yandell M, Feschotte C. Transposable elements are major contributors to the origin, diversification, and regulation of vertebrate long noncoding RNAs. PLoS Genet 2013; 9:e1003470; PMID:23637635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res 2009; 69:7557-68; PMID:19773443; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oshiro MM, Watts GS, Wozniak RJ, Junk DJ, Munoz-Rodriguez JL, Domann FE, Futscher BW. Mutant p53 and aberrant cytosine methylation cooperate to silence gene expression. Oncogene 2003; 22:3624-34; PMID:12789271; http://dx.doi.org/ 10.1038/sj.onc.1206545 [DOI] [PubMed] [Google Scholar]
- 34.Rice JC, Massey-Brown KS, Futscher BW. Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene 1998; 17:1807-12; PMID:9778046; http://dx.doi.org/ 10.1038/sj.onc.1202086 [DOI] [PubMed] [Google Scholar]
- 35.R_Development_Core_Team R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing, 2015 [Google Scholar]
- 36.Carvalho B, Bengtsson H, Speed TP, Irizarry RA. Exploration, normalization, and genotype calls of high-density oligonucleotide SNP array data. Biostatistics 2007; 8:485-99; PMID:17189563; http://dx.doi.org/ 10.1093/biostatistics/kxl042 [DOI] [PubMed] [Google Scholar]
- 37.Smyth GK. Limma: linear models for microarray data In: Gentleman RCV, Huber W, Irizarry R, Dudoit S, ed. Bioinformatics and Computational Biology Solutions using R and Bioconductor. New York: Springer, 2005:397-420 [Google Scholar]
- 38.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics 2007; 23:257-8; PMID:17098774; http://dx.doi.org/ 10.1093/bioinformatics/btl567 [DOI] [PubMed] [Google Scholar]
- 39.Dinger ME, Pang KC, Mercer TR, Mattick JS. Differentiating protein-coding and noncoding RNA: challenges and ambiguities. PLoS Comput Biol 2008; 4:e1000176; PMID:19043537; http://dx.doi.org/ 10.1371/journal.pcbi.1000176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novak P, Jensen TJ, Garbe JC, Stampfer MR, Futscher BW. Stepwise DNA methylation changes are linked to escape from defined proliferation barriers and mammary epithelial cell immortalization. Cancer Res 2009; 69:5251-8; PMID:19509227; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vrba L, Junk DJ, Novak P, Futscher BW. p53 induces distinct epigenetic states at its direct target promoters. BMC Genomics 2008; 9:486; PMID:18922183; http://dx.doi.org/ 10.1186/1471-2164-9-486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 2009; 25:1105-11; PMID:19289445; http://dx.doi.org/ 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009; 25:2078-9; PMID:19505943; http://dx.doi.org/ 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014; 15:R29; PMID:24485249; http://dx.doi.org/ 10.1186/gb-2014-15-2-r29 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.