

Abstract
Maternal infection during pregnancy increases the risk of neurodevelopmental disorders in the offspring. In addition to its influence on other neuronal systems, this early-life environmental adversity has been shown to negatively affect cortical γ-aminobutyric acid (GABA) functions in adult life, including impaired prefrontal expression of enzymes required for GABA synthesis. The underlying molecular processes, however, remain largely unknown. In the present study, we explored whether epigenetic modifications represent a mechanism whereby maternal infection during pregnancy can induce such GABAergic impairments in the offspring. We used an established mouse model of prenatal immune challenge that is based on maternal treatment with the viral mimetic poly(I:C). We found that prenatal immune activation increased prefrontal levels of 5-methylated cytosines (5mC) and 5-hydroxymethylated cytosines (5hmC) in the promoter region of GAD1, which encodes the 67-kDa isoform of the GABA-synthesising enzyme glutamic acid decarboxylase (GAD67). The early-life challenge also increased 5mC levels at the promoter region of GAD2, which encodes the 65-kDa GAD isoform (GAD65). These effects were accompanied by elevated GAD1 and GAD2 promoter binding of methyl CpG-binding protein 2 (MeCP2) and by reduced GAD67 and GAD65 mRNA expression. Moreover, the epigenetic modifications at the GAD1 promoter correlated with prenatal infection-induced impairments in working memory and social interaction. Our study thus highlights that hypermethylation of GAD1 and GAD2 promoters may be an important molecular mechanism linking prenatal infection to presynaptic GABAergic impairments and associated behavioral and cognitive abnormalities in the offspring.
Keywords: Autism; chromatin; DNA methylation; epigenetics; GABA, maternal infection; poly(I:C); prefrontal cortex; prenatal immune activation; schizophrenia
Abbreviations
- 5hmC, 5-hydroxymethylated cytosines; 5mC, 5-methylated cytosines; CNS, central nervous system; DNMT, DNA methyltransferase; GABA, γ-aminobutyric acid; GAD, glutamate decarboxylase; Gadd45-β, growth arrest and DNA Damage-inducible 45 β; MeCP2, methyl CpG-binding protein 2; mPFC, medial prefrontal cortex; poly(I:C), polyriboinosinic-polyribocytidilic acid; TET, ten-eleven translocation hydroxylase
Introduction
Maternal immune activation during pregnancy has been repeatedly implicated in the etiology of developmental neuropsychiatric disorders, including schizophrenia,1,2 autism,3,4 and bipolar disorder.5,6 Additional support for these epidemiological associations has been obtained by various translational rodent models demonstrating disease-relevant brain and behavioral abnormalities following prenatal exposure to infection and/or immune activation.7-8 Accumulating evidence suggests that cytokine-associated inflammatory events, together with downstream pathophysiological effects such as oxidative stress and (temporary) macronutrient and micronutrient deficiency, are critical in mediating the adverse effects of maternal infection on the fetal system.9-11 Disruption of normal fetal development,12 together with subsequent changes in brain maturation,1,2 may then confer an increased risk of behavioral and cognitive dysfunctions in later life.
In keeping with the crucial role of epigenetic remodeling in early development,13,14 epigenetic modifications may represent an important mechanism whereby maternal infection during pregnancy can alter brain and behavioral development in the offspring. Initial evidence for this hypothesis stems from rodent models of prenatal viral-like infection, demonstrating altered DNA methylation, histone modifications, and microRNA (miRNA) expression in the brains of infected offspring.15-18 Interestingly, some of the differentially methylated gene sequences or altered miRNA clusters found in prenatally infected mice show association with neurodevelopmental disorders, including genes in the glutamate receptor signaling pathway,16 the methyl CpG-binding protein 2 (MeCP2) gene,17 and the imprinted Dlk1-Dio3 miRNA cluster.18
In the present study, we sought to identify possible epigenetic modifications in the prefrontal γ-aminobutyric acid (GABA) system of mice exposed to prenatal immune activation. Impaired cortical GABAergic signaling is one of the hallmark pathologies in developmental neuropsychiatric disorders, including schizophrenia,19,20 autism,21 and bipolar disorder.20,22 These GABAergic modifications may impair neuronal synchronization and inhibition, thereby facilitating the emergence of disease-relevant cognitive deficits such as disruption of executive functions, cognitive flexibility and working memory.19,23 Besides other pre- and post-synaptic changes, GABAergic abnormalities in these disorders involve reduced mRNA expression of glutamic acid decarboxylase 67 (GAD67).23 GAD67 is the 67-kDa isoform of the rate-limiting enzyme for GABA synthesis, which is encoded by the GAD1 gene. It is responsible for most of the basal GABA synthesis in the central nervous system (CNS).24 The other main isoform, GAD65, is encoded by the GAD2 gene and appears to be important especially for GABA synthesis during sustained periods of neuronal activity.25,26 In the context of neuropsychiatric disease, impairments in GAD65 levels seem to occur primarily in schizoaffective disorder, but not other psychosis-associated disorders such as schizophrenia or bipolar disorder.27
Accumulating evidence suggests that epigenetic modifications at the GAD1 promoter critically contribute to the deficits in GAD67 as seen in schizophrenia, autism, and bipolar disorder.28 The promoter region of GAD1 (and GAD2) contains large cytosine-phosphodiester-guanine (CpG) dinucleotide islands, which in turn can facilitate promoter methylation and formation of transcriptionally inactive chromatin.28,29 Consistent with this notion, decreased GAD67 mRNA expression in the prefrontal cortex of schizophrenic or bipolar subjects has been found to correlate with GAD1 promoter hypermethylation.30,31 These changes are further associated with altered expression and/or promoter binding of enzymes that are critical for the maintenance of the DNA methylation/demethylation equilibrium of cytosines positioned within a CpG context in gene promoter regions, including DNA methyltransferase 1 and 3a (DNMT1 and DNMT3a), ten-eleven translocation hydroxylase (TET), growth arrest and DNA Damage-inducible 45 β (Gadd45-β), and MeCP2.32-34 Methylation-related GAD1 promoter remodeling thus appears to be a plausible epigenetic mechanism leading to impaired GAD67 mRNA expression in subjects with developmental neuropsychiatric disorders. The underlying etiological mechanisms, however, remain elusive and warrant further investigations.
Against these backgrounds, we explored the effects of prenatal immune activation on GAD1 and GAD2 promoter remodeling using a well-established mouse model of maternal treatment with the viral mimic polyriboinosinic-polyribocytidilic acid [poly(I:C)].7,8 This model is known to capture a wide spectrum of behavioral and cognitive abnormalities relevant to psychiatric disorders, especially schizophrenia and autism.7,8 Importantly, adult offspring of poly(I:C)-treated mice also display various GABAergic abnormalities pertinent to such neurodevelopmental disorders, including reduced GAD67 and GAD65 mRNA and protein expression in the medial prefrontal cortex (mPFC).35,36 The prenatal poly(I:C) administration model is highly suitable for our primary objective to explore epigenetic abnormalities at the prefrontal GAD1 and GAD2 promoters, and to correlate these putative changes with behavioral and cognitive deficits.
Results
Effects of prenatal immune activation on working memory and social interaction
First, we ascertained the deleterious effects of prenatal immune challenge on adult behavioral and cognitive functions. To this end, we exposed pregnant mice on gestation day 17 to poly(I:C) [5 mg/kg, intravenous (i.v.)] or control vehicle (pyrogen-free 0.9% NaCl) solution according to established protocols.35,36 The selected gestational window [i.e., gestation day (GD)17] in mice corresponds roughly to the middle-to-late second trimester of human pregnancy, with respect to developmental biology and percentage of gestation from mice to humans.37 The gestational stage was selected based on our previous findings showing that GD17 poly(I:C) treatment leads to impairments in prefrontal GABA neurocircuitries, working memory, and social interaction.35,36,38 The resulting offspring of poly(I:C)- or vehicle-exposed mothers (N = 8 in each group) were then tested in two paradigms relevant to human neuropsychiatric disease, namely working memory and social interaction.
Consistent with previous studies,36,38 we found that prenatal poly(I:C) treatment impaired working memory as assessed using a spatial recognition memory test in the Y-maze. In this test, the critical measure of working memory is the relative time spent in the novel (previously unexplored) arm during the choice phase.36,38 As expected, control offspring displayed a noticeable preference toward the novel arm, indicating intact working memory (Fig. 1A). Poly(I:C) offspring exhibited a significant (F(1,14) = 4.70, P < 0.05) reduction in this measure and performed below chance level (Fig. 1A). Poly(I:C) and control offspring did not differ with respect to the total distance moved during the choice phase of the test (Fig. 1A), indicating that the negative effects of prenatal immune activation on working memory are not confounded by possible differences in basal locomotor activity.
Figure 1.
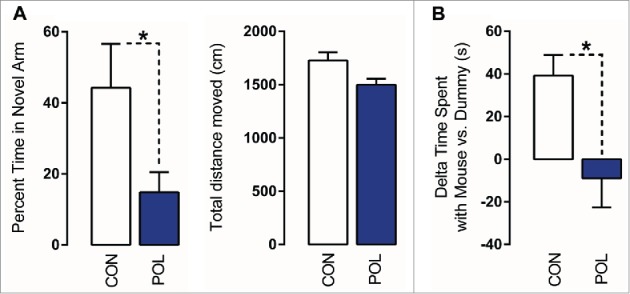
Prenatal immune activation produces working memory and social interaction deficits. (A) Working memory was assessed in the Y-maze spatial recognition paradigm. Left: The bar plot depicts the percent time spent in the novel (previously unexplored) arm during the choice phase of the test in control (CON) and poly(I:C) (POL) offspring. *P < 0.05 as determined by a Student t-test. Right: The bars plot shows the total distance moved during the choice phase of the test. (B) Social interaction was assessed by analyzing the relative exploration time between an unfamiliar congenic mouse (‘mouse’) and an inanimate dummy object (‘dummy’). The bar plots show the relative exploration time between the mouse and the dummy for the CON and POL offspring. *P < 0.05 as determined by a Student t-test. N(CON) = 8, N(POL) = 8. All values are means ± SEM.
Maternal immune activation in mice has previously been shown to cause social interaction deficits in the adult offspring.36,39 Here, we confirm this phenotype in our cohort of poly(I:C)-exposed mice relative to control mice using an established social approach test.36 In this test, the relative exploration time between an unfamiliar congenic mouse and an inanimate dummy object [(time spent with mouse) - (time spent with dummy object)] was used to assess social approach behavior. As shown in (Fig. 1B), control offspring displayed a clear preference toward the unfamiliar live mouse. Such social approach behavior was significantly (F(1,14) = 8.41, P < 0.05) impaired in poly(I:C)-exposed offspring. Indeed, they did not display a preference toward the unfamiliar live mouse, such that relative time spent with the mouse was negative (∼ −10 s; Fig. 1B).
Effects of prenatal immune activation on GAD1 and GAD2 promoter methylation and hydroxymethylation
Using the behaviorally characterized cohort of poly(I:C)-exposed and control offspring (see Fig. 1), we then sought to determine possible differences in the methylation patterns at the GAD1 and GAD2 promoter regions. In a first series of experiments, we used methylated and hydroxymethylated DNA immunoprecipitation (MeDIP and hMeDIP, respectively) assays to determine the ratio of 5-methylated cytosines (5mC) or 5-hydroxymethylated cytosines (5hmC) to the total cytosines of GAD1 and GAD2 CpG-enriched promoter fragments in mPFC tissue of poly(I:C) and control mice. To this end, sonicated genomic DNA that did not undergo immunoprecipitation (input DNA) and DNA immunoprecipitated with anti-5mC and anti-5hmC antibodies (IP DNA) were used as templates for quantitative real-time PCR (qRT-PCR) with primers specific against the GAD1 and GAD2 promoter regions (sequences given in Table 1). We chose these procedures for two main reasons: First, unlike bisulfite conversion and most enzyme-dependent methods, MeDIP is capable of distinguishing 5mC from 5hmC, the latter of which contains approximately 20% of all methylcytosines in the brain and likely has very different functions and genomic locations.40-42 Second, the MeDIP and hMeDIP procedures are sensitive methods to capture altered methylation patterns at the GAD1 promoter in mouse brain tissue.43 A schematic representation of the GAD1 and GAD2 promoters, including CpG islands, CpG shores, transcriptional start site (TSS) and genomic positions of interest, is depicted in (Fig. 2).
Figure 2.
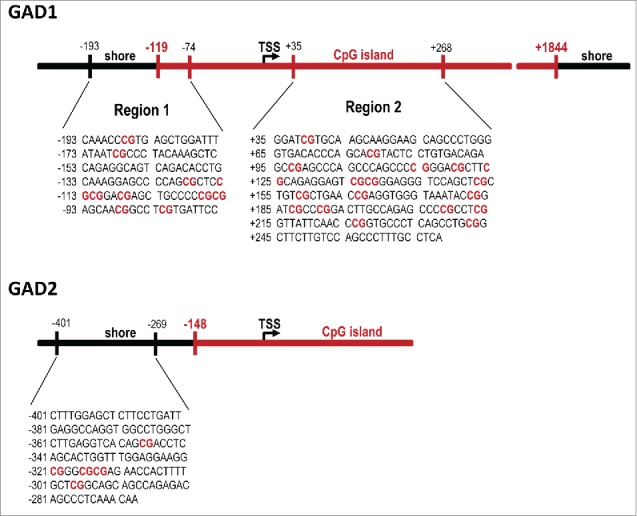
Sequences and genomic locations of the GAD1 and GAD2 promoter regions of interest. Two assays (before and after the transcriptional start site, TSS) were performed on the GAD1 promoter based on previous studies looking at sequences within both these regions,43,45 while one assay (before the TSS) was performed on the GAD2 gene.
Table 1.
Correlation analyses between behavioral performance and epigenetic markers at the GAD1 (Regions 1 and 2) and GAD2 promoter regions in the medial prefrontal cortex (mPFC) of control (CON) and poly(I:C) (POL) offspring. Behavioral performance variables included in the analysis were the percent time spent in the novel arm in the Y-maze test and the delta time spent with mouse vs. dummy in the social interaction test. Epigenetic markers included in the analysis were 5-methylcytosine (5mC) binding, 5-hydroxymethylcytosine (5hmC) binding, and methyl CpG-binding protein 2 (MeCP2) binding at the GAD1 (regions 1 and 2) and GAD2 promoter regions. The correlations were performed using Pearson's product moment correlations. The coefficients of correlation r are given in the table for each correlative comparison.
| GAD1 Promoter - Region 1 | 5mC enrichment | 5hmC enrichment | MeCP2 enrichment |
|---|---|---|---|
| Working Memory Score | −0.51* | −0.57* | −0.14 |
| Social Interaction Score |
−0.51* |
−0.55* |
−0.62* |
|
GAD1 Promoter - Region 2 |
5mC enrichment |
5hmC enrichment |
MeCP2 enrichment |
| Working Memory Score | −0.39 | −0.48 | −0.31 |
| Social Interaction Score |
−0.42 |
−0.50* |
−0.65* |
|
GAD2 Promoter |
5mC enrichment |
5hmC enrichment |
MeCP2 enrichment |
| Working Memory Score | −0.25 | −0.34 | −0.44 |
| Social Interaction Score | −0.44 | −0.44 | −0.35 |
*P < 0.05 signifies a significant Pearsons product correlation. N = 14-16 (7-8 CON, 7-8 POL) for each correlation.
We found that prenatally immune-challenged offspring displayed an overall ∼40% (F(1,14) = 4.80, P < 0.05) increase in 5mC modifications at the GAD1 promoter, which was apparent at two distinct regions of the GAD1 CpG islands spanning the TSS, namely (−193 to −74 bp) and (+35 to +268 bp) (Fig. 3A). Similarly, prenatal immune activation increased the levels of 5hmC at these GAD1 promoter regions by ∼20% (F(1,14) = 4.80, P < 0.05; Fig. 3B). 5mC and 5hmC levels in the CpG island region located outside the promoter region (i.e., within the gene body) were not significantly different between poly(I:C)-exposed and control mice (Fig. S1), suggesting that the effects of prenatal immune activation on GAD1 methylation and hydroxymethylation are specific to the promoter region.
Figure 3.
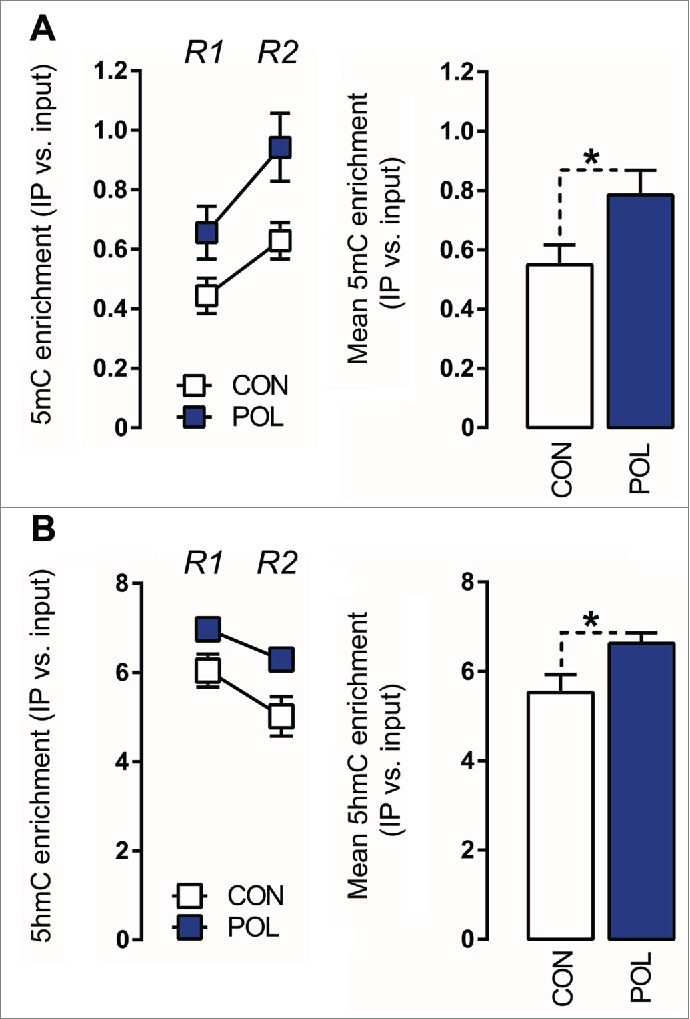
Prenatal immune activation increases 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) binding at the GAD1 promoter region in the medial prefrontal cortex (mPFC) (A) 5mC binding was assessed by comparing the ratio of 5mC to the total cytosines in genomic DNA samples of control (CON) and poly(I:C) (POL) offspring. Left: The line plot shows 5mC ratios at both promoter regions of interest (Region 1, R1; Region 2, R2) for the CON and POL offspring. Right: The bar plot shows the mean 5mC ratios for both regions R1 and R2. *P < 0.05 as determined by a main effect of treatment arising in the repeated-measures ANOVA analysis over the two regions R1 and R2 of interest. (B) 5hmC binding was assessed by comparing the ratio of 5hmC to the total cytosines in genomic DNA samples of CON and POL offspring. Left: The line plot shows 5hmC ratios at both promoter regions of interest R1 and R2. Right: The bar plot shows the mean 5hmC ratios for R1 and R2. *P < 0.05 as determined by a main effect of treatment arising in the repeated-measures ANOVA analysis over the two regions R1 and R2 of interest. N(CON) = 8, N(POL) = 8. All values are means ± SEM.
Prenatal immune activation also led to hypermethylation of the GAD2 promoter: The 5mC modifications at the targeted GAD2 promoter region (-401 to -269 bp) were significantly (F(1,14) = 6.78, P < 0.05) increased by ∼40% in offspring born to immune-challenged mothers relative to controls (Fig. 4A). In contrast to its effects on GAD1 (see Fig. 3), however, prenatal immune activation did not affect the levels of 5hmC at the GAD2 promoter region.
Figure 4.
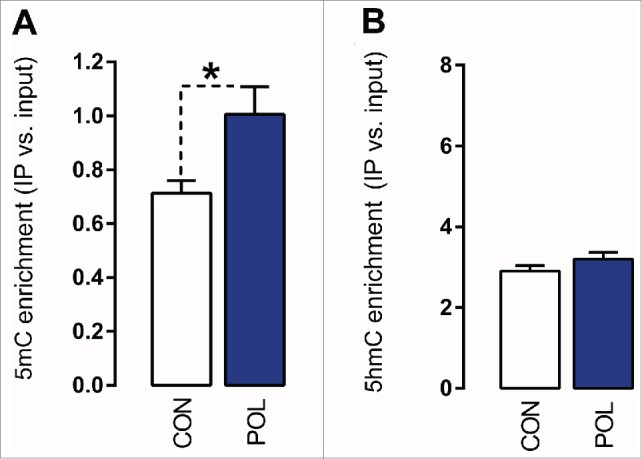
Prenatal immune activation increases 5-methylcytosine (5mC) binding at the GAD2 promoter region in the medial prefrontal cortex (mPFC). (A) 5mC binding was assessed by comparing the ratio of 5mC to the total cytosines in genomic DNA samples of control (CON) and poly(I:C) (POL) offspring. The bar plot shows 5mC ratios within the region of interest. *P < 0.05 as determined by a Student t-test. (B) 5hmC binding was assessed by comparing the ratio of 5hmC to the total cytosines in genomic DNA samples of CON and POL offspring. The bar plot shows 5hmC ratios within the region of interest. N(CON) = 8, N(POL) = 8. All values are means ± SEM.
Effects of prenatal immune activation on MeCP2 binding at GAD1 and GAD2 promoters
Hypermethylation of the GAD1 promoter has been associated with increased promoter binding of MeCP2, both in subjects with neuropsychiatric disorders and in disease-relevant animal models.29,43 MeCP2 is one of several methyl-binding domain (MBD) proteins with high affinity for methylated DNA.28 In view of the altered methylation patterns found in offspring of immune-challenged mothers (see Figs. 3,4), we explored whether prenatal immune activation would increase MeCP2 binding at the GAD1 and GAD2 promoters. To this end, we performed chromatin immunoprecipitation (ChIP) using a MeCP2 antibody according to protocols that were previously validated in mouse brain tissue.43,44 We found that prenatal immune activation markedly affected MeCP2 binding at GAD1 and GAD2 promoter regions: As depicted in Fig. 5A, offspring born to immune-challenged mothers displayed a ∼35% (F (1,12) = 6.36, P < 0.05) increase in MeCP2 binding at the GAD1 promoter, which was apparent at both regions of interest (−193 to −74 bp and +35 to +268 bp). Similarly, prenatal immune activation increased MeCP2 binding at the GAD2 promoter by ∼50% (F(1,12) = 14.01, P < 0.01; Fig. 5B). Our findings thus suggest that prefrontal GAD1 and GAD2 remodeling following prenatal immune challenge involves increased MeCP2 promoter binding (Fig. 5).
Figure 5.
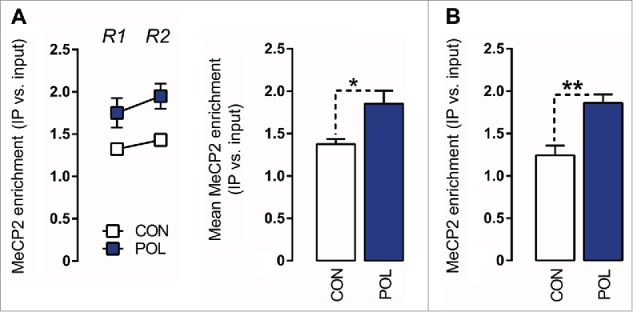
Prenatal immune activation increases methyl CpG-binding protein 2 (MeCP2) binding at the GAD1 and GAD2 promoter regions in the medial prefrontal cortex (mPFC) (A) MeCP2 binding at the GAD1 promoter. Left: the line plot shows MeCP2 binding at both promoter regions of interest (Region 1, R1; Region 2, R2) for the control (CON) and poly(I:C) (POL) offspring. Right: the bar plot shows the mean MeCP2 for both regions R1 and R2. *P < 0.05 as determined by a main effect of treatment arising in the repeated-measures ANOVA analysis. (B) MeCP2 binding at the GAD2 promoter. The bar plot shows the MeCP2 binding values in the region of interest in CON and POL offspring. **P < 0.01 as determined by a Student t-test. N(CON) = 7, N(POL) = 7. All values are means ± SEM.
Correlation between epigenetic markers and behavioral performance
To examine a possible relationship between the animals’ behavioral performance and the epigenetic markers of interest, we correlated the 5mC, 5hmC, and MeCP2 enrichment scores at the GAD1 and GAD2 promoters with working memory and social interaction scores of poly(I:C)-exposed and control offspring. We found that 5mC, 5hmC, and MeCP2 enrichment scores at the pre-TSS promoter region of the GAD1 gene negatively correlated with working memory and social interaction scores (Table 1 and Fig. 6). On the other hand, there were no correlations between 5mC, 5hmC, and MeCP2 enrichment scores at the GAD2 promoter and working memory or social interaction (Table 1). These data indicate that methylation-related epigenetic parameters at the prefrontal GAD1 promoter may be more readily involved in working memory and social interaction as compared with epigenetic changes occurring at the GAD2 promoter.
Figure 6.
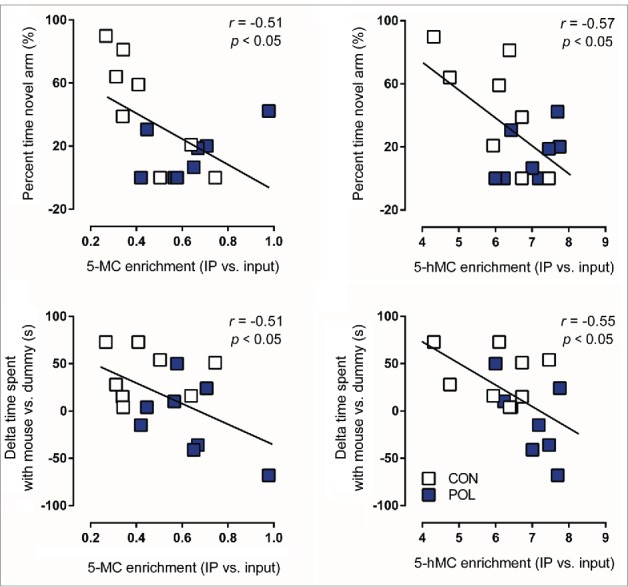
Significant correlations between behavioral performance and epigenetic markers at the GAD1 pre-TSS (transcriptional start site) promoter region in the medial prefrontal cortex (mPFC) of control (CON) and poly(I:C) (POL) offspring. Behavioral performance variables included in the analysis were the percent time spent in the novel arm in the Y-maze test and the delta time spent with mouse vs. dummy in the social interaction test. Epigenetic markers included in the analysis were 5-methylcytosine (5mC) binding or 5-hydroxymethylcytosine (5hmC) binding at the GAD1 pre-TSS promoter region. The correlations were performed using Pearson's product moment correlations. The regression lines represent the correlative analyses, r is the slope of the regression lines and P is the P-value. N = 16 (8 CON, 8 POL) for each correlation.
Effects of prenatal immune activation on prefrontal GAD65 and GAD67 mRNA expression
In a last series of experiments, we aimed to examine whether the infection-induced epigenetic modifications at the GAD1 and GAD2 promoters are associated with reduced GAD67 and GAD65 mRNA expression, respectively. Because of the limited quantities of prefrontal specimens in mice, we used mPFC tissue from a second cohort of adult poly(I:C)-exposed and control mice that were generated and maintained under identical conditions to the previous cohort of animals. Consistent with previous studies,35,36 quantitative real-time polymerase chain reaction (qRT-PCR) revealed that poly(I:C)-exposed offspring displayed a ∼20% (F(1,15) = 8.71, P < 0.01) and ∼15% (F(1,15) = 5.66, P < 0.05) reduction in the prefrontal mRNA levels of GAD67 and GAD65, respectively, compared to control offspring (Fig. 7).
Figure 7.
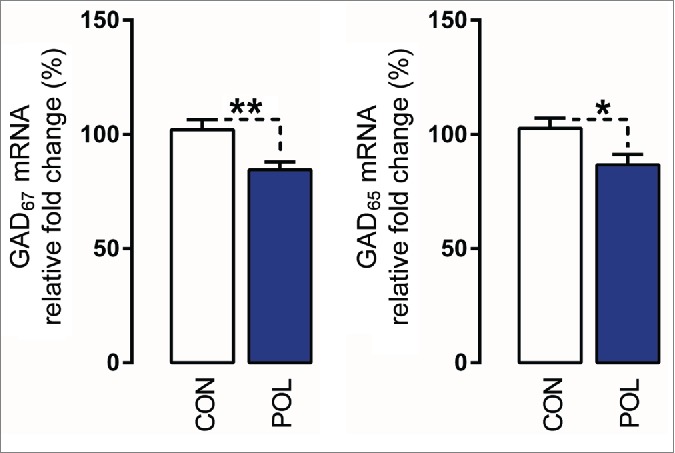
Prenatal immune activation reduces the mRNA levels of GAD67 and GAD65 in the medial prefrontal cortex (mPFC). The graphs depict the levels of normalized mRNA expression of GAD67 and GAD65 in control (CON) and poly(I:C)-exposed (POL) offspring. N(CON) = 10 and N(POL) = 7. *P< 0.05 and **P < 0.01. All values are means ± SEM.
Discussion
Our study demonstrates for the first time that prenatal viral-like immune activation results in altered promoter methylation of specific GABAergic genes, namely GAD1 and GAD2, in the offspring. Similar epigenetic aberrancies can be caused by other early-life adversities such as exposure to prenatal stress or deficient maternal care in mice or rats.43,45 Hence, the promoter regions of these GABAergic genes seem to be highly sensitive to the epigenetic effects of various environmental insults operating at early stages of brain development. The capacity of early-life adversities to affect GAD1 and GAD2 promoter methylation may be explained, at least in part, by the fact that the expression of these two genes in prefrontal regions is first detectable during early fetal life and continues throughout adolescence and adulthood.46,47 Furthermore, the promoter regions of both genes contain large CpG dinucleotide islands (i.e., they both span regions of over 1.5 kb extending from upstream the TSS through the first exon48), which are known to facilitate promoter hypermethylation and formation of transcriptionally inactive chromatin.20,28 The susceptibility of the GAD1 and GAD2 promoters to hypermethylation may be further explained by the fact that the expression of methylating enzymes, such as MeCP2, DNMT1, and DNMT3A, is particularly high in (cortical) GABAergic neurons.28 Hence, the developmentally regulated pattern of GAD1 and GAD2 transcription, along with the molecular composition of their promoter regions, would make it plausible that exposure to environmental adversities during sensitive periods of brain development and/or maturation can have a long-term impact on GAD1 and GAD2 functions.28,49
Another main finding of our study is the demonstration of increased MeCP2 binding at GAD1 and GAD2 promoters following prenatal immune challenge. In view of the high affinity of MeCP2 to methylated DNA,28 the increase in MeCP2 promoter binding further supports our interpretations that prenatal immune activation causes hypermethylation of GAD1 and GAD2 promoters in the mPFC. Compared to other cell types residing in the brain parenchyma, MeCP2 is most highly expressed in neurons, especially in GABAergic cells, where it acts as an important transcriptional regulator.28,50-52 When bound to methylated DNA, MeCP2 can interact with histone deacetylases (HDACs) and/or other transcription factors to form a complex that suppresses transcriptional activity.28,51 In addition to its transcriptional repressor functions, however, MeCP2 may also act as a transcriptional activator through the recruitment of the transcription factor CREB1.52 The exact mechanism by which MeCP2 represses or activates gene transcription is an area of intensive research and may be influenced by various factors, including gene length.53 In our model of prenatal infection, the increase in MeCP2 binding at the GAD1 and GAD2 promoters may be more readily associated with transcriptional repression as this effect was further accompanied by reduced GAD67 and GAD65 mRNA levels.
Despite the similarities between the effects of prenatal immune activation on GAD1 and GAD2 methylation and MeCP2 binding, the prenatal manipulation had a differential impact on promoter hydroxymethylation. Whereas it markedly increased 5hmC levels at the GAD1 promoter, it failed to do so at the GAD2 promoter. These differential effects suggest that the promoter regions of the 2 GABAergic genes may be under the control of partly distinct epigenetic regulatory mechanisms, at least in offspring with prenatal infectious histories. It remains debatable, however, whether 5hmC modifications serve as a stable epigenetic mark that directly regulates gene expression, or alternatively, whether they represent a transient intermediate state that marks sites of active CG demethylation.33,48 The latter possibility would be supported by findings showing that DNA demethylating enzymes, including TET enzymes, can bind to 5hmC and catalyze its oxidation to 5-formylcytosine and 5-carboxylcytosine.50 The latter yield substrates for thymine-DNA glycosylase, which in turn promotes the generation of non-methylated cytosines.49,54 Whatever functions 5hmC modifications may have, converging evidence suggests that altered hydroxymethylation can contribute to behavioral and cognitive deficits.55-57 Moreover, increased cortical 5hmC levels in GABAergic neurons are linked to reduced transcription of GAD1 in schizophrenia and schizophrenia-relevant animal models.58 Our data are consistent with these findings by showing altered prefrontal hydroxymethylation in an established infection-based neurodevelopmental disruption model that is characterized by disease-relevant behavioral and cognitive impairments.7-8,34-37
Future studies should attempt to identify the developmental mechanisms linking prenatal immune activation to adult GABAergic abnormalities. We have previously demonstrated that immune challenge in late pregnancy leads to a post-pubertal emergence of reduced GAD67 and GAD65 mRNA and protein expression in the mPFC.38 Therefore, it would be interesting to determine whether GAD1/GAD2 promoter abnormalities may follow a similar developmental pattern, that is, whether more extensive epigenetic changes may be present in adulthood as compared to pre-pubertal stages. In addition, future studies are required to identify the mechanisms mediating the epigenetic effects of prenatal immune activation. One possibility would be that poly(I:C)-induced inflammatory events directly affect the expression of epigenetic enzymes, which in turn would modify the methylation levels of GAD genes at early neonatal stages.43 Evidence for this possibility arises from studies demonstrating that pro-inflammatory cytokines such as IL-6 can induce expression of DNMT1 and in turn elicit changes in CpG DNA methylation at several genes.59,60 We and others have shown that prenatal immune activation by administration of poly(I:C) leads to a rapid increase in IL-6 levels in the fetal brain.11,61,62 IL-6 appears to assume a key role in mediating the effects of maternal immune activation on fetal brain development.61,62 Thus, it would be interesting to characterize the expression levels of epigenetic enzymes (such as DNMT1) and to quantify GAD1/GAD2 promoter methylation at distinct developmental stages following prenatal immune challenge in general, and following more specific manipulations leading to a selective increase of fetal IL-6 levels.
It should also be pointed out, however, that changes in DNA methylation (in response to early-life stress) do not necessarily correlate with changes in gene expression.63 Prevalent theories in fact suggest that DNA hypermethylation can lead to poised states that will determine future transcriptional responses.64 For example, it is thought that a large number of promoters are kept in a temporarily “dormant” hypermethylated state by DNMT1 or TET1 binding, while the subsequent recruitment of other epigenetic enzymes or transcription repressors at later stages would cement this dormant state into transcriptional repression by facilitating de novo methylation.28,65 It is thus feasible that prenatal immune activation would lead to immediate effects on DNMTs/TETs expression and GAD1/GAD2 promoter methylation, whereas noticeable downregulation of GAD67 and GAD65 would only emerge later, e.g., in late adolescence or early adulthood.
Finally, a last possibility would be that changes in the expression of DNMTs (and/or of other epigenetic regulators) following the prenatal immune activation may occur only at late maturational stages, that is, at times when reduced GAD67 and GAD65 mRNA and protein expression are evident.38 The dynamic and age-dependent expression of DNMTs across postnatal life would readily support this possibility. Indeed, brain levels of DNMTs are known to undergo substantial reductions across childhood and peri-adolescence in rodents.66 At the same time, global methylation patterns in the human frontal cortex are remodeled across postnatal life in a cell-specific manner,13 and in particular within the GAD1 gene.67 Thus, in view of the highly dynamic nature of DNMTs expression and activity, it is likely that numerous regulatory mechanisms converge to control epigenetic changes across postnatal life. Although these regulatory mechanisms remain largely unknown, miRNAs represent important candidates. Indeed, several in vitro studies have identified a number of candidate miRNAs capable of regulating the expression levels of DNMT1, DNMT3a, and DNMT3b mRNAs, respectively.59,68 In particular, a recent study has demonstrated that DNMT3a expression can be reduced by mir-132, a miRNA that is also downregulated in the PFC of patients with schizophrenia.69 Expression of mir-132 also increases during critical periods of brain maturation, where it plays important roles in the regulation of plasticity.70 Interestingly, mir-132 expression participates to the recruitment of anti-inflammatory responses in the brain and periphery following LPS administration.71 Changes in miRNA expression following prenatal immune activation may thus represent another possible mechanism whereby this early-life insult can lead to promoter remodeling of GAD and other genes.
In our study, we examined methylation-related epigenetic marks in mice that were previously subjected to behavioral testing. One possible limitation of this approach is that the animals were exposed to a certain degree of stress during behavioral testing. Hence, the previous testing history itself might have introduced some changes in the levels of promoter 5mC, 5hmC, and/or MeCP2 binding. Based on our long-standing expertise with behavioral examination in mice, however, we deem this possibility rather unlikely for two main reasons. First, the chosen behavioral tests were associated only with minimal stress for the animals, as they did not involve any major aversive events such as foot-shock, food- or water-deprivation, or long-term behavioral training. Second, a 1-week resting period was imposed between the completion of behavioral testing and collection of brain samples. This minimized the potential confounds that might arise from short-term exposure to stress. On the other hand, one clear advantage of our approach to examine epigenetic marks in behaviorally characterized animals is that it allowed us to perform correlations between the molecular and behavioral readouts. By doing so, we revealed that epigenetic modifications at the GAD1 promoter, but not at the GAD2 promoter, correlated with social interaction and working memory. Even though these findings do not allow us to draw any conclusions regarding possible causal relationships, they are consistent with the notion that molecular abnormalities within GAD1 may play a more prominent role than GAD2 in neuropsychiatric disorders that are characterized by social impairments and working memory deficiency.20,23,27-29 Furthermore, our results are consistent with the findings of reduced social behavior in transgenic mice harboring a conditional CNS-specific deletion of GAD1.72
As extensively reviewed elsewhere,7-8 the prenatal poly(I:C) administration model in mice (and other species) captures a wide variety of behavioral, anatomical, neurochemical and neuromorphological abnormalities pertinent to neuropsychiatric disorders, especially schizophrenia and autism. The present findings provide an important extension to this model by identifying epigenetic phenotypes that are reminiscent of those found in prefrontal cortical structures of patients with schizophrenia and related disoders.20,28,73 The etiological mechanisms underlying the epigenetic modifications in these disorders remain largely unknown. Against the background of the epidemiological association between prenatal infection and subsequent risk of neuropsychiatric disorders in humans,1-6 our findings suggest that maternal infection during pregnancy may be a significant environmental risk factor for GABA-related epigenetic abnormalities in these disorders. In broader context, our findings add to the emerging evidence suggesting that epigenetic remodeling is one of the critical processes by which early-life environmental experiences can be translated into altered neuronal and behavioral functions.74 This form of epigenetic plasticity is an essential contributor to phenotypic plasticity in developing and post-mitotic neurons and critically shapes experience-dependent neuronal adaptions required for normal brain development and functioning.14,75,76 Disruption of these processes, be it by early-life infection or by other developmental stressors, can therefore be expected to contribute to neurodevelopmental disorders by promoting global or specific epigenetic reconfigurations and subsequent changes in neuronal gene expression and functions.
It should be also be noted, that our findings are somewhat contrary to those reported by Connor et al.,15 showing that late pregnancy immune activation (gestational day 17.5) leads to only subtle non-significant changes in genome-wide promoter trimethylated histone H3 at lysine 4 (H3K4me3) marks in the cerebral cortex. Hence, whereas our findings indicate that prenatal immune activation can lead to altered promoter methylation patterns, those of Connor et al.15 suggest that these effects do not extend to all forms of chromatin remodeling, but rather induce specific changes within particular classes of epigenetic marks. Future studies shall attempt determining whether prenatal immune activation affects other types of histone modifications known to regulate transcriptional activity such as H3K27me3 and H3K9me2/3 known to closely associate with gene repression or H3K4me3 that associates with the promoter regions of active genes.77 Related to this, it would also be interesting to extend our own findings by assessing methylation levels at individual CpGs of the GAD1/GAD2 genes, using high-resolution methods, such as bisulfite cloning or pyrosequencing, and possibly also extend our targeted approach to a genome-wide level.
Our findings may have implications for attempts to ameliorate the behavioral and cognitive impairments that emerge following prenatal exposure to infection. Indeed, it will be intriguing to explore whether demethylating pharmacological agents may be effective in restoring such impairments. To the best of our knowledge, there have been no attempts yet to normalize behavioral and cognitive abnormalities using primary epigenetic pharmacotherapy in models of prenatal infection. Such attempts, however, have been proven useful in other preclinical models relevant to schizophrenia, including pharmacological models and models of prenatal stress exposure.74,78-80 The GAD1 promoter region seems particularly sensitive to epigenetic-based pharmacotherapy, as supported by previous work using DNMT or HDAC inhibitors.80-82 Related to this, it is interesting to note that prenatal infection-induced working memory deficits can be mitigated by chronic treatment with the atypical antipsychotic drug clozapine.83 Besides other neurochemical properties, clozapine has DNA-demethylation activity and is capable of reversing GAD1 promoter hypermethylation in animal models.84 Whether the effectiveness of clozapine to mitigate infection-induced working memory deficits is somehow related to epigenetic effects at the GAD1 promoter is still unknown and thus awaits direct examination.
In conclusion, our findings lend further support to the hypothesis that exposure to early-life adversities can regulate the transcriptional regulation of genes that are critically implicated in adult brain disorders. Specifically, our study demonstrates that prenatal viral-like immune activation in mice induces GAD1 and GAD2 promoter remodeling, which is characterized by promoter hypermethylation and increased MeCP2 promoter binding. These epigenetic effects are associated with reduced expression of the corresponding mRNA transcripts (GAD67 and GAD65) and, in the case of GAD1, correlate with behavioral and cognitive functions. Epigenetic modifications at presynaptic GABAergic systems may thus represent an important mechanism whereby maternal infection during pregnancy can induce long-term behavioral and cognitive impairments in the offspring.
Materials and Methods
Animals
C57BL6/N mice were used throughout the study. Female and male breeders were obtained from Charles River Laboratories (Germany) at the age of 10–14 weeks. Breeding began after 2 weeks of acclimatization to the animal holding rooms, which were temperature- and humidity-controlled (21 ± 1°C, 55 ± 5%) facilities under a reversed light–dark cycle (lights off: 8:00 A.M. to 8:00 P.M.). All animals had ad libitum access to food (Kliba, #3430) and water. All procedures described in the present study had been previously approved by the Cantonal Veterinary Office of Zurich. All efforts were made to minimize the number of animals used and their suffering.
Maternal immune activation during pregnancy
For the purpose of the maternal immunological manipulation in late gestation, female mice were subjected to a timed mating procedure as described previously.40-43 Pregnant dams on GD17 received either a single injection of poly(I:C) (potassium salt; Sigma-Aldrich, # P9582) or vehicle. Poly(I:C) (5 mg/kg) was dissolved in sterile pyrogen-free 0.9% NaCl (vehicle) solution to yield a final concentration of 1 mg/ml and was administered via i.v. route at the tail vein under mild physical constraint. The dose of poly(I:C) was chosen based on our previous studies.35-38 All solutions were freshly prepared on the day of administration and injected with a volume of 5 ml/kg. Offspring born to poly(I:C)- or vehicle-treated mothers were allocated to behavioral and molecular investigations of interest, as detailed in the Supplementary Material.
Behavioral analyses
Working memory was assessed using a spatial recognition test in the Y-maze using protocols established and validated before.36,38 Social interaction was assessed by analyzing the relative exploration time between an unfamiliar congenic mouse and an inanimate dummy object using methods established before.36 A detailed description of the test apparatus and procedures is given in the Supplementary Material.
Collection of medial prefrontal cortex samples
Following decapitation of the animals, the brains were immediately extracted from the skull and allowed to freeze on dry ice before storage at −80°C until further use. Frozen brains were placed dorsal side up on a dry ice-chilled plate for micro-dissection of the medial prefrontal cortices (mPFC) using methods described in detail elsewhere.35,36,38 Micropunches of the mPFC were collected in 1.5-mL Eppendorf tubes and stored at −80°C for subsequent epigenetic or mRNA analyses (see below).
Methylated and hydroxymethylated DNA immunoprecipitation (MeDIP and hMeDIP)
We analyzed the ratio of 5mC or 5hmC to the total cytosines of mouse GAD1 and GAD2 CpG-enriched promoter fragments according to protocols established and validated before.43,44 A schematic representation of the GAD1 and GAD2 promoters, including CpG islands, CpG shores, TSS and genomic positions of interest, is depicted in (Fig. 2). Mouse monoclonal 5mC (MagMeDIP kit, Diagenode, #C02010021) or rabbit polyclonal 5hmC (hMeDIP kit, Diagenode, #C02010031) antibodies were used for the immunoprecipitations. Genomic DNA (gDNA) was extracted from approximately 16 mg mPFC tissue using phenol-chloroform extraction and ethanol precipitation procedures and then quantified by spectrophotometric analysis (NanoDrop, Thermoscientific). gDNA (13 μg) were sonicated for 1min at 20°C (Duty factor 10%; Cycles/burst 200) to produce a fragment size of 200-600 bp (as verified by gel electrophoresis) using the SonoLab #M-220 sonicator (Covaris). Sonicated DNA (1.2 μg) were heat-denatured and used for the 5mC or 5hmC immunoprecipitations (IP) according to the manufacturer's instructions. Ten percent of the solution was removed and stored at 4°C overnight to be used to quantify the total amount of DNA before immunoprecipitation (input samples). The remaining solution was incubated overnight at 4°C with mouse anti-5mC or anti-5hmC antibodies provided by the kit (IP samples). The immunoprecipitated DNA was washed and released from the antibody complex by proteinase-K digestion and resuspended in 100 μL of DNA-immunoprecipitation buffer (DIB) provided by the kit. An immunoprecipitation negative control (IgG antibody at the same concentration) was included in each assay and did not produce any detectable signal.
MeCP2 binding to GAD1 and GAD2 promoters by chromatin immunoprecipitation (ChIP) measurements
The MeCP2 ChIP assays were performed using the Magna ChIP G Tissue Kit (Merck-Millipore, #17-20000) as described in detail elsewhere.43,44 Briefly, approximately 16 mg of mPFC tissue was used for this procedure. Tissue microcubes (1 mm3) were homogenized and washed into 500 μl tissue stabilizing solution supplemented with a protease inhibitor (PI) cocktail. Tissue pellets were then incubated with 900 μL of PBS containing 1% formaldehyde at 37°C for 7 min, after which the fixation was terminated with 100 μl 10x glycine. After three washes with cold PBS containing PI, the tissue was homogenized in 500 μL SDS lysis buffer containing PI, incubated at 4°C for 20 min and resuspended into 130 μl dilution buffer containing PI. To obtain consistent chromatin fragmentation, the lysates were sonicated for 15 min at 7°C (Duty factor 10%; Cycles/burst 200) to produce a fragment size of 200-600 bp (as verified by gel electrophoresis) using the SonoLab #M-220 sonicator (Covaris). The samples were then centrifuged to remove insoluble material and resuspended into 2mL dilution buffer with PI. An aliquot of 500 μl was incubated overnight at 4°C with magnetic beads (provided by the kit) and 1.5 μg MeCP2 antibody (rabbit polyclonal; Merck-Millipore, #07-013) using protocols established and validated before.49,50 An aliquot (1%) of the sonicated lysate without antibody (input) was used to quantify the total amount of DNA in sample extracts before immunoprecipitation. At the end of the ChIP procedure, the protein/DNA cross-linked nucleosomal chromatin complex immunoprecipitated with MeCP2 antibody was washed, and then reverse cross-linked at 62°C for 2 h and 95°C for 10 min with proteinase-K. Protein-free DNA was further purified using spin filter columns (provided by the kit). The extract was used for detection and quantification of GAD1 and GAD2 gene promoters. An immunoprecipitation negative control (IgG antibody at the same concentration) was included in each assay and did not produce any detectable signal.
Quantitative real-time PCR analysis of ChIP, MeDIP and hMeDIP fragments
CpG-rich GAD1 and GAD2 gene promoters and gene bodies were measured by qRT-PCR as fully described elsewhere.43,44 In brief, the samples were run in 96-well formats using Sybr green (Applied Biosystems, #K0222) on a Agilent Mx3005P qRT-PCR system. DNA samples (5 μl) were run in duplicates for the regions of interest in 20 μl reactions. Primer sequences were designed manually with assistance from Integrated DNA Technologies (IDT) software and purchased from IDT. The assays were validated on input and IP DNA samples to verify the unicity of the PCR product (one-peak melting curve) and sufficient amplification. The primer sequences are summarized in Table 2. Thermal cycling was initiated with incubation at 95°C for 10 min (TaqMan polymerase activation). After this initial step, 40 cycles of PCR were performed. Each PCR cycle consisted of heating the samples at 95°C for 30 s to enable the melting process and then for 60 s at 60°C and 60 s at 72°C for the annealing and extension reactions. The PCR reaction was followed by a final melting curve step. The percent (hydroxy-)methylated vs. total cytosines was calculated by the following equation: % (meDNA-IP/total input) = 2^[(Ct(10% input) - 3.32) - Ct(meDNA-IP)] x 100%.43,44 The percent MeCP2-bound DNA vs. total DNA was calculated by the following equation: % (MeCP2-DNA-IP/total input) = 2^[(Ct(1% input) - 3.32) - Ct(MeCP2-DNA -IP)] x 100%.43,44
Table 2.
Primers used in the methylation assays and real-time PCR analyses. Top: Primers for the epigenetic assays. The three columns give the sequences of the forward and reverse primers, as well as the genomic location of the amplicon (*) as compared to the transcriptional start site (TSS) defined using the UCSC Genome Browser, positions chr2:70562163 (TGTAGTTTAA…) for GAD1 and chr2:22,622,326 (CTCCCTAAAA…) for GAD2. Bottom: Primers for the real-time PCR analyses given the forward and reverse primers for GAD1, GAD,2 and β-Actin assays.
| Primer Sequences for MeDIP, hMeDIP and ChIP assays | Forward Primer | Reverse Primer | Amplicon Position* |
|---|---|---|---|
| GAD1 Promoter - Region 1 | 5'-CAAACCCGTGAGCTGGATTTA-3' | 5'-GGAATCACGAGGCCGTTG-3' | −193 to −74bp |
| GAD1 Promoter - Region 2 | 5'-GGATCGTGCAAGCAAGGAAGCA-3' | 5'-TGAGGCAAAGGGCTGGACAAG-3' | +35 to +268bp |
|
GAD2 Promoter |
5'-CTTTGGAGCTCTTCCTGATTGA-3' |
5'-TTGTTTGAGGGCTGTCTCTG-3' |
−401 to −269bp |
|
Primer Sequences for gene expression assays |
Forward Primer |
Reverse Primer |
|
| GAD1 | 5'-ATGATACTTGGTGTGGCGTAG-3' | 5'-GACTCTTCTCTTCCAGGCTATTG-3' | |
| GAD2 | 5'-GCTTTTGGTCCTTCGGATCT-3' | 5'-GAACTTTTGGGCCACCTG-3' | |
| β-Actin | 5'-TGTGATGGTGGGAATGGGTCAGAA-3' | 5'-TGTGGTGCCAGATCTTCTCCATGT-3' |
Quantification of GAD67 and GAD65 mRNA by quantitative real-time PCR
Total RNA from mPFC specimen (see above) was isolated using the Qiagen RNeasy Mini kit (Qiagen, #74104) according to the manufacturer's instructions, and quantified by spectrophotometric analysis (NanoDrop, Thermoscientific). Each sample was treated with DNase to avoid DNA contamination. One μg total RNA was then converted to cDNA in a 20 μL reaction using the High Capacity cDNA Reverse transcription kit (Applied Biosystems, #4374966) according to the manufacturer's instructions, and then frozen at -20°C until further use. cDNA samples were diluted 1:10 and used for subsequent quantitative real-time PCR (qRT-PCR) assays. The samples were run in 384-well formats using Fast-Sybr green (Applied Biosystems, #4385612) on an Applied Biosystems ViiATM7 qRT-PCR system. Samples were run in triplicates for the gene transcripts of interest (GAD67 and GAD65) and a normalizing control (β-actin) in 5 μL reactions. Primer sequences were designed using the Roche online Universal Probe Library and purchased from Microsynth AG. Assays were validated using 1:5 dilution standard curves of the same cDNA samples to verify the unicity of the PCR product (one-peak melting curve) and sufficient amplification. The primer sequences of interest demonstrated high amplification efficiency across a wide range of serial dilutions of our samples. The primer sequences are summarized in Table 2. Thermal cycling was initiated with incubation at 95°C for 5min (TaqMan polymerase activation). After this initial step, 40 cycles of PCR were performed. Each PCR cycle consisted of heating the samples at 95°C for 3 s to enable the melting process and then for 30 s at 60°C for the annealing and extension reactions. The PCR reaction was followed by a final melting curve step. Relative target gene expression was calculated according to the 2[-delta Ct] method.85
Statistical analyses
All data were analyzed using parametric analysis of variance (ANOVA). Correlative analyses between epigenetic and behavioral variables of interest were performed using Pearson's product moment correlations. Statistical significance was set at P < 0.05. All statistical analyses were conducted using the statistical software StatView (version 5.0) and SPSS (version 13.0).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Ying Chen for helpful advice and Wolfgang Langhans for continuous infrastructural support and for comments on the final version of the manuscript.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
Funding
The present study was supported by grants from the Swiss National Science Foundation (Grant Nr. 310030_146217) and the European Union Seventh Framework Program (FP7/2007–2011; Grant Agreement Nr. 259679) awarded to UM and by grants from the National Institutes of Health (Grants Nr. 5R01MH093348-03 and NIMH2013-00362) awarded to AG. Additional funding was provided by a European Molecular Biology Organization (EMBO) Short-Term Fellowship awarded to MAL.
References
- 1. Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry 2010; 167:261–80; PMID:20123911; http://dx.doi.org/ 10.1176/appi.ajp.2009.09030361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Canetta S, Sourander A, Surcel HM, Hinkka-Yli-Salomäki S, Leiviskä J, Kellendonk C, McKeague IW, Brown AS. Elevated maternal C-reactive protein and increased risk of schizophrenia in a national birth cohort. Am J Psychiatry 2014; 171:960–8; PMID:24969261; http://dx.doi.org/ 10.1176/appi.ajp.2014.13121579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Atladóttir HO, Thorsen P, Østergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J Autism Dev Disord 2010; 40:1423–30; http://dx.doi.org/ 10.1007/s10803-010-1006-y [DOI] [PubMed] [Google Scholar]
- 4. Brown AS, Sourander A, Hinkka-Yli-Salomäki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry 2014; 19:259–64; PMID:23337946; http://dx.doi.org/ 10.1038/mp.2012.197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Parboosing R, Bao Y, Shen L, Schaefer CA, Brown AS. Gestational influenza and bipolar disorder in adult offspring. JAMA Psychiatry 2013; 70:677–85; PMID:23699867; http://dx.doi.org/ 10.1001/jamapsychiatry.2013.896 [DOI] [PubMed] [Google Scholar]
- 6. Canetta SE, Bao Y, Co MD, Ennis FA, Cruz J, Terajima M, Shen L, Kellendonk C, Schaefer CA, Brown AS. Serological documentation of maternal influenza exposure and bipolar disorder in adult offspring. Am J Psychiatry 2014; 171:557–63; PMID:24480930; http://dx.doi.org/ 10.1176/appi.ajp.2013.13070943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harvey L, Boksa P. Prenatal and postnatal animal models of immune activation: relevance to a range of neurodevelopmental disorders. Dev Neurobiol 2012; 72:1335–48; PMID:22730147; http://dx.doi.org/ 10.1002/dneu.22043 [DOI] [PubMed] [Google Scholar]
- 8. Meyer U. Prenatal poly(I:C) exposure and other developmental immune activation models in rodent systems. Biol Psychiatry 2014; 75:307–15; PMID:23938317; http://dx.doi.org/ 10.1016/j.biopsych.2013.07.011 [DOI] [PubMed] [Google Scholar]
- 9. Meyer U. Developmental neuroinflammation and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42:20–34; PMID:22122877; http://dx.doi.org/ 10.1016/j.pnpbp.2011.11.003 [DOI] [PubMed] [Google Scholar]
- 10. Miller BJ, Culpepper N, Rapaport MH, Buckley P. Prenatal inflammation and neurodevelopment in schizophrenia: a review of human studies. Prog Neuropsychopharmacol Biol Psychiatry 2013; 42:92–100; PMID:22510462; http://dx.doi.org/ 10.1016/j.pnpbp.2012.03.010 [DOI] [PubMed] [Google Scholar]
- 11. Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006; 26:4752–62; PMID:16672647; http://dx.doi.org/ 10.1523/JNEUROSCI.0099-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piontkewitz Y, Arad M, Weiner I. Abnormal trajectories of neurodevelopment and behavior following in utero insult in the rat. Biol Psychiatry 2011; 70:842–51; PMID:21816387; http://dx.doi.org/ 10.1016/j.biopsych.2011.06.007 [DOI] [PubMed] [Google Scholar]
- 13. Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013; 341:1237905; PMID:23828890; http://dx.doi.org/ 10.1126/science.1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 2003; 33Suppl:245–54; PMID:12610534; http://dx.doi.org/ 10.1038/ng1089 [DOI] [PubMed] [Google Scholar]
- 15. Connor CM, Dincer A, Straubhaar J, Galler JR, Houston IB, Akbarian S. Maternal immune activation alters behavior in adult offspring, with subtle changes in the cortical transcriptome and epigenome. Schizophr Res 2012; 140:175–84; PMID:22804924; http://dx.doi.org/ 10.1016/j.schres.2012.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tang B, Jia H, Kast RJ, Thomas EA. Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav Immun 2013; 30:168–75; PMID:23402795; http://dx.doi.org/ 10.1016/j.bbi.2013.01.086 [DOI] [PubMed] [Google Scholar]
- 17. Basil P, Li Q, Dempster EL, Mill J, Sham PC, Wong CC, McAlonan GM. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Transl Psychiatry 2014; 4:e434; PMID:25180573; http://dx.doi.org/ 10.1038/tp.2014.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hollins SL, Zavitsanou K, Walker FR, Cairns MJ. Alteration of imprinted Dlk1-Dio3 miRNA cluster expression in the entorhinal cortex induced by maternal immune activation and adolescent cannabinoid exposure. Transl Psychiatry 2014; 4:e452; PMID:25268256; http://dx.doi.org/ 10.1038/tp.2014.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 2005; 6:312–24; PMID:15803162; http://dx.doi.org/ 10.1038/nrn1648 [DOI] [PubMed] [Google Scholar]
- 20. Guidotti A, Auta J, Chen Y, Davis JM, Dong E, Gavin DP, Grayson DR, Matrisciano F, Pinna G, Satta R, et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacol 2011; 60:1007–16; PMID:21074545; http://dx.doi.org/ 10.1016/j.neuropharm.2010.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blatt GJ, Fatemi SH. Alterations in GABAergic biomarkers in the autism brain: research findings and clinical implications. Anat Rec 2011; 294:1646–5; http://dx.doi.org/ 10.1002/ar.21252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sibille E, Morris HM, Kota RS, Lewis DA. GABA-related transcripts in the dorsolateral prefrontal cortex in mood disorders. Int J Neuropsychopharmacol 2011; 14:721–34; PMID:21226980; http://dx.doi.org/ 10.1017/S1461145710001616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev 2006; 52:293–304; PMID:16759710; http://dx.doi.org/ 10.1016/j.brainresrev.2006.04.001 [DOI] [PubMed] [Google Scholar]
- 24. Asada H, Kawamura Y, Maruyama K, Kume H, Ding RG, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Cleft palate and decreased brain gamma-aminobutyric acid in mice lacking the 67-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A 1997; 94:6496–9; PMID:9177246; http://dx.doi.org/ 10.1073/pnas.94.12.6496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A 1999; 96:12911–6; PMID:10536022; http://dx.doi.org/ 10.1073/pnas.96.22.12911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Patel AB, de Graaf RA, Martin DL, Battaglioli G, Behar KL. Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo. J Neurochem 2006; 97:385–96; PMID:16539672; http://dx.doi.org/ 10.1111/j.1471-4159.2006.03741.x [DOI] [PubMed] [Google Scholar]
- 27. Glausier JR, Kimoto S, Fish KN, Lewis DA. Lower glutamic acid decarboxylase 65-kDa isoform messenger RNA and protein levels in the prefrontal cortex in schizoaffective disorder but not schizophrenia. Biol Psychiatry 2015; 77:167–76; PMID:24993056; http://dx.doi.org/ 10.1016/j.biopsych.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology 2013; 38:138–166; PMID:22948975; http://dx.doi.org/ 10.1038/npp.2012.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhubi A, Cook EH, Guidotti A, Grayson DR. Epigenetic mechanisms in autism spectrum disorder. Int Rev Neurobiol 2014; 115:203–44; PMID:25131546; http://dx.doi.org/ 10.1016/B978-0-12-801311-3.00006-8 [DOI] [PubMed] [Google Scholar]
- 30. Huang HS, Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS One. 2007; 2:e809; PMID:17726539; http://dx.doi.org/ 10.1371/journal.pone.0000809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dong E, Ruzicka WB, Grayson DR, Guidotti A. DNA-methyltransferase1 (DNMT1) binding to CpG rich GABAergic and BDNF promoters is increased in the brain of schizophrenia and bipolar disorder patients. Schizophr Res 2014; 167(1-3):35–41, in press; Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A, Grayson DR. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl Psychiatry 2014; 4:e349; PMID:24448211; http://dx.doi.org/ 10.1038/tp.2013.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong E, Gavin DP, Chen Y, Davis J. Upregulation of TET1 and downregulation of APOBEC3A and APOBEC3C in the parietal cortex of psychotic patients. Transl Psychiatry 2012; 2:e159; PMID:22948384; http://dx.doi.org/ 10.1038/tp.2012.86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gavin DP, Sharma RP, Chase KA, Matrisciano F, Dong E, Guidotti A. Growth arrest and DNA-damage-inducible, β (GADD45b)-mediated DNA demethylation in major psychosis. Neuropsychopharmacology 2012; 37:531–42; PMID:22048458; http://dx.doi.org/ 10.1038/npp.2011.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Richetto J, Calabrese F, Riva MA, Meyer U. Prenatal immune activation induces maturation-dependent alterations in the prefrontal GABAergic transcriptome. Schizophr Bull 2014; 40:351–61; PMID:23328159; http://dx.doi.org/ 10.1093/schbul/sbs195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Richetto J, Labouesse MA, Poe MM, Cook JM, Grace AA, Riva MA, Meyer U. Behavioral effects of the benzodiazepine-positive allosteric modulator SH-053-2'F-S-CH₃ in an immune-mediated neurodevelopmental disruption model. Int J Neuropsychopharmacol 2015; 18(4); PMID:25636893; http://dx.doi.org/ 10.1093/ijnp/pyu055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Meyer U, Feldon J, Fatemi SH. In-vivo rodent models for the experimental investigation of prenatal immune activation effects in neurodevelopmental brain disorders. Neurosci Biobehav Rev 2009; 33:1061–79; PMID:19442688; http://dx.doi.org/ 10.1016/j.neubiorev.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 38. Richetto J, Calabrese F, Meyer U, Riva MA. Prenatal versus postnatal maternal factors in the development of infection-induced working memory impairments in mice. Brain Behav Immun 2013; 33:190–200; PMID:23876745; http://dx.doi.org/ 10.1016/j.bbi.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 39. Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun 2012; 26:607–16; PMID:22310922; http://dx.doi.org/ 10.1016/j.bbi.2012.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, Brückl T, Biel M, Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 2010; 5:e15367; PMID:21203455; http://dx.doi.org/ 10.1371/journal.pone.0015367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res 2011; 39:5015–24; PMID:21378125; http://dx.doi.org/ 10.1093/nar/gkr120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guo X, Wu X, Ren L, Liu G, Li L. Emerging roles of TET proteins and 5-hydroxymethylcytosines in active DNA demethylation and beyond. Cell Cycle 2011; 10:2662–8; PMID:21811096; http://dx.doi.org/ 10.4161/cc.10.16.17093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, Nicoletti F, Guidotti A. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 2013; 68:184–94; PMID:22564440; http://dx.doi.org/ 10.1016/j.neuropharm.2012.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dong E, Dzitoyeva SG, Matrisciano F, Tueting P, Grayson DR, Guidotti A. Brain-derived neurotrophic factor epigenetic modifications associated with schizophrenia-like phenotype induced by prenatal stress in mice. Biol Psychiatry 2015; 77:589–96; PMID:25444166; http://dx.doi.org/ 10.1016/j.biopsych.2014.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang TY, Hellstrom IC, Bagot RC, Wen X, Diorio J, Meaney MJ. Maternal care and DNA methylation of a glutamic acid decarboxylase 1 promoter in rat hippocampus. J Neurosci 2010; 30:13130–7; PMID:20881131; http://dx.doi.org/ 10.1523/JNEUROSCI.1039-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, et al. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci 2011; 31:11088–95; PMID:21795557; http://dx.doi.org/ 10.1523/JNEUROSCI.1234-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Catts VS, Fung SJ, Long LE, Joshi D, Vercammen A, Allen KM, Fillman SG, Rothmond DA, Sinclair D, Tiwari Y, et al. Rethinking schizophrenia in the context of normal neurodevelopment. Front Cell Neurosci. 2013; 7:60; PMID:23720610; http://dx.doi.org/ 10.3389/fncel.2013.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002; 99:3740–5; PMID:11891299; http://dx.doi.org/ 10.1073/pnas.052410099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kinde B, Gabel HW, Gilbert CS, Griffith EC, Greenberg ME. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A 2015; 112:6800–6; PMID:25739960; http://dx.doi.org/ 10.1073/pnas.1411269112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chao HT, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu HC, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature 2010; 468:263–9; PMID:21068835; http://dx.doi.org/ 10.1038/nature09582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol 2011; 27:631–52; PMID:21721946; http://dx.doi.org/ 10.1146/annurev-cellbio-092910-154121 [DOI] [PubMed] [Google Scholar]
- 52. Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008; 320:1224–9; PMID:18511691; http://dx.doi.org/ 10.1126/science.1153252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sugino K, Hempel CM, Okaty BW, Arnson HA, Kato S, Dani VS, Nelson SB. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci 2014; 34:12877–83; PMID:25232122; http://dx.doi.org/ 10.1523/JNEUROSCI.2674-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011; 333:1303–7; PMID:21817016; http://dx.doi.org/ 10.1126/science.1210944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kaas GA, Zhong C, Eason DE, Ross DL, Vachhani RV, Ming GL, King JR, Song H, Sweatt JD. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron 2013; 79:1086–93; PMID:24050399; http://dx.doi.org/ 10.1016/j.neuron.2013.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rudenko A, Dawlaty MM, Seo J, Cheng AW, Meng J, Le T, Faull KF, Jaenisch R, Tsai LH. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013; 79:1109–22; PMID:24050401; http://dx.doi.org/ 10.1016/j.neuron.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li X, Wei W, Zhao QY, Widagdo J, Baker-Andresen D, Flavell CR, D'Alessio A, Zhang Y, Bredy TW. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci U S A 2014; 111:7120–5; PMID:24757058; http://dx.doi.org/ 10.1073/pnas.1318906111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guidotti A, Grayson DR. DNA methylation and demethylation as targets for antipsychotic therapy. Dialogues Clin Neurosci 2014; 16:419–29; PMID:25364290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Braconi C, Huang N, Patel T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology. 2010; 51:881–90; PMID:20146264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Foran E, Garrity-Park MM, Mureau C, Newell J, Smyrk TC, Limburg PJ, Egan LJ. Upregulation of DNA methyltransferase-mediated gene silencing, anchorage-independent growth, and migration of colon cancer cells by interleukin-6. Mol Cancer Res 2010; 8:471–81; PMID:20354000; http://dx.doi.org/ 10.1158/1541-7786.MCR-09-0496 [DOI] [PubMed] [Google Scholar]
- 61. Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 2007; 27:10695–702; PMID:17913903; http://dx.doi.org/ 10.1523/JNEUROSCI.2178-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lipina TV, Zai C, Hlousek D, Roder JC, Wong AH. Maternal immune activation during gestation interacts with Disc1 point mutation to exacerbate schizophrenia-related behaviors in mice. J Neurosci 2013; 33:7654–66; PMID:23637159; http://dx.doi.org/ 10.1523/JNEUROSCI.0091-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, Kobor MS. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci U S A 2012; 109:17253–60; PMID:23045638; http://dx.doi.org/ 10.1073/pnas.1121249109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Klengel T, Binder EB. Epigenetics of Stress-Related Psychiatric Disorders and Gene × Environment Interactions. Neuron 2015; 86:1343–57; PMID:26087162; http://dx.doi.org/ 10.1016/j.neuron.2015.05.036 [DOI] [PubMed] [Google Scholar]
- 65. Szulwach KE, Li X, Li Y, Song CX, Wu H, Dai Q, Irier H, Upadhyay AK, Gearing M, Levey AI, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci 2011; 14:1607–16; PMID:22037496; http://dx.doi.org/ 10.1038/nn.2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Simmons RK, Stringfellow SA, Glover ME, Wagle AA, Clinton SM. DNA methylation markers in the postnatal developing rat brain. Brain Res 2013; 1533:26–36; PMID:23954679; http://dx.doi.org/ 10.1016/j.brainres.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, Jaenisch R, Laird PW, Akbarian S. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One 2007; 2:e895; PMID:17878930; http://dx.doi.org/ 10.1371/journal.pone.0000895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A 2007; 104:15805–10; PMID:17890317; http://dx.doi.org/ 10.1073/pnas.0707628104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J, Willoughby D, Kenny PJ, Elsworth JD, Lawrence MS, et al. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci U S A 2012; 109:3125–30; PMID:22315408; http://dx.doi.org/ 10.1073/pnas.1113793109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mellios N, Sugihara H, Castro J, Banerjee A, Le C, Kumar A, Crawford B, Strathmann J, Tropea D, Levine SS, et al. miR-132, an experience-dependent microRNA, is essential for visual cortex plasticity. Nat Neurosci 2011; 14:1240–2; PMID:21892155; http://dx.doi.org/ 10.1038/nn.2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Shaked I, Meerson A, Wolf Y, Avni R, Greenberg D, Gilboa-Geffen A, Soreq H. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 2009; 31:965–73; PMID:20005135; http://dx.doi.org/ 10.1016/j.immuni.2009.09.019 [DOI] [PubMed] [Google Scholar]
- 72. Fujihara K, Miwa H, Kakizaki T, Kaneko R, Mikuni M, Tanahira C, Tamamaki N, Yanagawa Y. Glutamate decarboxylase 67 deficiency in a subset of GABAergic neurons induces schizophrenia-related phenotypes. Neuropsychopharmacology 2015; 40(10):2475–86, in press; [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mitchell AC, Jiang Y, Peter C, Akbarian S. Transcriptional regulation of GAD1 GABA synthesis gene in the prefrontal cortex of subjects with schizophrenia. Schizophr Res 2015; 167(1-3):28–34, in press; [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gapp K, Woldemichael BT, Bohacek J, Mansuy IM. Epigenetic regulation in neurodevelopment and neurodegenerative diseases. Neuroscience 2014; 264:99–111; PMID:23256926; http://dx.doi.org/ 10.1016/j.neuroscience.2012.11.040 [DOI] [PubMed] [Google Scholar]
- 75. Sweatt JD. Experience-dependent epigenetic modifications in the central nervous system. Biol Psychiatry 2009; 65:191–7; PMID:19006788; http://dx.doi.org/ 10.1016/j.biopsych.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Roadmap Epigenomics Consortium . Integrative analysis of 111 reference human epigenomes. Nature 2015; 518:317–30; PMID:25693563; http://dx.doi.org/ 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sims RJ, 3rd, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet 2003; 19:629–39; PMID:14585615; http://dx.doi.org/ 10.1016/j.tig.2003.09.007 [DOI] [PubMed] [Google Scholar]
- 78. Tremolizzo L, Doueiri MS, Dong E, Grayson DR, Davis J, Pinna G, Tueting P, Rodriguez-Menendez V, Costa E, Guidotti A. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry 2005; 57:500–9; PMID:15737665; http://dx.doi.org/ 10.1016/j.biopsych.2004.11.046 [DOI] [PubMed] [Google Scholar]
- 79. Dong E, Agis-Balboa RC, Simonini MV, Grayson DR, Costa E, Guidotti A. Reelin and glutamic acid decarboxylase67 promoter remodeling in an epigenetic methionine-induced mouse model of schizophrenia. Proc Natl Acad Sci U S A 2005; 102:12578–83; PMID:16113080; http://dx.doi.org/ 10.1073/pnas.0505394102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dong E, Guidotti A, Grayson DR, Costa E. Histone hyperacetylation induces demethylation of reelin and 67-kDa glutamic acid decarboxylase promoters. Proc Natl Acad Sci U S A 2007; 104:4676–81; PMID:17360583; http://dx.doi.org/ 10.1073/pnas.0700529104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kundakovic M, Chen Y, Costa E, Grayson DR. DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol Pharmacol 2007; 71:644–53; PMID:17065238; http://dx.doi.org/ 10.1124/mol.106.030635 [DOI] [PubMed] [Google Scholar]
- 82. Kundakovic M, Chen Y, Guidotti A, Grayson DR. The reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol 2009; 75:342–54; PMID:19029285; http://dx.doi.org/ 10.1124/mol.108.051763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Meyer U, Knuesel I, Nyffeler M, Feldon J. Chronic clozapine treatment improves prenatal infection-induced working memory deficits without influencing adult hippocampal neurogenesis. Psychopharmacology 2010; 208:531–43; PMID:20041229; http://dx.doi.org/ 10.1007/s00213-009-1754-6 [DOI] [PubMed] [Google Scholar]
- 84. Dong E, Nelson M, Grayson DR, Costa E, Guidotti A. Clozapine and sulpiride but not haloperidol or olanzapine activate brain DNA demethylation. Proc Natl Acad Sci U S A 2008; 105:13614–9; PMID:18757738; http://dx.doi.org/ 10.1073/pnas.0805493105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402–8; PMID:11846609; http://dx.doi.org/ 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.