

Abstract
Loss of 3p11-p14 is a frequent event in epithelial cancer and a candidate prognostic biomarker in cervical cancer. In addition to loss, promoter methylation can participate in gene silencing and promote tumor aggressiveness. We have performed a complete mapping of promoter methylation at 3p11-p14 in two independent cohorts of cervical cancer patients (n = 149, n = 121), using Illumina 450K methylation arrays. The aim was to investigate whether hyperm-ethylation was frequent and could contribute to gene silencing and disease aggressiveness either alone or combined with loss. By comparing the methylation level of individual CpG sites with corresponding data of normal cervical tissue, 26 out of 41 genes were found to be hypermethylated in both cohorts. The frequency of patients with hypermethylation of these genes was found to be higher at tumor stages of 3 and 4 than in stage 1 tumors. Seventeen of the 26 genes were transcriptionally downregulated in cancer compared to normal tissue, whereof 6 genes showed a significant correlation between methylation and expression. Integrated analysis of methylation, gene dosage, and expression of the 26 hypermethylated genes identified 3 regulation patterns encompassing 8 hypermethylated genes; a methylation driven pattern (C3orf14, GPR27, ZNF717), a gene dosage driven pattern (THOC7, PSMD6), and a combined methylation and gene dosage driven pattern (FHIT, ADAMTS9, LRIG1). In survival analysis, patients with both hypermethylation and loss of LRIG1 had a worse outcome compared to those harboring only hypermethylation or none of the events. C3orf14 emerged as a novel methylation regulated suppressor gene, for which knockdown was found to promote invasive growth in human papilloma virus (HPV)-transformed keratinocytes. In conclusion, hypermethylation at 3p11-p14 is common in cervical cancer and may exert a selection pressure during carcinogenesis alone or combined with loss. Information on both events could lead to improved prognostic markers.
Keywords: 3p, cervical cancer, chromosomal loss, gene expression, integrative genomic profiling, promoter methylation, tumor suppressor genes
Introduction
Chromosomal loss and hypermethylation of CpG sites in promoter regions are major mechanisms of gene silencing in cancer.1-3 Strong evidences for an important role of both events in the carcinogenesis of cervical cancer have been presented.4-8 A connection between the 2 events has been hypothesized and was recently supported by a genome wide study in breast cancer, showing significant enrichment of differentially methylated regions at chromosomal breakpoints like fragile sites.9 Hence, promoter methylation may cooperate with loss in gene silencing and possibly enhance the malignant phenotype associated with the chromosomal aberration or promote chromosomal instability and loss of the unaffected allele.10,11 This has been demonstrated in experimental studies where loss of gene function was associated with hypermethylation of one allele coupled with mutation of the other allele.12,13 Promoter methylation and loss may also be alternative routes toward increased malignancy where gene silencing is caused by methylation in some tumors or tumor clones and loss in others. To fully understand how the 2 events contribute in the carcinogenesis of cervical cancer, their possible individual and combined effects on gene silencing must be clarified.
Loss on chromosome 3p encompassing the fragile site FRA3B at p14.2 is a frequent event in many epithelial cancers and a candidate prognostic biomarker of cervical cancer.5,14,15 These findings indicate an important role of the loss in cancer development, and efforts have been undertaken to understand its biological meaning. In cervical cancer, the loss seems to constitute a selection advantage early in the invasive phase of the disease, promoting a treatment resistant phenotype.16 By a complete transcript mapping of the prognostic 3p11.2-p14.2 region, we have identified 8 candidate targets of the loss; THOC7, PSMD6, SLC25A26, TMF1, RYBP, SHQ1, EBLN2, and GBE1, for which silencing was associated with activation of tumorigenic pathways in proliferation and anti-apoptosis.16 However, the region harbors other potential suppressor genes, like ROBO1, FHIT, and FAM19A4, which might primarily be regulated by methylation.17-19 More comprehensive studies, integrating promoter methylation and chromosomal loss of the entire prognostic region, would reveal a possible interplay between the two events in gene silencing and thereby provide a better understanding of the aggressive phenotype predicted by the loss.
In the present study we have performed a complete mapping of promoter methylation at 3p11.2-p14.2 in cervical cancer using the Illumina 450K methylation arrays, which detect the methylation level of more than 485 000 CpG sites and cover 96% of CpG islands and 99% of all genes in the RefSeq database.20 The aim was to investigate whether hypermethylation was a frequent event and could contribute to gene silencing and disease aggressiveness, either alone or in combination with loss. The methylation data was found to be highly consistent at the level of individual CpG sites in two independent patient cohorts, demonstrating considerable robustness in the technology. Hypermethylated sites were identified by comparing the methylation level to the corresponding level in normal cervical tissue. The methylation data were further integrated with gene dosage and expression data. This approach proposed individual as well as combined effects of methylation and loss on gene silencing and suggested that the regulation pattern could influence the aggressive phenotype of the disease. Moreover, a novel methylation regulated suppressor candidate was discovered, for which downregulation was shown to promote invasive growth in human papilloma virus (HPV)-transformed keratinocytes.
Results
Hypermethylated CpG sites on 3p gene promoters
Based on an investigation cohort of 149 patients (cohort 1; Table S1), 559 CpG sites were mapped to the promoter region of 48 genes at 3p11.2-p14.2. After data preprocessing, including exclusion of sites with low variation in their β-value across the samples, 229 CpGs on 41 genes remained. To identify hypermethylated sites that were differentially methylated between normal and tumor samples, the β-value of each site and tumor was compared to the β-distribution of the corresponding site in an external methylation data set of 20 normal cervical samples (GSE46306). A CpG site of a tumor sample was defined as hypermethylated if β exceeded the median plus 2 times the standard deviation of the corresponding site in the normal samples. This resulted in 150 CpGs on 26 genes which were hypermethylated in at least 10% of the patients (Fig. 1). Frequency of chromosomal loss on the same region ranged from 55% to 60% (Fig. 1). When stratifying the patients based on FIGO stage, a gradual increase in the average hypermethylation frequency of the 26 genes with increasing stage, which reached statistical significance when comparing stage 1 and stage 3/4 tumors, was seen (P = 0.04; Fig. S1). For chromosomal loss, the frequency was higher for stage 1 compared to stage 2, 3, and 4 (P < 0.001).
Figure 1.
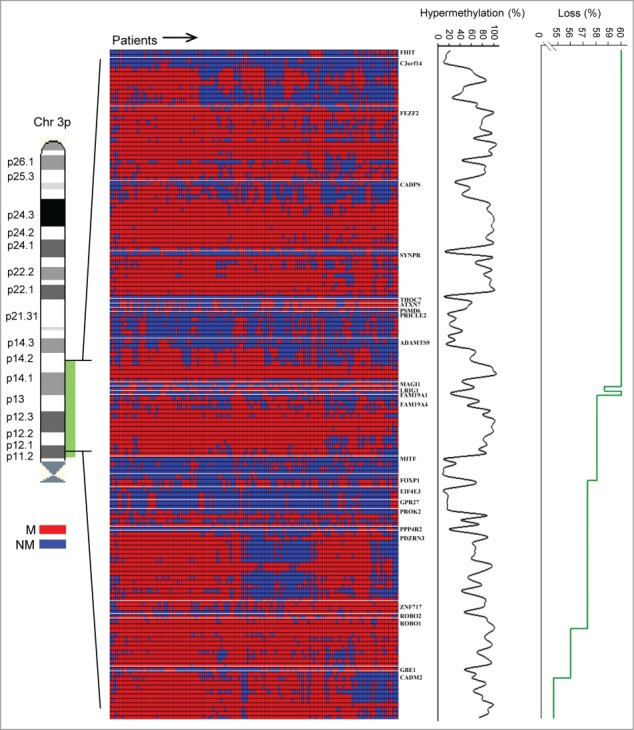
Hypermethylated CpG sites. Methylation status of 150 CpG sites on 26 genes for 149 cervical cancer patients in cohort 1. Patients are shown in columns and CpG sites found to be hypermethylated in tumors compared to normal tissue in at least 10% of the patients are ordered by chromosomal location in rows with gene symbols indicated. Hypermethylated (M) and not hypermethylated (NM) sites are indicated with red and blue color, respectively. Frequency of patients with hypermethylation and chromosomal loss is shown for each site by the black and green curves, respectively.
The number of hypermethylated sites differed considerably among the 26 genes, ranging from a single to 17 CpGs. To ensure reliability in our results, we performed a probe based validation in an independent cohort of 121 patients (cohort 2; Table S1). All 150 sites were found to be hypermethylated in at least 4 patients (3.3%) in cohort 2 and a highly significant correlation was found between the hypermethylation frequencies of individual CpG sites in the 2 cohorts (Fig. 2; Table S2). This supports the findings in Figure 1 and indicates that the observed frequencies are characteristic of cervical cancer. The 26 genes were candidates for methylation controlled silencing and were selected for the further analyses (Table 1).
Figure 2.
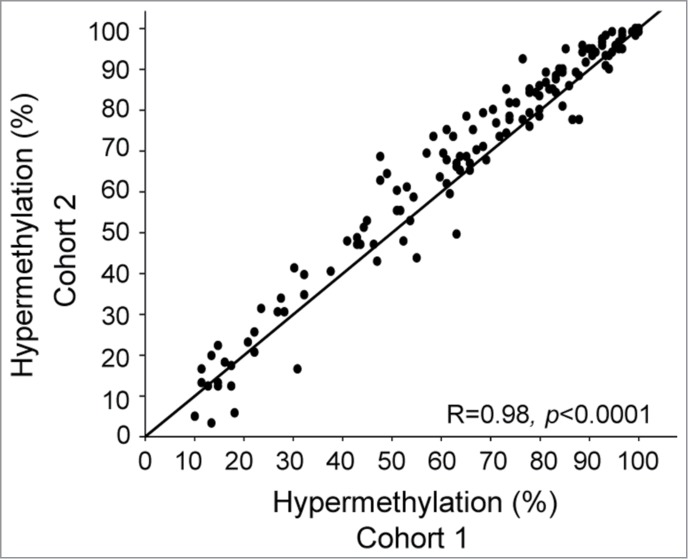
Validation of hypermethylation frequency in cohort 2. Frequency of 121 cohort 2 patients with hypermethylation versus the corresponding frequency for 149 cohort 1 patients. In total, 150 CpG sites found to be hypermethylated in cohort 1 are shown, and each dot represents the hypermethylation frequency of an individual site. Pearson's correlation coefficient and P-value are indicated. Line of unity is included.
Table 1.
Hypermethylated genes on 3p11.2-p14.2 in cervical cancer
| Gene symbola | Gene name | Hypermethylation frequency (%)b | Regulation patternc |
|---|---|---|---|
| FHIT | Fragile histidine triad | 18.5 | Combined |
| C3orf14 | Chromosome 3 open reading frame 14 | 48.6 | Methylation |
| FEZF2 | FEZ family zinc finger 2 | 81.4 | ND |
| CADPS | Ca2+-dependent secretion activator | 78.4 | ND |
| SYNPR | Synaptoporin | 74.8 | ND |
| THOC7 | THO complex 7 homolog (Drosophila) | 55.0 | Gene dosage |
| ATXN7 | Ataxin 7 | 55.0 | ND |
| PSMD6 | Proteasome (prosome, macropain) 26S subunit, non-ATPase, 6 | 46.3 | Gene dosage |
| PRICKLE2 | Prickle homolog 2 (Drosophila) | 26.7 | ND |
| ADAMTS9 | ADAM metallopeptidase with thrombospondin type 1 motif, 9 | 63.6 | Combined |
| MAGI1 | Membrane associated guanylate kinase, WW and PDZ domain containing 1 | 51.0 | ND |
| LRIG1 | Leucine-rich repeats and immunoglobulin-like domains 1 | 63.1 | Combined |
| FAM19A1 | Family with sequence similarity 19 (chemokine (C-C motif)-like), member A1 | 23.5 | ND |
| FAM19A4 | Family with sequence similarity 19 (chemokine (C-C motif)-like), member A4 | 82.4 | ND |
| MITF | Microphthalmia-associated transcription factor | 16.6 | ND |
| FOXP1 | Forkhead box P1 | 45.2 | ND |
| EIF4E3 | Eukaryotic translation initiation factor 4E family member 3 | 15.4 | ND |
| GPR27 | G protein-coupled receptor 27 | 16.8 | Methylation |
| PROK2 | Prokineticin 2 | 57.9 | ND |
| PPP4R2 | Protein phosphatase 4, regulatory subunit 2 | 20.8 | ND |
| PDZRN3 | PDZ domain containing ring finger 3 | 70.0 | ND |
| ZNF717 | Zinc finger protein 717 | 69.4 | Methylation |
| ROBO2 | Roundabout, axon guidance receptor, homolog 2 (Drosophila) | 81.2 | ND |
| ROBO1 | Roundabout, axon guidance receptor, homolog 1 (Drosophila) | 89.3 | ND |
| GBE1 | Glucan (1,4-α-), branching enzyme 1 | 47.7 | ND |
| CADM2 | Cell adhesion molecule 2 | 79.0 | ND |
Genes are listed by chromosomal location.
Based on cohort 1of 149 patients.
Combined, combined methylation and gene dosage driven regulation; Methylation, methylation driven regulation; Gene dosage, gene dosage driven regulation; ND, none detected.
Silencing of hypermethylated genes
Relationships between hypermethylation and silencing of the genes were investigated by analyzing their expression in normal tissues and cancer. First, the expression was compared between tumors and normal cervical samples in 3 external datasets (GSE6791, GSE7803, GSE9750). In total, 17 genes had a mean expression that was lower in tumors than in normal tissues and significantly downregulated at an adjusted (adj) P ≤ 0.10 in at least one of the data sets (Fig. 3A). The downregulated genes included many of the most frequently hypermethylated genes from Figure 1, showing a hypermethylation frequency in the range from 16.6 to 89.3 in cohort 1 (Table 1).
Figure 3.
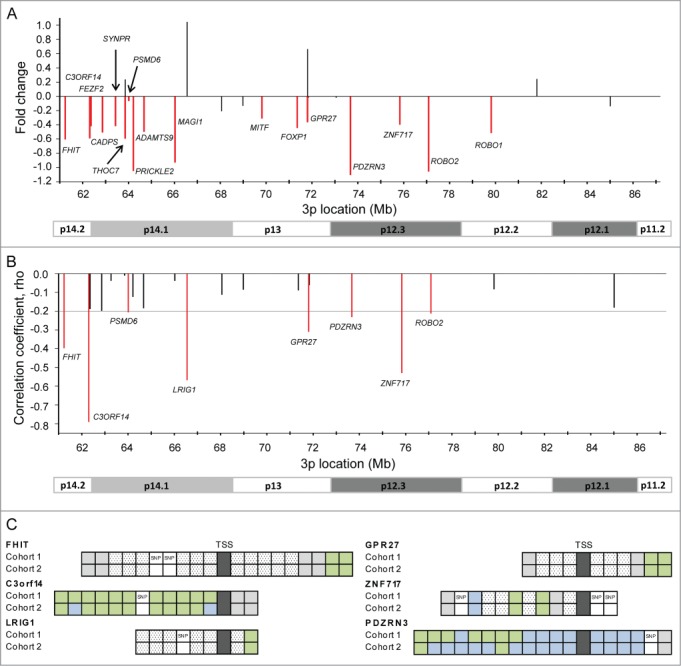
Identification and validation of silenced genes. (A) Difference in expression of 26 hypermethylated genes between tumors and normal cervical samples. The mean fold change based on 3 external datasets (GSE6791, GSE7803, GSE9750) is shown for each gene. Seventeen genes which were significantly downregulated at an adjusted P ≤ 0.10 in at least one of the data sets are indicated in red and their symbol is listed. (B) Correlation coefficients (rho) from Spearman's rank correlation analysis of methylation (β-value) against expression for 26 hypermethylated genes in 147 cervical tumors (cohort 1). Only negative correlations are shown, and in cases of several methylation and expression probes for the same gene, the most significant probes are presented. The horizontal line indicates the cut-off significance level, corresponding to rho = −0.20 (adj P ≤ 0.10). Eight significant genes are indicated in red and their symbol is listed. (C) Schematic illustration of the CpG sites for 6 significant genes in (B), which were validated in cohort 2. Significant CpG sites are indicated in green (adj P ≤ 0.10) and not significant sites in blue for 147 patients in cohort 1 and 121 patients in cohort 2. Sites in white were filtered during preprocessing due to their location closer than 10 bp from a SNP or low variation across the patients (hatched white; IQR < 0.08). Gray sites were hypermethylated in <10% of the patients. TSS: transcription start site.
Second, a possible correlation between methylation and expression was investigated in our tumor data sets. In cohort 1, 8 genes showed a highly significant negative correlation between the β-value of at least one CpG site and expression (adj P ≤ 0.10; Fig. 3B). For LRIG1, PSMD6, and ROBO2, significant correlation was found for the only CpG, whereas the others had 2 (FHIT, GPR27, ZNF717), 7 (PDZRN3), and 11 (C3orf14) significant CpGs (Fig. 3C; Table S2). The reproducibility of the results was investigated in cohort 2, where the expression data were based on another Illumina beadarray version. A similar correlation was found for all but 9 sites, using the same strict significance level and the same expression probe in cases of several probes representing the same gene (Fig. 3C). The six most significant genes in cohort 1 had at least one significant CpG in both cohorts, and for C3orf14, the correlation was confirmed for 9 CpGs, suggesting a high degree of reproducibility across patient cohorts. All together, these analyses indicate a relationship between promoter hypermethylation and silencing of specific genes within the region.
Combined effect of hypermethylation and loss on gene expression
To investigate a possible combined effect of hypermethylation and loss on the expression of the hypermethylated genes in Table 1, we divided 73 tumors with pairwise methylation, gene dosage, and expression data into 4 groups with different combinations of gene dosage (loss, no loss) and methylation status (hypermethylated, not hypermethylated) and performed a systematic comparison of the expression between the groups. By claiming an adjusted P ≤ 0.10, 3 different gene regulation patterns involving 8 genes were suggested; methylation driven regulation, gene dosage driven regulation, and combined methylation and gene dosage driven regulation (Table 1). In the methylation driven pattern, only hypermethylation, and not loss, was associated with downregulation. This pattern was seen for C3orf14, where downregulation by methylation was found in both the loss and no loss group, and for GPR27 and ZNF717, where downregulation was seen only in the loss group (Fig. 4A). In the gene dosage driven pattern, on the other hand, only loss was associated with downregulation, whereas hypermethylation seemed to have no effect. The strongly gene dosage regulated genes identified in previous work,16 THOC7 and PSMD6, showed this pattern. Downregulation by loss was seen in both the hypermethylated and not hypermethylated group for THOC7, but only in the not hypermethylated group for PSMD6 (Fig. 4B). Individual effect of either methylation or gene dosage on the expression level was thus suggested for 5 genes within the region.
Figure 4.
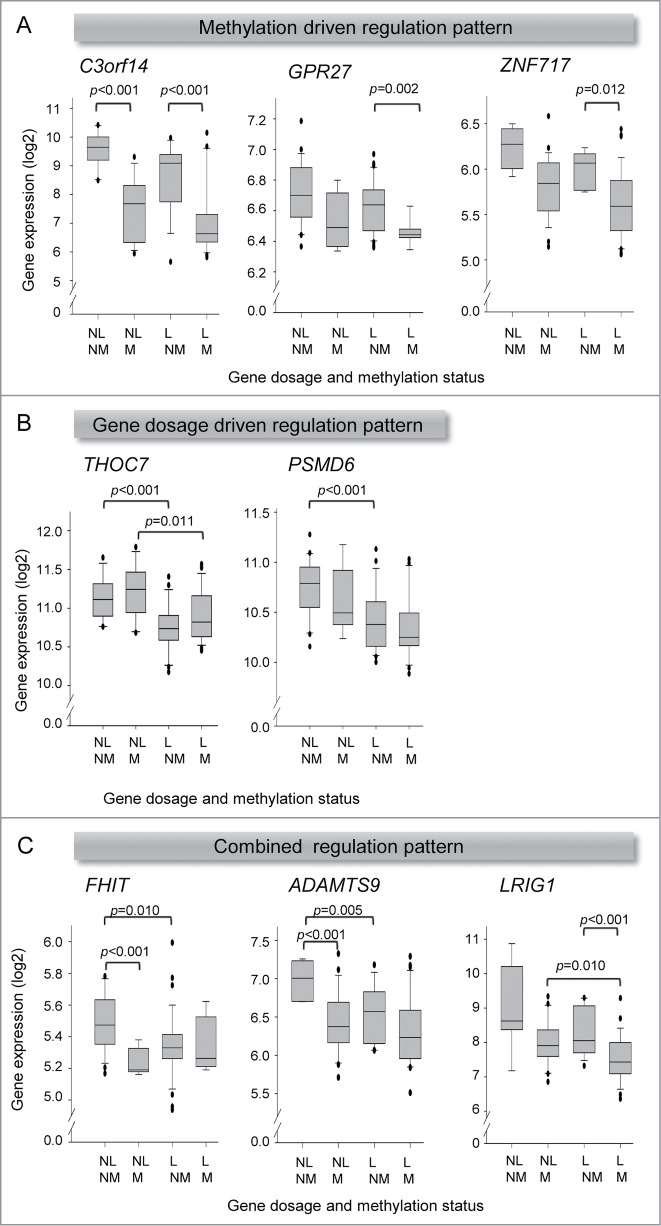
Individual and combined effect of loss and hypermethylation on gene expression. Box plots of gene expression in 4 groups of tumors with different combination of gene dosage and methylation status, demonstrating individual effect of methylation (A), individual effect of gene dosage (B), and combined effect of methylation and gene dosage (C) on gene expression. In total, 73 tumors from cohort 1 were included, for which gene expression, gene dosage, and methylation data were available. NL: no loss; L: loss; NM: not hypermethylated; M: hypermethylated. The median expression value of each group is indicated by the horizontal lines, and the edges of the boxes represent the first and third quartiles. P-values from Welch's t-test are indicated. All indicated differences had an adjusted P ≤ 0.10.
For FHIT, ADAMTS9, and LRIG1 a combined regulation pattern was observed where both hypermethylation and loss were associated with downregulation. For FHIT and ADAMTS9, downregulation by methylation was seen in the no loss group only, whereas downregulation by loss was seen in the not hypermethylated group (Fig. 4C). Hence, although both events were suggested to influence expression, only an individual effect depending on gene dosage and methylation status could be detected. Another variant of this pattern was seen for LRIG1, where significant downregulation occurred only when both events where present in the same tumors (Fig. 4C). The downregulation was not significant when only hypermethylation or loss was found. The two events combined may therefore possibly enhance the transcriptional repression, leading to a significant downregulation.
Since many tumors were aneuploid and therefore could have unaffected alleles in cases of both hypermethylation and loss, the analysis was also performed for the 38 near diploid tumors separately, for the 8 genes with a defined regulation pattern (Table S3). The same regulation patterns emerged, except for THOC7, which was on the borderline of significance for one of the combinations (data not shown). For GPR27, ZNF717, and FHIT, some tumor groups were too small (n ≤ 3) for statistical analysis. Moreover, the expression levels were similar regardless of whether only the near diploid or all tumors were considered, also for the combined loss and hypermethylated group.
Relationship to clinical outcome
Loss of 3p11.2-p14.2 has previously been shown to be associated with poor outcome for the patients included in the present study.16 To investigate whether inclusion of methylation data could add to the prognostic value of the loss, we selected the 8 genes with a defined regulation pattern (Fig. 4, Table 1) and generated survival curves for patients with different combinations of gene dosage and methylation status. For LRIG1, both loss and hypermethylation were associated with poor outcome in separate survival analyses (Fig. 5A, B). Moreover, a large difference in outcome between hypermethylated and not hypermethylated tumors was seen within the no loss group, where patients with hypermethylation had a poor survival compared to the patients without hypermethylation (Fig. 5C). The lowest survival probability was observed for the patients with loss, but in these cases the methylation status seemed to have only a minor effect on the outcome. In accordance with this, we also found that reduced gene expression of LRIG1 was associated with poor survival (Fig. 5D). For the other genes, no relationship to outcome was seen when the methylation status or the combined gene dosage and methylation status was considered.
Figure 5.
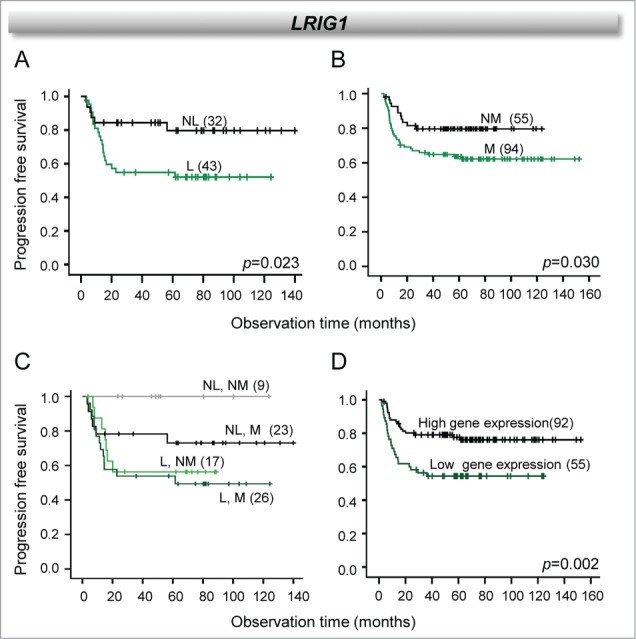
Individual and combined effect of LRIG1 loss and hypermethylation on clinical outcome. Kaplan-Meier curves of progression free survival for cervical cancer patients (cohort 1) with and without loss of LRIG1 (A), with and without hypermethylation of LRIG1 (B), the 2 combined (C), and high and low LRIG1 expression (D). P-values in log-rank test and number of patients are indicated. NL: no loss; L: loss; NM: not hypermethylated; M: hypermethylated.
C3orf14 methylation and suppressor function
The finding of genes with a methylation driven regulation pattern, opened for the possibility to discover tumor suppressor genes that have not been identified in the search for targets of 3p loss. C3orf14, GPR27, and ZNF717 emerged as novel methylation regulated suppressor candidates with unknown function. C3orf14 had the highest number of hypermethylated CpG sites and its expression was more strongly associated with methylation. The gene was therefore selected for further investigations. To better understand the regulation pattern of C3orf14, the methylation data of the 11 CpG sites retained in the data set after data preprocessing were analyzed in more detail. The frequency of hypermethylation differed considerably across the sites, ranging from 13% to 86% in cohort 1 and from 3% to 86% in cohort 2 (Fig. 6A; Table S2). The methylation level of the sites also differed and was strongly correlated to the total number of hypermethylated sites in both cohorts (Fig. 6B and data not shown). This heterogeneous methylation pattern could be clearly seen when considering the β-distribution across all patients, where a distinct group of hypermethylated tumors with a wide range of methylation levels appeared for the most hypermethylated sites (Fig. 6C). Both number of hypermethylated sites and their methylation level were associated with expression in both cohorts (Fig. 6D, E), suggesting that the highly heterogeneous methylation across CpG sites and tumors is of relevance for gene silencing.
Figure 6.
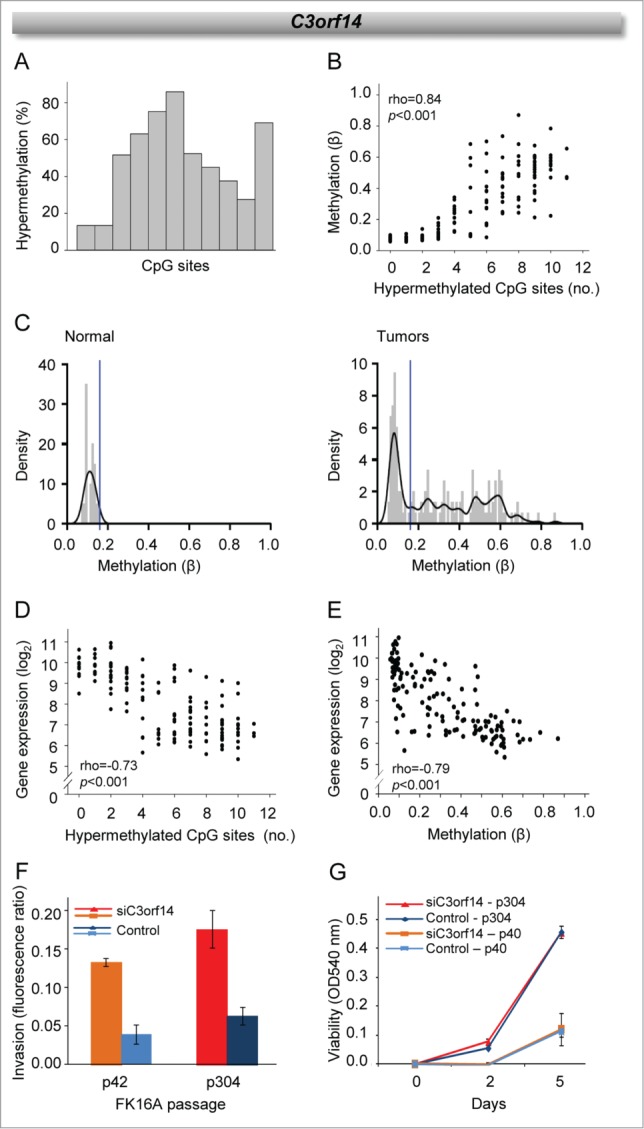
Methylation characteristics and suppressor function of C3orf14. (A) Frequency of cervical cancer patients with hypermethylation of individual CpG sites ordered by location. (B) Methylation (β-value) against number of hypermethylated CpG sites. (C) Density plots (kernel density estimation with band width 0.02, black line; histogram, gray bars) of C3orf14 methylation in normal cervical tissue from GSE46306 (left) and tumors in cohort 1 (right). The blue lines indicate cut-off β-value for scoring hypermethylation. (D) C3orf14 expression against number of hypermethylated CpG sites. (E) C3orf14 expression against methylation (β-value). Invasion (F) and cell viability (G) of control and C3orf14 siRNA treated FK16A cells at early (p40/42) and late (p304) passages. The columns and bars show the mean and standard deviation of triplicates for one representative experiment for each passage. The difference between siRNA treated and control cells was significant for invasion (p42: P = 0.010, p304: P = 0.006, t-test), but not for cell viability. In B, C, and E, methylation data of the CpG site correlating most strongly with gene expression were used. In B, D, and E, correlation coefficient (rho) and P-value in Spearman's rank correlation analysis are indicated. In A-E, data from cohort 1 are presented; similar results were found for cohort 2.
The role of C3orf14 silencing was further studied in early- and late-passage FK16A cells, which mimic preinvasive stages in cervical carcinogenesis. C3orf14 knockdown led to increased invasive capacity in both passages, as shown for two representative experiments in Figure 6F. Cell viability and, hence, the proliferation rate was not affected by the knockdown (Fig. 6G). Downregulation of C3orf14 may therefore possibly promote tumor invasiveness at early stages of the disease, in line with a suppressor function of the gene.
Discussion
The complete methylation mapping of promoter CpG sites performed in our work showed that the 3p11.2-p14.2 region was subjected to differential methylation in cervical cancer, both when comparing tumors and normal cervical tissue and different tumors in patient cohorts. By combining the data with gene dosage and expression, we identified genes for which methylation and loss were suggested to cooperate in the transcriptional regulation, and other genes for which an individual effect of the events was likely. Considerable efforts have been made in previous studies to reveal how genes within the region are regulated, by focusing on individual genes21 or large scale gene dosage data.16 Our study demonstrates a potential of integrating large scale methylation and gene dosage data to achieve a more complete understanding of the regulatory mechanisms. Moreover, the detailed information provided by the Illumina technology opened for the discovery of novel methylation regulated tumor suppressor genes, which may play a role in cervical carcinogenesis.
Promoter hypermethylation at 3p11.2-p14.2 was found to be a frequent event, affecting more than half of the 48 genes with promoter associated CpG-sites in two independent data sets. For many genes, hypermethylation was seen in the majority of patients and was even more common than chromosomal loss. Moreover, a small, but significant, increase in the overall hypermethylation frequency was found with increasing tumor stage. The hypermethylated state may therefore pose a selection pressure on the way from normal epithelial cells to invasive cancer, as indicated by previous studies based on individual genes, such as FHIT, ROBO1, and FAM19A4,17-19 and possibly toward increased malignancy. It should be emphasized that although most islands were represented on the array, not all sites within the islands were included. It was therefore not possible to judge whether the CpG sites included were representative of the whole island.
More than half of the identified hypermethylated genes (17 out of 26) were found to be downregulated in tumors compared to normal tissues. In addition, an association between methylation and expression level was seen in tumors for some of the genes. It is therefore likely that the methylation plays a direct role in gene silencing at this region and participates in generation of the transcriptional program that drives cervical carcinogenesis. Moreover, the finding of candidate targets of the recurrent 3p11.2-p14.2 loss, i.e., GBE1, THOC7, and PSMD6 ,16 among the hypermethylated genes, points to the necessity to integrate gene dosage into the analysis to better understand the role of methylation in gene regulation. The exact transcript variant which is regulated by methylation could not be determined from our data, since the expression probes often cover several variants. Reverse transcription (RT) PCR with primers specific for each individual variant would clarify this.
In the methylation driven regulation pattern, which was suggested for C3orf14, GPR27, and ZNF717, only hypermethylation and not loss was associated with downregulation. Promoter methylation therefore seemed to play a major role in gene regulation, and the hypermethylated state probably exerts a selection pressure during carcinogenesis. This is in contrast to the findings for THOC7 and PSMD6, where a gene dosage driven regulation pattern was suggested. For these genes, methylation seemed to have no effect on the expression level, although hypermethylation was found in more than half of the tumors. The meaning of methylation is not clear for these genes.
A combined methylation and gene dosage driven regulation pattern was suggested for FHIT, ADAMTS9, and LRIG1. For FHIT and ADAMTS9, methylation seemed to be an important regulation mechanism in cases without loss, while loss is probably important in non-methylated tumors. Both genes have been shown to be regulated by promoter methylation in previous work,17,22 and their downregulation has been associated with suppressed proliferation and increased apoptosis in experimental studies.23-25 Our data indicate that both loss and hypermethylation can lead to downregulation of FHIT and ADAMTS9 and possibly be alternative routes in tumor progression. No significant increase in downregulation could, however, be seen in cases with both loss and hypermethylation, as compared to those with only one of the events.
For LRIG1, on the other hand, the lowest expression was observed in the loss group in cases with hypermethylation. The presence of the 2 events in the same tumors therefore apparently poses a strong selection pressure. This hypothesis was strengthened by the results from the survival analyses, where loss and methylation were suggested to have prognostic impact when analyzed separately, and the worse prognosis was seen for patients with both methylation and loss. In addition, patients with hypermethylation without loss had an intermediate outcome that could probably be explained because the downregulation in this group was on the borderline of significance. LRIG1 has been proposed to be an important tumor suppressor in many cancer types,26 and low protein expression has been associated with poor prognosis of early stage cervical cancer27 in accordance with our gene expression data. Gene dosage alteration has been suggested as a major regulation mechanism of LRIG1 and a prognostic factor in breast cancer.28 Our study indicates a role of methylation in addition to loss in gene regulation and proposes a possible use of methylation combined with loss as a prognostic factor in cervical cancer.
C3orf14 appeared to be the most strongly methylation regulated gene in both patient cohorts, showing the largest number of CpG sites with a highly significant correlation between methylation and expression. The number of hypermethylated CpG sites as well as their methylation level varied considerably across the tumors, and both factors were associated with gene expression. These observations are in accordance with a model describing gene silencing by methylation as a dynamic process where methylation is gradually spread to more CpG sites along the DNA molecule, to more alleles and to a larger part of the tumor.10,29
The methylation driven downregulation of C3orf14, GPR27, and ZNF717 implies a suppressor function of the genes. The genes have not been studied in cervical cancer before and emerge as novel suppressor candidates in the disease. GPR27 encodes a G protein-coupled receptor, which probably has a role in insulin production and/or secretion,30 whereas the zinc finger protein ZNF717 may be involved in transcriptional regulation and has been found to be affected by recurrent mutations in gastric tumors.31 C3orf14 encodes an uncharacterized protein and was listed as one out of many hypermethylated and downregulated genes in a previous whole-genome study of glioblastomas32 in accordance with our results. We further found that knockdown of C3orf14 increased the invasiveness of early and late passages of FK16A cells, which are models of intraepithelial cervical lesions. Silencing of C3orf14 therefore possibly contributes to invasive growth. Loss of 3p11.2-p14.2 apparently emerges early during the invasive stage of tumor development, and affects genes involved in cell proliferation and anti-apoptosis.16 The fact that C3orf14 is located at the most frequently lost region, but is not regulated by the loss, may imply that C3orf14 methylation occurs first. Hence, it is tempting to speculate that C3orf14 methylation and consequently downregulation pose a selection pressure prior to the loss by being directly involved in the invasion process.
In conclusion, methylation and loss seem to cooperate in silencing of specific genes within the 3p11.2-p14.2 region. To better understand the regulation of these genes during tumor progression and develop robust prognostic markers based on genetic or epigenetic events, combined information of both events would be required. For other genes, methylation and loss probably play individual roles in gene silencing. In particular, novel methylation regulated candidates, like C3orf14, GPR27, and ZNF717, were suggested. Further exploration of these genes may lead to a better understanding of cervical carcinogenesis.
Materials and Methods
Patients
Tumor specimens were achieved from 270 patients with locally advanced squamous cell carcinoma (SCC) of the uterine cervix who were prospectively recruited to our chemoradiotherapy protocol. The patients were divided into an investigation cohort of 149 patients (cohort 1) and a validation cohort of 121 patients (cohort 2), based on the Illumina beadarray version used for gene expression profiling (Table S1). The primary tumor and elective pelvic areas were treated with 50 Gy of external irradiation in 25 fractions, and metastatic lymph nodes received additional 14 Gy. Brachytherapy, 25 Gy, to the primary tumor was delivered in 5 fractions of 5 Gy. Concomitant chemotherapy with cisplatin (40 mg per m2) was administered according to tolerance. Standard procedures including imaging and clinical examination were applied for follow up. One to 4 biopsies were taken at different locations of the tumor at the time of diagnosis, immediately frozen in liquid nitrogen, stored at −80°C, and used for DNA copy number, gene expression, and methylation analyses. DNA and RNA from different biopsies of the same tumor were pooled. The clinical protocol was approved by the Regional Committee for Medical Research Ethics in southern Norway (REK no. S-01129). Written informed consent was obtained from all patients.
DNA methylation
DNA methylation profiling of 270 tumors was performed using the Illumina Infinium Human Methylation450 BeadArrays (Illumina Inc., San Diego, CA).20 DNA was isolated according to a standard protocol with proteinase K, phenol, chloroform, and isoamylalcohol (cohort 1),33 or by the use of PureLink™ Genomic DNA Mini Kit (Life Technologies, Carlsbad, CA, USA) (cohort 2). Purified DNA quality and concentration were assessed using the Quant-iT™PicoGreen® dsDNA Assay Kit (Life Technologies, Carlsbad, CA, USA) prior to bisulfite conversion. One µg of DNA was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA) according to the manufacturer's protocol. The use of different DNA isolation methods in the two cohorts did not influence the quality of bisulfite converted DNA, as demonstrated by the equal fragment length produced by methylation-specific PCR for selected primers (Fig. S2). Bisulfite converted sample (4 μl) was whole-genome amplified, enzymatically digested, and hybridized to the array before single nucleotide extension was performed. The array data are available in the Gene Expression Omnibus (GEO) through accession number GSE68339.
Preprocessing of methylation data
Preprocessing of the methylation data, involving quality control, probe filtering, signal correction, and normalization was performed in R (version 2.15.1) using the pipeline developed by Touleimat and Tost.34 In brief, for quality control, probes with less than 3 functional arraybeads were considered non-functional and assigned a detection P-value of 1. Samples having > 80% high quality probes (detection P-value < 0.01) were defined as of “good quality.” All samples complied with this criterion. Poorly performing probes with a detection P-value > 0.05 in more than 20% of the samples, probes located 10 bp or closer to known genetic variants and allosomal probes on the Y chromosome were removed. A color balance adjustment between the 2 channels was performed followed by a separate color background adjustment based on negative control probes provided by Illumina. Finally, subset based quantile normalization was applied to correct for the shift in signal between Infinium I and Infinium II probes and normalize between samples. For probe category construction, CpG-categories were built using the “relationToCpG” annotation from the Illumina file. Only CpGs within the 3p11.2-p14.2 region mapping to promoter regions, which we defined as located closer than ±2 kb from the transcription start site, were included in the further analyses.
As a measure of methylation, β-values were used. β represents fraction of methylated DNA molecules at a specific CpG site and ranges from 0 (no methylation at any allele) to 1.0 (complete methylation of all alleles). An interquartile range (IQR) > 0.08 for each site was claimed to ensure sufficient dynamics in β across patients.
DNA copy number
DNA copy number profiling of 75 tumors in cohort 1 was performed by array comparative genomic hybridization (aCGH) and has been presented previously.16 Absolute DNA copy numbers were calculated from the aCGH ratios by the GeneCount algorithm, and were corrected for tumor ploidy, as determined by flow cytometry, and normal cell fraction of the samples.35 Samples showing 2 distinct G1 peaks in the DNA histogram by flow cytometry were classified as aneuploid, and the ploidy was determined from the position of the G1 peak of the aneuploid cells relative to the corresponding peak of the diploid cells. Samples with a single G1 peak were classified as near diploid. Normal cell fraction was estimated by GeneCount prior to copy number calculations. Absolute gene dosages were calculated by dividing the copy numbers with ploidy. Gains and losses were scored by using gene dosage thresholds of 1.1 and 0.9, respectively, taking into account an uncertainty in the ploidy measurement of approximately 10%. The array data are available from the ArrayExpress repository through the accession number E-MTAB-3531.
Gene expression
Gene expression profiling of 147 tumors in cohort 1 and 121 tumors in cohort 2 was performed using the Illumina HumanWG-6 v3 (cohort 1) and HumanHT-12 v4 (cohort 2) expression beadarrays with approximately 48 000 transcripts (Illumina Inc., San Diego, CA). All samples had more than 50% tumor cells in hematoxylin and eosin stained sections. This selection may have led to some bias in the results, but was chosen to reduce the influence of different normal cell proportion across the samples. Total RNA was isolated by the use of Trizol reagent (Life Technologies, Carlsbad, CA) followed by LiCl precipitation (cohort 1) or miRNeasy mini kit (Qiagen, Germantown, MD) (cohort 2). Hybridization, scanning, signal extraction and normalization were performed as described.5 The array data are available in GEO (GSE68339).
External data sets
Four data sets, GSE6791, GSE7803, GSE9750, and GSE46306, were retrieved from the GEO database. The GSE46306 data set included methylation (β-values) of 20 human papillomavirus (HPV) negative normal cervical samples determined with the Illumina Infinium Human Methylation450 BeadArrays (Illumina Inc.),36 and was used to determine methylation status of the tumors in cohorts 1 and 2 as compared to normal tissue. The GSE6791,37 GSE7803,38 and GSE9750.39 data sets included gene expression of normal and cancerous cervical samples determined with Affymetrix U133 arrays (Affymetrix, Santa Clara, CA) and were used to investigate expression changes of hypermethylated genes in tumors as compared to normal tissue. The data sets were based on 8, 10, and 21 normal samples and 20, 21, and 32 tumor samples in GSE6791, GSE7803, and GSE9750, respectively. Using the online tool GEO2R (www.ncbi.nlm.nih.-gov/geo/geo2r), the differences in gene expression between normal and cancer samples were estimated. An adjusted P-value ≤ 0.10 was considered statistically significant. The normal cervical samples in all data sets were collected from pap smears or hysterectomy specimens and were enriched for epithelial cells by microdissection.
Cell lines and siRNA transfection
The HPV16-immortalised keratinocyte cell line FK16A was used to explore the functional meaning of C3orf14 silencing. The morphology of the FK16A cells in 3D cultures is reminiscent of dysplastic precancerous lesions of the cervix.40 The cells are immortalized and telomerase positive, like a subset of CIN3 lesions,41 but not tumorigenic. Early- and late-passage cells, FK16A passage 40/42 and FK16A passage 304, respectively, were included and cultured as described previously.40 Both passages still express C3orf14. For knockdown of C3orf14, FK16A cells were seeded in a 6-well plate and transfected with a pool of C3orf14 specific siRNAs (#L-017892-02-0005, GE Healthcare Dharmacon Inc., Lafayette, CO, USA) (Fig. S3) and controls, using DharmaFECT2 transfection reagent (Thermo Fisher Scientific, Waltham, MA, USA). Control cells were transfected with ON-TARGETplus Non-targeting Pool (#D-001810-10-05, GE Healthcare Dharmacon Inc.). The transfected cells were harvested after 48 hours for RNA isolation, or re-plated for invasion and proliferation analysis.
Knockdown was confirmed with RT PCR, using the Primer Express software v3.0 (Applied Biosystems) to select primers for C3orf14 (Fig. S3). The U1 small nuclear ribonucleoprotein A (snRNP U1A).42 was used as endogenous control. Total RNA was isolated using TRIzol reagent (Life Technologies), according to the manufacturer's instructions. cDNA synthesis from 200 ng RNA and PCR amplification in 40 cycles were performed essentially as described,42 with annealing temperatures of 58°C and 60°C for snRNP U1A and C3orf14, respectively. The PCR products were separated on a 2% agarose gel.
Functional assays
Cellular invasion was determined using transwells containing a fluorescence blocking filter (HTS FluoroBlok; Falcon, BD Biosciences, Breda, The Netherlands). The bottom of the transwells was coated with fibronectin (2 µg/ml solution in PBS; MP Biomedicals, Illkrich, France) and the top compartment was coated with collagen (50 µg/ml solution in PBS; Collagen Type 1 rat tail, BD Biosciences) to mimic tumor matrices in vivo. FK16A cells in medium without supplements were added to the top compartment, and complete medium was added to the bottom. Calcein AM (Molecular Probes, Invitrogen, Leiden, the Netherlands) was used for fluorescent labeling of the cells. The fluorescence intensity of the bottom relative to the top compartment after 48 h was used as a measure of invasion.
Cell proliferation was measured using a colorimetric (MTT-tetrazolium) assay (MP Biomedicals) as described.7 In this assay, the amount of dye conversion, as measured by the optical density at a wavelength of 540 nm, is directly related to the number of viable cells in each well. In total, 2500 FK16A cells were seeded in triplicate in 96-well plates and assayed for MTT conversion at day 0, 2, and 5. The proliferation rate was determined by subtracting the measurement of day 0 from that of the other time points.
Statistics
Spearman's rank correlation analysis was applied to search for correlations between methylation (β-values), gene dosage, and expression. The methylation probes were linked to the gene expression probes by using the HumanHT-12v4 Methyl450K lookup table provided by Illumina, where missing probes were added manually. For comparison of patient groups with different combinations of loss and methylation, Welch's t-test was used. Both analyses included adjustment of the P-values by the algorithm developed by Benjamini and Hochberg to control the false discovery rate (FDR),43 and an adjusted P ≤ 0.10 was used for selection of significant correlations and differences. For analyses of individual CpG sites, Spearman's rank or Pearson's correlation analysis was performed with a significance level of P < 0.05. Survival curves were generated by Kaplan-Meier analysis and compared using log-rank test. Progression free survival, i.e., the time between diagnosis and the first event of locoregional and/or distant relapse, was used as end point. In total, 20 patients died of causes not related to cancer and were therefore censored.
Disclosure of Potential Conflicts of Interest
R Steenbergen has a minority stock in Self-Screen BV, The Netherlands, a spin-off company of VU University Medical Center. All other authors declare that they have no conflicts of interest.
Acknowledgment
We are grateful to Annelieke Jaspers for excellent technical assistance.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Funding
The study was supported by The South-Eastern Norway Regional Health Authority (project 2012015), The Research Council of Norway (project 226120/O30), The Dutch Cancer Society (VU2010-4668), and The European Research Council (ERC advanced 2012-AdG, proposal 322986 Mass-care).
References
- 1.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143-53; PMID:14732866; http://dx.doi.org/ 10.1038/nrc1279 [DOI] [PubMed] [Google Scholar]
- 2.Herman JG. Hypermethylation of tumor suppressor genes in cancer. Semin.Cancer Biol 1999; 9:359-67; http://dx.doi.org/ 10.1006/scbi.1999.0138 [DOI] [PubMed] [Google Scholar]
- 3.Knuutila S, Aalto Y, Autio K, Bjorkqvist AM, El-Rifai W, Hemmer S, Huhta T, Kettunen E, Kiuru-Kuhlefelt S, Larramendy ML. et al.. DNA copy number losses in human neoplasms. Am.J.Pathol 1999; 155:683-94; PMID:10487825; http://dx.doi.org/ 10.1016/S0002-9440(10)65166-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Strooper LM, van, Zummeren M, Steenbergen RD, Bleeker MC, Hesselink AT, Wisman GB, Snijders PJ, Heideman DA, Meijer CJ. CADM1, MAL and miR124-2 methylation analysis in cervical scrapes to detect cervical and endometrial cancer. J Clin Pathol 2014; 67:1067-71; PMID:25281766; http://dx.doi.org/ 10.1136/jclinpath-2014-202616 [DOI] [PubMed] [Google Scholar]
- 5.Lando M, Holden M, Bergersen LC, Svendsrud DH, Stokke T, Sundfor K, Glad IK, Kristensen GB, Lyng H. Gene dosage, expression, and ontology analysis identifies driver genes in the carcinogenesis and chemoradioresistance of cervical cancer. PLoS Genet 2009; 5:e1000719; PMID:19911042; http://dx.doi.org/ 10.1371/journal.pgen.1000719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steenbergen RD, Kramer D, Braakhuis BJ, Stern PL, Verheijen RH, Meijer CJ, Snijders PJ. TSLC1 gene silencing in cervical cancer cell lines and cervical neoplasia. J Natl Cancer Inst 2004; 96:294-305; PMID:14970278; http://dx.doi.org/ 10.1093/jnci/djh031 [DOI] [PubMed] [Google Scholar]
- 7.Overmeer RM, Henken FE, Bierkens M, Wilting SM, Timmerman I, Meijer CJ, Snijders PJ, Steenbergen RD. Repression of MAL tumour suppressor activity by promoter methylation during cervical carcinogenesis. J Pathol 2009; 219:327-36; PMID:19662663; http://dx.doi.org/ 10.1002/path.2598 [DOI] [PubMed] [Google Scholar]
- 8.Mine KL, Shulzhenko N, Yambartsev A, Rochman M, Sanson GF, Lando M, Varma S, Skinner J, Volfovsky N, Deng T. et al.. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun 2013; 4:1806; PMID:23651994; http://dx.doi.org/ 10.1038/ncomms2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tang MH, Varadan V, Kamalakaran S, Zhang MQ, Dimitrova N, Hicks J. Major chromosomal breakpoint intervals in breast cancer co-localize with differentially methylated regions. Front Oncol 2012; 2: 197; PMID:23293768; http://dx.doi.org/ 10.3389/fonc.2012.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet 2002; 3:415-28; PMID:12042769; http://dx.doi.org/ 10.1038/nrg962 [DOI] [PubMed] [Google Scholar]
- 11.Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer 2006; 6:107-16; PMID:16491070; http://dx.doi.org/ 10.1038/nrc1799 [DOI] [PubMed] [Google Scholar]
- 12.Grady WM, Willis J, Guilford PJ, Dunbier AK, Toro TT, Lynch H, Wiesner G, Ferguson K, Eng C, Park JG. et al.. Methylation of the CDH1 promoter as the second genetic hit in hereditary diffuse gastric cancer. Nat Genet 2000; 26:16-7; PMID:10973239; http://dx.doi.org/ 10.1038/79120 [DOI] [PubMed] [Google Scholar]
- 13.Myohanen SK, Baylin SB, Herman JG. Hypermethylation can selectively silence individual p16ink4A alleles in neoplasia. Cancer Res 1998; 58:591-3; PMID:9485004 [PubMed] [Google Scholar]
- 14.Pandis N, Bardi G, Mitelman F, Heim S. Deletion of the short arm of chromosome 3 in breast tumors. Genes Chromosomes Cancer 1997; 18:241-5; PMID:9087563; http://dx.doi.org/ 10.1002/(SICI)1098-2264(199704)18:4%3c241::AID-GCC1%3e3.0.CO;2-0 [DOI] [PubMed] [Google Scholar]
- 15.Zabarovsky ER, Lerman MI, Minna JD. Tumor suppressor genes on chromosome 3p involved in the pathogenesis of lung and other cancers. Oncogene 2002; 21:6915-35; PMID:12362274; http://dx.doi.org/ 10.1038/sj.onc.1205835 [DOI] [PubMed] [Google Scholar]
- 16.Lando M, Wilting SM, Snipstad K, Clancy T, Bierkens M, Aarnes EK, Holden M, Stokke T, Sundfor K, Holm R. et al.. Identification of eight candidate target genes of the recurrent 3p12-p14 loss in cervical cancer by integrative genomic profiling. J Pathol 2013; 230:59-69; PMID:23335387; http://dx.doi.org/ 10.1002/path.4168 [DOI] [PubMed] [Google Scholar]
- 17.Ki KD, Lee SK, Tong SY, Lee JM, Song DH, Chi SG. Role of 5′-CpG island hypermethylation of the FHIT gene in cervical carcinoma. J Gynecol Oncol 2008; 19:117-22; PMID:19471558; http://dx.doi.org/ 10.3802/jgo.2008.19.2.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Narayan G, Goparaju C, rias-Pulido H, Kaufmann AM, Schneider A, Durst M, Mansukhani M, Pothuri B, Murty VV. Promoter hypermethylation-mediated inactivation of multiple Slit-Robo pathway genes in cervical cancer progression. Mol Cancer 2006; 5: 16; PMID:16700909; http://dx.doi.org/ 10.1186/1476-4598-5-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Strooper LM, Meijer CJ, Berkhof J, Hesselink AT, Snijders PJ, Steenbergen RD, Heideman D. A. Methylation analysis of the FAM19A4 gene in cervical scrapes is highly efficient in detecting cervical carcinomas and advanced CIN2/3 lesions. Cancer Prev Res (Phila) 2014; 7:1251-7; PMID:25281488; http://dx.doi.org/ 10.1158/1940-6207.CAPR-14-0237 [DOI] [PubMed] [Google Scholar]
- 20.Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, Gunderson KL. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics 2009; 1:177-200; PMID:22122642; http://dx.doi.org/ 10.2217/epi.09.14 [DOI] [PubMed] [Google Scholar]
- 21.Choi CH, Lee KM, Choi JJ, Kim TJ, Kim WY, Lee JW, Lee SJ, Lee JH, Bae DS, Kim BG. Hypermethylation and loss of heterozygosity of tumor suppressor genes on chromosome 3p in cervical cancer. Cancer Lett 2007; 255:26-33; PMID:17467893; http://dx.doi.org/ 10.1016/j.canlet.2007.03.015 [DOI] [PubMed] [Google Scholar]
- 22.Peng L, Yang Z, Tan C, Ren G, Chen J. Epigenetic inactivation of ADAMTS9 via promoter methylation in multiple myeloma. Mol Med Rep 2013; 7:1055-61; PMID:23358566 [DOI] [PubMed] [Google Scholar]
- 23.Huang Q, Liu Z, Xie FM, Liu C, Shao F, Zhu CL, Hu S. Fragile histidine triad (FHIT) suppresses proliferation and promotes apoptosis in cholangiocarcinoma cells by blocking PI3K-Akt pathway. Scientific World J 2014; 2014: 179698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingvarsson S. FHIT alterations in breast cancer. Semin Cancer Biol 2001; 11:361-6; PMID:11562178; http://dx.doi.org/ 10.1006/scbi.2001.0391 [DOI] [PubMed] [Google Scholar]
- 25.Lung HL, Lo PH, Xie D, Apte SS, Cheung AK, Cheng Y, Law EW, Chua D, Zeng YX, Tsao SW. et al.. Characterization of a novel epigenetically-silenced, growth-suppressive gene, ADAMTS9, and its association with lymph node metastases in nasopharyngeal carcinoma. Int J Cancer 2008; 123:401-8; PMID:18449890; http://dx.doi.org/ 10.1002/ijc.23528 [DOI] [PubMed] [Google Scholar]
- 26.Lindquist D, Kvarnbrink S, Henriksson R, Hedman H. LRIG and cancer prognosis. Acta Oncol 2014; 53:1135-42; PMID:25180912; http://dx.doi.org/ 10.3109/0284186X.2014.953258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lindstrom AK, Ekman K, Stendahl U, Tot T, Henriksson R, Hedman H, Hellberg D. LRIG1 and squamous epithelial uterine cervical cancer: correlation to prognosis, other tumor markers, sex steroid hormones, and smoking. Int J Gynecol Cancer 2008; 18:312-7; PMID:17624990; http://dx.doi.org/ 10.1111/j.1525-1438.2007.01021.x [DOI] [PubMed] [Google Scholar]
- 28.Thompson PA, Ljuslinder I, Tsavachidis S, Brewster A, Sahin A, Hedman H, Henriksson R, Bondy ML, Melin BS. Loss of LRIG1 locus increases risk of early and late relapse of stage I/II breast cancer. Cancer Res 2014; 74:2928-35; PMID:24879564; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turker MS. Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 2002; 21:5388-93; PMID:12154401; http://dx.doi.org/ 10.1038/sj.onc.1205599 [DOI] [PubMed] [Google Scholar]
- 30.Ku GM, Pappalardo F, Luo CC, German MS, McManus MT. An siRNA screen in pancreatic β cells reveals a role for Gpr27 in insulin production. PLoS Genet 2012; 8:e1002449; PMID:22253604; http://dx.doi.org/ 10.1371/journal.pgen.1002449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui J, Yin Y, Ma Q, Wang G, Olman V, Zhang Y, Chou WC, Hong CS, Zhang C, Cao S. et al.. Comprehensive characterization of the genomic alterations in human gastric cancer. Int J Cancer 2015; 137:86-95; PMID:25422082; http://dx.doi.org/ 10.1002/ijc.29352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Etcheverry A, Aubry M, Tayrac M de, Vauleon E, Boniface R, Guenot F, Saikali S, Hamlat A, Riffaud L, Menei P. et al.. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genomics 2010; 11:701; PMID:21156036; http://dx.doi.org/ 10.1186/1471-2164-11-701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Angelis PM, Clausen OP, Schjolberg A, Stokke T. Chromosomal gains and losses in primary colorectal carcinomas detected by CGH and their associations with tumour DNA ploidy, genotypes and phenotypes. Br J Cancer 1999; 80:526-35; PMID:10408863; http://dx.doi.org/ 10.1038/sj.bjc.6690388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Touleimat N, Tost J. Complete pipeline for Infinium((R)) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012; 4:325-41; PMID:22690668; http://dx.doi.org/ 10.2217/epi.12.21 [DOI] [PubMed] [Google Scholar]
- 35.Lyng H, Lando M, Brovig RS, Svendsrud DH, Johansen M, Galteland E, Brustugun OT, Meza-Zepeda LA, Myklebost O, Kristensen GB. et al.. GeneCount: Genome-wide calculation of absolute tumor DNA copy numbers from array CGH data. Genome Biol 2008; 9:R86; PMID:18500990; http://dx.doi.org/ 10.1186/gb-2008-9-5-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farkas SA, Milutin-Gasperov N, Grce M, Nilsson TK. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 2013; 8:1213-25; PMID:24030264; http://dx.doi.org/ 10.4161/epi.26346 [DOI] [PubMed] [Google Scholar]
- 37.Pyeon D, Newton MA, Lambert PF, den Boon JA, Sengupta S, Marsit CJ, Woodworth CD, Connor JP, Haugen TH, Smith EM. et al.. Fundamental differences in cell cycle deregulation in human papillomavirus-positive and human papillomavirus-negative head/neck and cervical cancers. Cancer Res 2007; 67:4605-19; PMID:17510386; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhai Y, Kuick R, Nan B, Ota I, Weiss SJ, Trimble CL, Fearon ER, Cho KR. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res 2007; 67:10163-72; PMID:17974957; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2056 [DOI] [PubMed] [Google Scholar]
- 39.Scotto L, Narayan G, Nandula SV, rias-Pulido H, Subramaniyam S, Schneider A, Kaufmann AM, Wright JD, Pothuri B, Mansukhani M. et al.. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: Potential role in progression. Genes Chromosomes Cancer 2008; 47:755-65; PMID:18506748; http://dx.doi.org/ 10.1002/gcc.20577 [DOI] [PubMed] [Google Scholar]
- 40.Steenbergen RD, Parker JN, Isern S, Snijders PJ, Walboomers JM, Meijer CJ, Broker TR, Chow LT. Viral E6-E7 transcription in the basal layer of organotypic cultures without apparent p21cip1 protein precedes immortalization of human papillomavirus type 16- and 18-transfected human keratinocytes. J.Virol 1998; 72:749-57; PMID:9420282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snijders PJ, van, Duin M, Walboomers JM, Steenbergen RD, Risse EK, Helmerhorst TJ, Verheijen RH, Meijer CJ. Telomerase activity exclusively in cervical carcinomas and a subset of cervical intraepithelial neoplasia grade III lesions: strong association with elevated messenger RNA levels of its catalytic subunit and high-risk human papillomavirus DNA. Cancer Res 1998; 58:3812-8; PMID:9731489 [PubMed] [Google Scholar]
- 42.Bijl J, van Oostveen JW, Kreike M, Rieger E, van der Raaij-Helmer LM, Walboomers JM, Corte G, Boncinelli E, van den Brule AJ, Meijer CJ. Expression of HOXC4, HOXC5, and HOXC6 in human lymphoid cell lines, leukemias, and benign and malignant lymphoid tissue. Blood 1996; 87:1737-45; PMID:8634419 [PubMed] [Google Scholar]
- 43.Benjamini Y, Hochberg Y. Controlling the false discoveryrate: a practical and powerful approach to multiple testing. J R Stat Soc B 1995; 57:289-300 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.