

Abstract
Amorphous calcium phosphate (ACP), a postulated precursor in the formation of biological hydroxyapatite, has been evaluated as a filler phase in bioactive polymeric composites that utilize dental monomers to form the matrix phase on polymerization. In addition to excellent biocompatibility, these composites provided sustained release of calcium and phosphate ions into simulated saliva milieus. In an effort to enhance the physicochemical and mechanical properties and extend the utility of remineralizing ACP composites to a greater variety of dental applications, we have focused on: a) hybridizing ACP by introducing silica and/or zirconia, b) assessing the efficacy of potential coupling agents, c) investigating the effects of chemical structure and compositional variation of the resin matrices on the mechanical strength and ion-releasing properties of the composites, and d) improving the intrinsic adhesiveness of composites by using bifunctional monomers with an affinity for tooth structure in resin formulations. Si- and Zr-modified ACPs along with several monomer systems are found useful in formulating composites with improved mechanical and remineralizing properties. Structure-property studies have proven helpful in advancing our understanding of the remineralizing behavior of these bioactive composites. It is expected that this knowledge base will direct future research and lead to clinically valuable products, especially therapeutic materials appropriate for the healing or even regeneration of defective teeth and bone structures.
Keywords: adhesion, amorphous calcium phosphate, dental composites, hydroxyapatite, mechanical strength, methacrylate conversion, tooth remineralization, volumetric contraction, water sorption
1. Introduction
Calcium phosphates (CaPs) are of special interest to oral biology, dentistry and medicine since they occur in normal skeletal tissues (enamel, dentin, bone) and in pathological (atheroschlerotic deposits, urinary and dental calculi) calcifications [1]. Systematic studies of their use as preventive or restorative dental materials only began in the 1980s [2]. CaPs of biological significance are listed in Table 1. Crystalline HAP is considered to be the final, stable product in the precipitation of calcium and phosphate ions from neutral or basic solutions. Over the broad range of solution conditions in which precipitation occurs spontaneously, ACP precedes the appearance of HAP [3]. The possible role that ACP may play as a precursor to HAP in biological calcification places it in the mainstream of calcium phosphate chemistry [3–6]. In material applications, however, the relatively high solubility of ACP and its ready conversion to HAP in aqueous environments might pose limitations where structural, mechanical and chemical stabilities are desired. However, these same properties may make ACP suitable as a mineralizing agent. When compounded with appropriate polymeric resins, ACP’s bioactivity may be particularly useful in enhancing the prophylactic performance of composites, sealants and adhesives by preventing tooth demineralization and by actively promoting remineralization.
Table 1.
Calcium phosphates (CaPs) of biomedical significance
| CaP | Compositional formula | Acronym |
|---|---|---|
| Amorphous calcium phosphate | Ca3(PO4)2⋅3H2Oa | ACP |
| Monocalcium phosphate | Ca(H2PO4)2 | MCP |
| Dicalcium phosphate anhydrous | CaHPO4 | DCPA |
| Dicalcium phosphate dihydrate | CaHPO4⋅2 H2O | DCPD |
| Tricalcium phosphate | Ca3(PO4)2 | TCP |
| Octacalcium phosphate | Ca8H2(PO4)6⋅3H2O | OCP |
| Hydroxyapatite | Ca10(PO4)2(OH)2 | HAP |
We have recently developed unique bioactive composites based on filler phases consisting of pyrophosphate-stabilized ACP embedded in certain types of polymer matrix phases derived from the ambient polymerization of acrylic monomers [7, 8]. Significant levels of calcium and phosphate ions were found to be released from these composites that were sustainable over long periods. It was found that they efficiently promoted the recovery of mineral-deficient tooth structures in vitro [9]. However, ACP does not act as a reinforcing filler in a manner similar to that of commonly used silanized glass fillers (Table 2). We are currently exploring protocols for hybridizing and surface-modifying ACP fillers and compounding hybrid and/or surface-modified ACPs with resins of different chemical compositions and structural characteristics in an effort to make stronger ACP composites. In this article we report on the effect(s) of the ACP filler type and/or the resin matrix structure on: 1) ACP’s stability and the rate of internal conversion to HAP, 2) visible light-polymerization efficacy, 3) volumetric contraction upon polymerization, 4) remineralization potential and 5) mechanical strength of the composites. Our current research is designed to lead to improved, remineralizing bioactive and biocompatible ACP composites with extended dental and potentially orthopedic applications.
Table 2.
Functional differences between biostable and bioactive dental materials
| Type of dental material | Components | Function/activity |
|---|---|---|
| Biostable restoratives | Monomer + initiator system | Provides polymeric matrix |
| Silanized glass/ceramic filler | Reinforces matrix phase, enhances modulus and strength | |
| Bioactive composites | Glass ionomers/resin modified ionomers/compomers | Release of fluoride ions from fluoride-containing filler |
| Amorphous calcium phosphate (ACP) composites | Release of calcium and phosphate ions from ACP filler phase |
2. Experimental
2.1 Formulation of Methacrylate Resins
The matrix resins were formulated from the commercially available dental monomers, coupling agents and components of the photoinitiator systems (Figs. 1a–c, Tables 3 and 4). Acronyms indicated in Tables 3 and 4 will be used throughout this manuscript. Bis-GMA-, EBPADMA- and UDMA-based resins were generally photoactivated by the inclusion of CQ and 4EDMAB as the photo-oxidant and photo-reductant, respectively. In the ETHM series, 1850 IRGACURE was utilized as the photoinitiator and for the PT resin a photoinitiator system consisting of 369 IRGACURE, 4265 DAROCUR and CQ was selected to enhance photopolymerization and storage stabiliy.
Fig. 1.
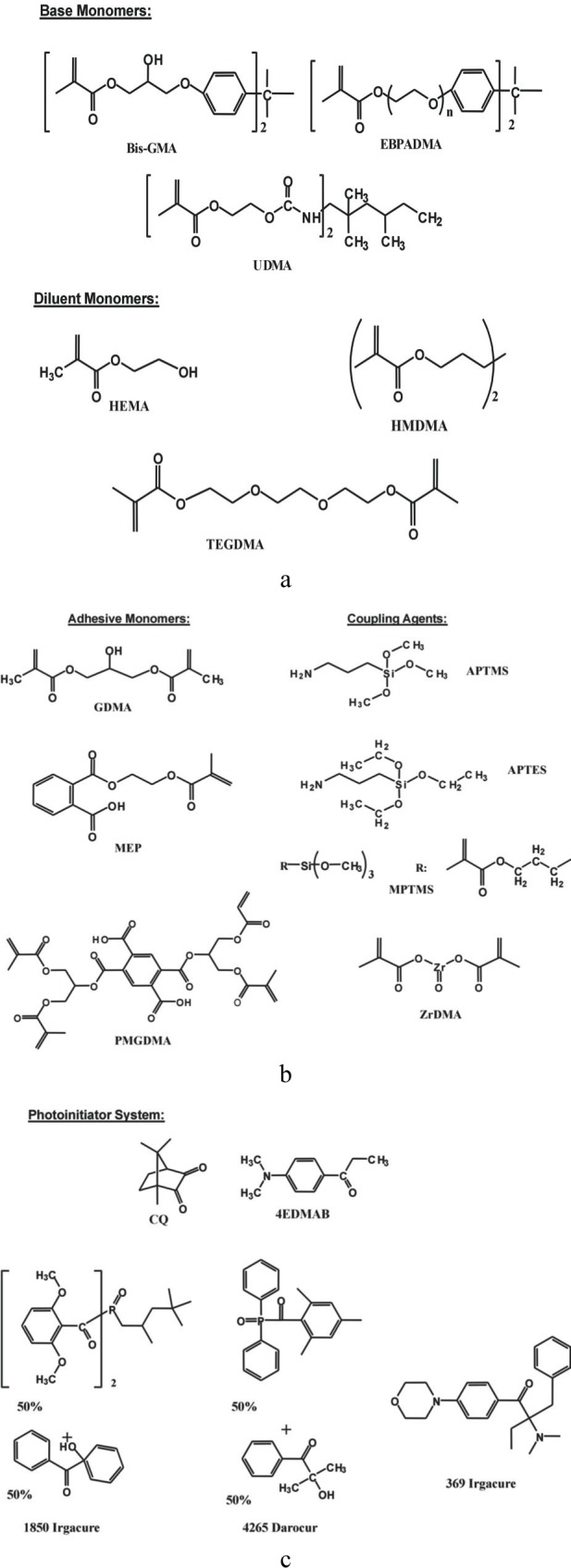
Chemical structure of the base and diluent monomers (a), adhesive monomers and coupling agents (b) and components of photoinitiator systems (c) used in the study.
Table 3.
Monomers, coupling agents and photonitiator system employed in resin formulations
| Component | Chemical nomenclature | Acronym |
|---|---|---|
| Base monomers | 2,2-bis[p-(2′-hydroxy-3′-methacryloxypropoxy)phenyl]propane | Bis-GMA |
| Ethoxylated bisphenol A dimethacrylate | EBPADMA | |
| Urethane dimethacrylate | UDMA | |
| Diluent monomers | 2-hydroxyethyl methacrylate | HEMA |
| Hexamethylene dimethacrylate | HMDMA | |
| Triethyleneglycol dimethacrylate | TEGDMA | |
| Adhesive monomers | Glycerol dimethacrylate | GDMA |
| Methacryloyloxyethyl phtalate | MEP | |
| Pyromellitic glycerol dimethacrylate | PMGDMA | |
| Coupling agents | 3-aminopropyltriethoxysilane | APTES |
| 3-aminopropyltrimethoxysilane | APTMS | |
| methacryloxypropyl trimethoxysilane | MPTMS | |
| zirconyl dimethacrylate | ZrDMA | |
| Photoinitiator system | Camphorquinone | CQ |
| Ethyl-4-N,N-dimethylaminobenzoate | 4EDMAB | |
| Bis(2,6-dimethoxybenzoyl)-2,4,4-trimethylpentylphosphine oxide & 1-hydroxycyclohexyl phenyl ketone | 1850 IRGACURE | |
| diphenyl (2,4,6-trimethylbenzoyl) phosphine oxide & 2-hydroxy-2-methyl-1-phenyl-1-propanone | 4265 DAROCUR | |
| 2-benzyl-2-(dimethylamino)-1-(4-(4-morpholinyl)phenyl)-1-butanone | 369 IRGACURE |
Table 4.
Composition (mass fraction %) of experimental resins evaluated in the study
| a. Bis-GMA-based resinsa | ||||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Resin/monomer | Bis-GMA | HEMA | HMDMA | GDMA | PDMA | pHEMA | TEGDMA | ZrDMA |
| BG | 55.00 | 44.00 | ||||||
| BH | 68.35 | 30.65 | ||||||
| BHm | 52.44 | 46.56 | ||||||
| BT | 49.50 | 49.50 | ||||||
| BHmH | 36.97 | 29.20 | 32.83 | |||||
| BTH | 35.50 | 28.00 | 35.50 | |||||
| BTHZr | 35.50 | 27.00 | 35.50 | 1.00 | ||||
| b. EBPADMA-based resinsb | |||||
|---|---|---|---|---|---|
|
| |||||
| Resin/monomer | EBPADMA | HEMA | HMDMA | MEP | TEGDMA |
| EHm | 49.57 | 49.43 | |||
| ET | 46.70 | 52.30 | |||
| EHmH | 34.33 | 30.44 | 34.23 | ||
| ETH | 32.90 | 29.17 | 36.93 | ||
| ETHM1.00c | 42.00 | 10.00 | 5.00 | 42.00 | |
| ETHM0.67 | 33.60 | 10.00 | 5.00 | 50.40 | |
| ETHM0.50 | 28.00 | 10.00 | 5.00 | 56.00 | |
| ETHM0.25 | 16.80 | 10.00 | 5.00 | 67.20 | |
| c. UDMA-based resinsd | ||||
|---|---|---|---|---|
|
| ||||
| Resin/monomer | UDMA | HEMA | HMDMA | TEGDMA |
| U | 99.00 | |||
| UH14.00e | 92.40 | 6.60 | ||
| UH6.47 | 85.50 | 13.20 | ||
| UHm | 51.75 | 47.25 | ||
| UT | 48.82 | 50.18 | ||
| UHmH | 36.24 | 29.46 | 33.30 | |
| UTH | 34.83 | 28.32 | 35.85 | |
| d. Otherf | ||
|---|---|---|
|
| ||
| Resin/monomer | PMGDMA | TEGDMA |
| PT | 48.65 | 48.65 |
Photoinitiator system consisted of a mass fraction of 0.20 % CQ and 0.80 % 4EDMAB.
Photoinitiator system consisted of a mass fraction of 0.20 % CQ and 0.80 % 4EDMAB except for the ETHM resins where a mass fraction of 1.00 % IRGACURE 1850 was utilized instead.
Numbers represent a mass ratio EBPADMA: TEGDMA of 1.00, 0.67, 0.50 and 0.25, respectively.
Photoinitiator system consisted of a mass fraction of 0.20 % CQ and 0.80 % 4EDMAB.
Numbers represent a mass ratio UDMA: HEMA of 14.00 and 6.47, respectively.
Photoinitiator system comprised a mass fraction of 0.40 % CQ, 0.80 % 4625 DAROCUR and 1.50 % 369 IRGACURE.
2.2 Synthesis and Characterization of ACP Fillers
The types of ACPs employed in the study are given in Table 5. Corresponding acronyms will be used in the manuscript hereafter. Syntheses of unmodified and hybrid ACPs were carried out according to a modified version of the preparation protocol proposed by Eanes et al. [10] (Fig. 2). Si-ACP was surface-treated as follows: APTMS, APTES or MPTMS was mixed into a slurry of ACP powder in cyclohexane containing a mass fraction of 2 % of n-propylamine (based on ACP). The suspension was then rotary evaporated (100 °C, moderate vacuum—2.7 kPa) to remove the solvents, cooled to room temperature (23 °C), washed with cyclohexane to remove residual silane and unattached products and re-dried under vacuum. ZrDMA was applied to Zr-ACP in a similar fashion from a methylene chloride solution.
Table 5.
ACP fillers employed in the study
| Type of ACP filler | Stabilizing ion | Hybridizing agent | Surface-modifying agent | Acronym |
|---|---|---|---|---|
| Untreated ACP | P2O74− | none | none | u-ACP |
| Hybridized ACP | P2O74− | Tetraethoxy silane | none | Si-ACP |
| (TEOS)a | ||||
| Zirconyl chloride | none | Zr-ACP | ||
| (ZrOCl2)a | ||||
| Surface- treated ACP | P2O74− | TEOS | APTMSb | Si/APTMS-ACP |
| TEOS | APTESb | Si/APTES-ACP | ||
| TEOS | MPTMSb | Si/MPTMS-ACP | ||
| ZrOCl2 | ZrDMAc | Zr/Zr-ACP |
TEOS or ZrOCl2 were introduced ab initio during the ACP synthesis as a mole fraction of 10 % relative to calcium reactant. A mixture with mass fractions of 10 % TEOS, 10 % ethanol, 10 % tartaric acid and 70 % water, proven to effectively prevent premature TEOS gelation, was used to introduce the TEOS during hybridization.
Si-ACP was surface-treated by introducing a mass fraction of 10 % of APTMS, APTES or MPTMS relative to ACP from a cyclohexane/n-propyl amine solution.
Zr-ACP was surface-treated by introducing a mass fraction of 2 % ZrDMA relative to ACP from a methylene chloride solution.
Fig. 2.
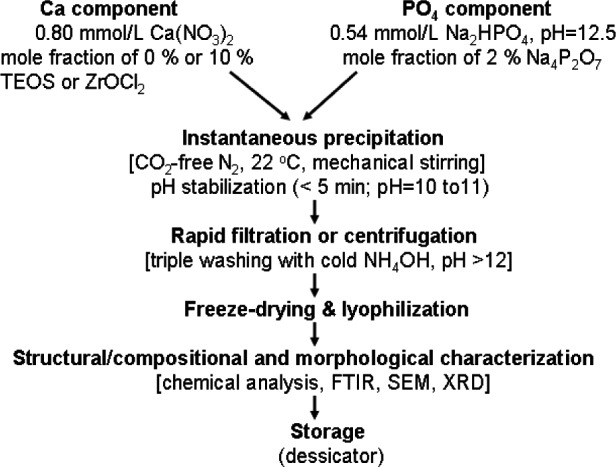
Schema of the experimental steps employed in the syntheses of ACP fillers.
The amorphous state of ACPs was verified by powder x-ray diffraction (XRD: Rigaku X-ray diffractometer1, Rigaku/USA Inc., Danvers, MA, USA) and Fourier-transform spectroscopy (FTIR: Nicolet Magna-IR FTIR System 550 spectrophotometer, Nicolet Instrument Corporation, Madison, WI, USA). The standard uncertainty of measuring the d-spacing values was 0.0013, and the measured d-values were within 0.05 % of the reported values of NIST SRM 640 (silicon powder, 2θ = 28.442, d = 3.1355). The wavelength accuracy of FTIR measurements was ≤ 0.01 cm−1 at 2000 cm−1. The particle size distribution (PSD) of the solids dispersed in isopropanol was determined by gravitational and centrifugal sedimentation analysis (SA-CP3 particle size analyzer, Shimadzu Scientific Instruments, Inc., Columbia, MD, USA) following 10 min ultrasonication. The Ca/PO4 ratio of the solids after dissolution in HCl was calculated from solution Ca2+ (atomic adsorption spectroscopy (AAS), Perkin Elmer Mo. 603 spectrophotometer (Perkin Elmer, Norwalk, CT, USA) and PO4 (UV/VIS Carey Model 219 spectrophotometer (Varian Analytical Instruments, Palo Alto, CA, USA, [11]) values. Additionally, AAS was employed to determine the amount of Si and/or Zr incorporated into hybrid fillers. Surface morphology of the fillers, after specimens were sputter-coated with gold, was determined by scanning electron microscopy (SEM: JSM-5400 instrument JEOL Inc., Peabody, MA, USA).
2.3 Physicochemical Evaluation of Resins, Pastes, and ACP Composites
The methodologies and techniques utilized to characterize and evaluate the methacrylate resins, the ACP fillers, and their composites are summarized in Table 6. The sequence of experimental steps employed in the physicochemical and mechanical evaluation of these bioactive ACP dental composites is schematically presented in Fig. 3. Composite pastes made up of various resins (Table 4; mass fraction 60 %) and ACP fillers (Table 5; mass fraction 40 %) were formulated by hand spatulation. The homogenized pastes were kept under a moderate vacuum (2.7 kPa) overnight to eliminate the air entrained during mixing. The pastes were molded into disks (15. 8 mm to 19.8 mm in diameter and 1.55 mm to 1.81 mm in thickness) by filling the circular openings of flat Teflon molds, covering each side of the mold with a Mylar film plus a glass slide, and then clamping the assembly together with a spring clip. The disks were photo-polymerized by irradiating sequentially each face of the mold assembly for 120 s with visible light (Triad 2000, Dentsply International, York, PA, US). After post-curing at 37 °C in air overnight, the disks were examined intact by XRD.
Table 6.
Methods and techniques employed in physicochemical characterization of the resins, fillers and ACP composites
| Method | Property/parameter | Application/information |
|---|---|---|
| Atomic absorption spectroscopy (AAS) | Calcium, silica or zirconia concentration | Chemical analysis of ACP fillers Levels of hybridizing ions incorporated in ACP fillers Calcium release from composite disks exposed to aqueous environment |
| Computer-controlled mercury dilatometry | Volumetric contraction profiles | Volumetric polymerization shrinkage (PS) of composite resins upon light-polymerization |
| Fourier-transform infrared (FTIR) spectroscopy and microspectroscopy (m-FTIR) | Short-range structural arrangement | Structural/compositional properties of the monomers, ACP fillers, uncured and cured composites Degree of methacrylate conversion (DC) upon polymerization as indirect measure of the leachability of unreacted monomeric species Intra-composite ACP to HAP conversion |
| Gravimetry | Water sorption | Kinetics of the water uptake (water sorption, WS) by unfilled resins and ACP-filled composites |
| Mechanical testing | Physical strength | Biaxial flexure strength (BFS) |
| Particle size analysis | Cumulative and differential particle size distribution | Size (range, median diameter) and specific surface area of ACP fillers |
| Scanning electron microscopy (SEM) | Morphology, topology | Characterization of ACP fillers and composites before and after exposure to saliva-like solutions (soaking) |
| Ultraviolet/visible (UV/VIS) spectrophotometry | Phosphate concentration | Chemical analysis of ACP fillers Phosphate release from composite disks exposed to aqueous environment |
| X-ray diffraction (XRD) | Long-range crystalline order | Characterization of the fillers and uncured pastes Stability of ACP fillers upon immersion |
Fig. 3.

Schematic presentation of the experimental protocols utilized in physicochemical and mechanical assessment of ACP composites. Acronyms defined in text and Table 6.
Volumetric contraction upon polymerization or polymerization shrinkage (PS) of the composites was measured by a computer-controlled mercury dilatometry (Fig. 4, [12]) that records the volume changes of the composite specimen, corrected for temperature fluctuations during the measurement, as a function of time and calculates the overall PS (volume fraction, %) based on the known mass of the sample and its density. Sample density was determined by means of the Archimedean principle using a water bath attachment to a microbalance (Sartorius YDK01 Density Determination Kit; Sartorius AG, Goettingen, Germany).
Fig. 4.

Schematic diagram of a computer-controlled mercury dilatometer used to determine volumetric contraction of the composites.
To determine the degree of methacrylate conversion (DC) attained after polymerization of the composites, a recently developed, non-destructive near infrared (NIR) spectroscopic technique for measuring the methacrylate conversion in dental resins was employed [13]. The absorption band at 6165 cm−1 in the overtone region was used to assess the DC in paired unpolymerized and polymer samples of known thickness. DC was calculated from the decrease in integrated peak area/sample thickness values in going from the unpolymerized to polymerized composites using the following expression:
| (1) |
The biaxial flexure strength (BFS) of each composite disk specimen was determined by using a computer-controlled Universal Testing Machine (Instron 5500R, Instron Corp., Canton, MA, US) operated by Instron Merlin Software Series 9. Detailed description of experimental protocols and calculations used in BFS screening are given in Ref. [14].
Mineral ion release from each individual composite disk specimen in a continuously stirred, HEPES-buffered (pH = 7.40) 240 mOsm/kg saline solution, was examined at 37 °C. Ca2+ and PO4 levels were determined by AAS and UV/VIS spectroscopy, respectively. Ion-release data were corrected for variations in the total area of disk surface exposed to the immersion solution using the simple relation for a given surface area, A: normalized value = (measured value) × (500/A).
To determine the water sorption (WS) profiles, a minimum of five replicate disks in each experimental group were initially dried over CaSO4 until a constant mass was achieved (± 0.1 mg). Specimens were then exposed to 75 % relative humidity (RH) at room temperature (23 °C) by keeping them over aqueous NaCl slurry in a closed system. Gravimetric mass changes were recorded at predetermined time intervals. Degree of water sorption (WS) of any individual specimen at any given time interval (t), expressed as a % mass fraction, was calculated using a simple equation:
| (2) |
where Wt represents sample mass at the time t, and W0 is the initial mass of a dry sample.
One standard deviation was given in this paper for comparative purposes as the estimated standard uncertainty of the measurements. These values should not be compared with data obtained in other laboratories under different conditions.
3. Results and Discussion
XRD patterns, FTIR spectra (representative scans are given in Fig. 5 a, b, respectively) and SEM images (Fig. 6) revealed no significant difference in structural and morphological features of unmodified, hybridized and surface-treated ACPs. All ACPs had heterogeneous PSDs with particle diameters (expressed as the equivalent spherical diameter) spanning from 0.1 µm to 80 µm. Apparent differences in the mean values of median diameters (dm; Fig. 7a) and the specific surface area (SSA; Fig. 7b) calculated from the corresponding PSDs of the powders were found to be greater than expected by chance (one-way ANOVA; P = 0.024). However, all pair-wise multiple comparisons (Tukey test) revealed that only the differences in dm and SSA between the control HAP powder and Si-hybridized ACP are of statistical significance (P = 0.014 and P = 0.011, respectively). The observed increase in the dm, and consequently the lower SSA of Si-ACP compared to HAP, may be explained by different degree (extent) of agglomeration of the Si-hybridized filler. While the Ca/PO4 molar ratio of u-ACP and Si-treated ACPs was practically unchanged, it was significantly higher (Tukey test) for all Zr-treated fillers (Table 7). A speculative explanation for the observed compositional difference is that the loss of PO4 was caused by the formation of soluble Zr-PO4 complexes that paralleled the ACP precipitation, and that these soluble complexes were removed during the later stages of Zr-ACP and/or Zr/Zr-ACP synthesis. A lower level of incorporated Si compared to Zr in hybrid ACPs suggests that TEOS was most likely bound on the surface of the particles and, therefore, was more easily removed during filtering and washing of the freshly precipitated solids.
Fig. 5.
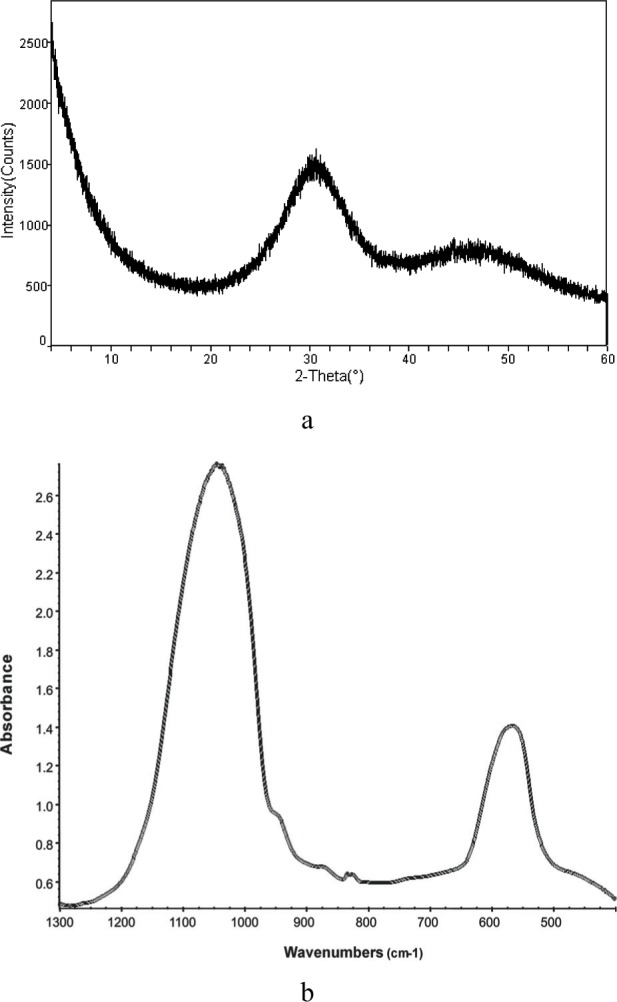
XRD pattern (a) and FTIR spectrum (b) of a representative ACP filler.
Fig. 6.

SEM microphotographs of unmodified, hybrid and surface-treated ACP powders.
Fig. 7.
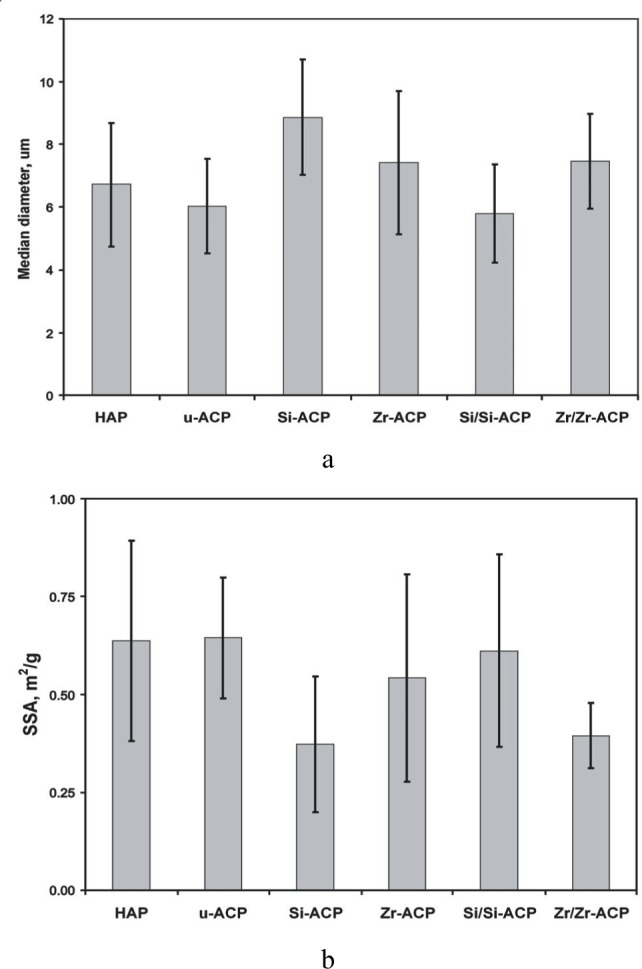
Mean values and standard deviations (indicated by bars) of the median particle diameter dm, (part a) and the corresponding SSA (part b) of various ACP fillers in comparison with HAP control. The standard deviation is taken as a measure of the standard uncertainty.
Table 7.
Elemental analysis of ACP fillers
| Filler | Ca/PO4 molar ratioa | Incorporated hybridizing ionb (mass fraction, %) |
|---|---|---|
| u-ACP | 1.50 ± 0.09 | |
| Si-ACP | 1.59 ± 0.06 | 3.1 ± 0.5 |
| Si/APTMS-ACP | 1.56 ± 0.11 | |
| Si/APTES-ACP | 1.63 ± 0.15 | |
| Si/MPTMS-ACP | 1.49 ± 0.12 | |
| Zr-ACP | 1.91 ± 0.09 | 8.6 ± 1.4 |
| Zr/Zr-ACP | 2.19 ± 0.19 |
All results are given as the mean values ± standard deviation.
Number of repetitive experiments in each group n ≥ 5.
Number of repetitive experiments in each group n = 5.
As evidenced by solution analysis, and by XRD and FTIR, the stability of ACPs exposed to different test solutions [15] decreased in the following order: Zr-ACP > Si-ACP > u-ACP (Fig. 8). Zr and Si retard the conversion of hybrid ACPs to HAP by their adsorption at HAP nucleation/growth sites. Since slower internal conversion into HAP is desirable in composites with remineralizing applications, these hybrid ACPs, especially Zr-ACPs, would appear to be the more suitable choice for use in bioactive dental materials.
Fig. 8.
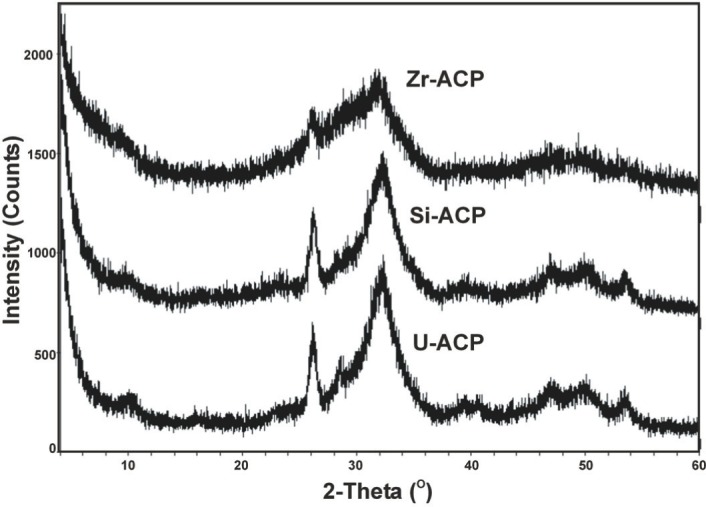
Stability of unhybridized (u-ACP) and hybrid ACPs (Si-ACP and Zr-ACP) in a test solutions containing 0.1 mol/L lactic acid and a mass fraction of 1 % hydroxymethyl cellulose (pH adjusted to 5.10 by NaOH solution). Time interval: 150 min. XRD patterns of Zr-ACP and Si-ACP show lesser conversion to hydroxyapatite than u-ACP (crystalline peaks marked by asterisk).
The results of the DC screening of experimental resins and ACP-composites are summarized in Fig. 9. Both unfilled Bis-GMA- and EBPADMA-based resins and to a lesser extent UDMA-based resins, as well as their ACP composites, achieved a higher methacrylate conversion when the hydrophilic, monofunctional HEMA was included as a co-monomer in the resin. Higher DCs for the resins with relatively high content of HEMA could be attributed to its high diffusivity and monofunctionality. High vinyl conversion, coupled with moderate contraction, was found in light-cured Bis-GMA resins containing hydroxypropyl methacrylate as a comonomer [16], a monomer homologous to HEMA. Regardless of the resin matrix composition the DCs of ACP-containing Bis-GMA- and EBPADMA-based composites were lower than UDMA-based composites. The UDMA monomer has already been been shown to be more reactive than Bis-GMA or EBPADMA [17]. No clear-cut trend could be established on the effect of filler type (u-, Si- or Zr-ACP) on the DC of the resin, except perhaps with regard to some of the composites formulated with HMDMA, e.g., BHm and EHm resins, which showed rather low DC with all ACP fillers. However, the following order of decreasing DC, independent of the filler type, can clearly be seen when the DC data are compared as shown in Figs. 10 a-d: XTH ≥ XHmH > XT ≥ XHm, with X being Bis-GMA, EBPADMA or UDMA. Furthermore, TEGDMA-containing matrices showed higher conversion that the corresponding HmDMA-containing ones (Student two-tail t-test; 95 % confidence interval). In conclusion, (Bis-GMA, EBPADMA or UDMA)/TEGDMA/HEMA formulations have the lowest potential for leaching out unreacted monomeric species. The significantly lower DC obtained with PT composites (most probably caused by the rigid aromatic core structure with practically no side-chain flexibility of PMGDMA) indicates a greater probability that the ACP-PT composites will release un-reacted monomers into the oral environment and consequently more likely will have a lower biocompatibility than Bis-GMA-, EBPADMA- and/or UDMA-based ACP composites.
Fig. 9.
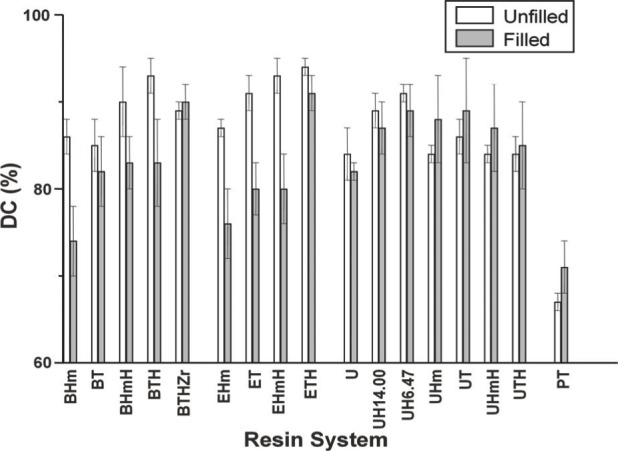
Mean values and standard deviations (indicated by bars) of the degree of methacrylate conversion, DC, of unfilled resins and corresponding ACP composites (average for u-ACP, Si-ACP and Zr-ACP fillers). Number of runs in each experimental group n ≥ 8. The standard deviation is taken as a measure of the standard uncertainty.
Fig. 10.
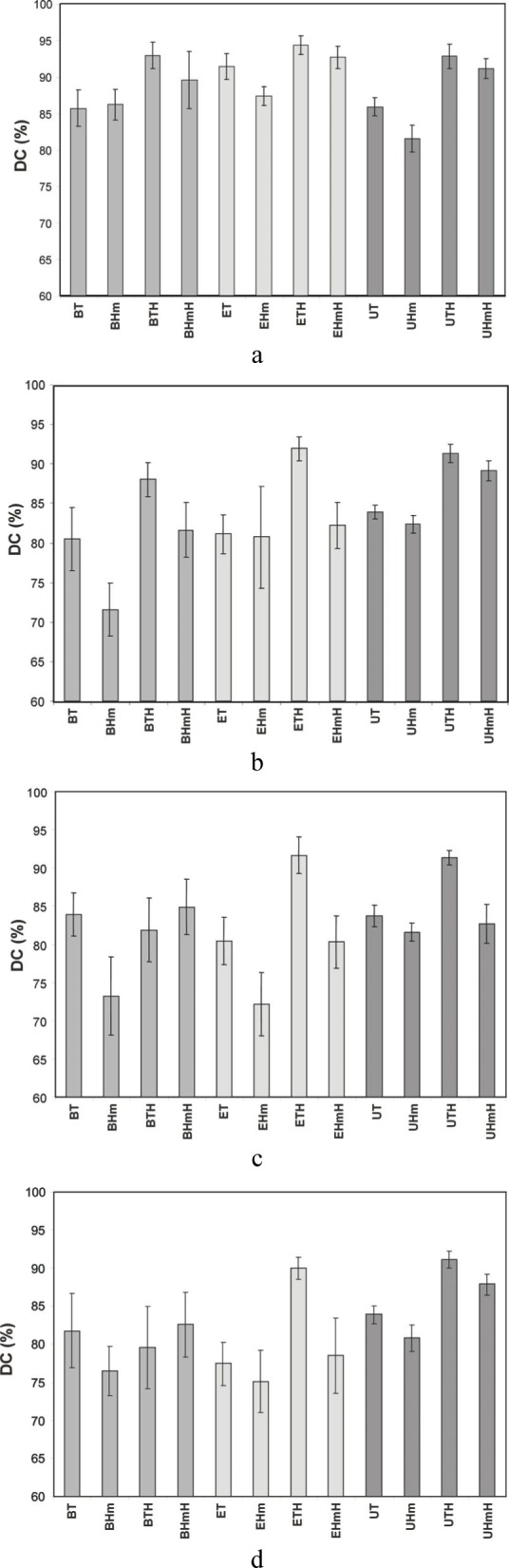
DC (mean value ± standard deviation (indicated by bars) of unfilled resins (a), u-ACP (b), Si-ACP (c) and Zr-ACP (d) composites as a function of resin composition. The standard deviation is taken as a measure of the standard uncertainty.
The PS results showed a very complex dependence on both the resin composition and the filler type (Fig. 11). The majority of the experimental composites shrank more than did the commercial composite materials (PS 1.9 % to 4.1 % [18,19]), most probably due to the lower filler load (mass fraction of only 40 % ACP compared to that of (77 to 85) % of silica-based fillers in highly-filled conventional composites) and to ACP's heterogeneous size distribution. The experimental PS fell into the category of either flowable composites or adhesive resins ((3.6 to 6.0) % and (6.7 to 13.5) %, respectively [18]). Reformulated ACP-filled experimental composites should be further studied to determine if additional adjustments in resin formulations (bulkier but relatively low viscosity resins or ringopening monomers as the resin matrix component [20,21] might lead to composites with lower PS and optimal DMC.
Fig. 11.
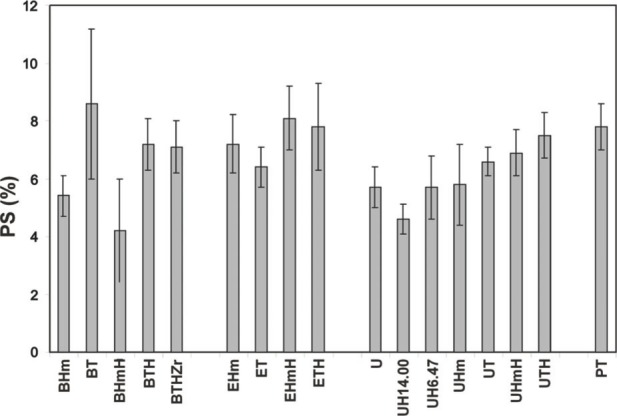
Mean values and standard deviations (indicated by bars) of the polymerization shrinkage of Bis-GMA-, EBPADMA- or UDMA-based ACP-filled composites (filler loading: 40 % mass fraction; average for u-ACP, Si-ACP and Zr-ACP) compared to TP composites. Number of runs in each experimental group n ≥ 9. The standard deviation is taken as a measure of the standard uncertainty.
Ion release from composites was affected by both the chemical structure and the composition of the monomer system as well as by the type of ACP filler (Table 8). Elevated Ca and PO4 concentrations were sustained in all but PT composites, which with increased time failed to maintain a favorable remineralizing potential due to the matrix uptake of released Ca via ion binding by the high concentration of carboxylic acid groups of PMGDMA [22]. Generally, the remineralizing capacity of ACP composites may be enhanced by a) introducing EBPADMA as a base monomer, b) elevating the level of HEMA in the resin formulation and c) by utilizing hybrid ACPs instead of u-ACP. The most probable mechanism by which the hydrophilic HEMA-enriched resins increased internal mineral saturation was by allowing the uptake of more water and/or better accessibility of hybrid ACP to the water already entrained. On the other hand, higher releases obtained with EBPADMA-based composites may partly be due to a more open cross-linked network structure of their resin matrix.
Table 8.
Maximum concentration of the mineral ions (mean value for all types of ACP fillers) released from composites after 336 h of immersion in buffered saline. Number of runs in each experimental group n ≥ 9. SDs of the reported values ranged from (0.02 to 0.20) mmol/L and from (0.02 to 0.14) mmol/L for Ca and PO4 values, respectively
| Resin | Ca (mmol/L) | PO4 mmol/L |
|---|---|---|
| BHm | 1.33 | 0.60 |
| BT | 0.98 | 0.49 |
| BHmH | 1.52 | 0.68 |
| BTH | 1.29 | 0.58 |
| BTHZr | 0.97 | 0.62 |
| EHm | 2.52 | 0.62 |
| ET | 3.34 | 0.81 |
| EHmH | 4.73 | 1.17 |
| ETH | 3.13 | 0.80 |
| PT | 0.72 | 0.47 |
| U | 1.08 | 0.89 |
| UH14.00 | 1.19 | 1.02 |
| UH6.47 | 1.55 | 1.01 |
| UHm | 0.33 | 0.25 |
| UT | 1.06 | 0.64 |
| UHmH | 1.14 | 0.82 |
| UTH | 1.09 | 0.78 |
Results of the BFS testing of dry (before immersion) and wet (after immersion in saline solutions) composites are presented in Table 9 a-d. The mechanical strength of unfilled Bis-GMA- and EBPADMA-based resins did not deteriorate upon soaking. Unfilled UDMA-based and PT resins, however, failed to maintain their strength upon exposure to an aqueous environment. Generally, dry ACP-filled composites had substantially lower BFS than unfilled specimens regardless of the type of filler or resin matrix. The strength of all BisGMA-based (except the BTHZr resin), UDMA- based and PT composites deteriorated further upon soaking. Comparison of the BFS values of hybrid vs u- ACP/BTHZr composites showed modest but significant increase in the mechanical strength of Si- and Zr-ACP specimens. However, surface-treated Zr/Zr ACP composites failed to maintain their strength upon immersion. Recent m-FTIR mapping of ACP composites [23] indicated the existence of the numerous defects/voids (resin-rich, phosphate-depleted regions) in polymerized Si/MPTMS-ACP and Zr/Zr-ACP specimens compared to u- and hybrid ACPs. Uneven distribution of highly agglomerated, surface-treated ACP particulates throughout the matrix is, most probably, responsible for inadequate filler/resin interlocking and the resulting adverse effect on the overall mechanical strength. No clear-cut trend could be established for dry vs. wet EHm, ET, EHmH and ETH composites. However, the BFS of ETHM composites (formulated with MEP, proven to effectively promote bonding to dentin due to its surface activity [24]) decreased significantly upon soaking. The effect was independent of the resins' EBPADMA:TEGDMA ratio or the type of ACP filler utilized, except that again the weakening was more pronounced with surface-treated ACPs.
Table 9.
Biaxial flexure strength (BFS) of dry (before immersion) and wet (after immersion in buffered saline for > 336 h) unfilled resins and ACP-filled composites. Results are indicated as mean value ± standard deviation with the number of specimens tested in each group given in parentheses
| a. Bis-GMA-based resins | |||
|---|---|---|---|
|
| |||
| Resin matrix | BFS (MPa) | ||
| Dry | Wet | ||
| BG | Unfilled copolymer | 155 ± 32 (6) | 131 ± 26 (6) |
| composite | 37 ± 4 (3) | 17 ± 2 (4) | |
| BH | Unfilled copolymer | 167 ± 41 (6) | 130 ± 27 (5) |
| composite | 42 ± 7 (4) | 20 ± 6 (5) | |
| BHm | Unfilled copolymer | 101 ± 26 (4) | 123 ± 26 (4) |
| composite | 53 ± 13 (12) | 55 ± 11 (13) | |
| BT | Unfilled copolymer | 132 ± 27 (27) | 123 ± 22 (17) |
| composite | 62 ± 15 (26) | 62 ± 13 (26) | |
| BHmH | Unfilled copolymer | 155 ± 45 (4) | 133 ± 36 (4) |
| composite | 71 ± 10 (12) | 48 ± 8 (13) | |
| BTH | Unfilled copolymer | 156 ± 40 (8) | 144 ± 52 (9) |
| composite | 56 ± 10 (28) | 40 ± 9 (28) | |
| BTHZr | Unfilled copolymer | 116 ± 23 (25) | 118 ± 30 (11) |
| composite | 69 ± 10 (118) | 60 ± 14 (120) | |
| b. EBPADMA-based resins | |||
|---|---|---|---|
|
| |||
| BFS (MPa) | |||
| Resin | Filler | Dry | Wet |
| EHm | Unfilled copolymer | 95 ± 18 (4) | 93 ± 24 (4) |
| composite | 59 ± 8 (12) | 53 ± 11(14) | |
| ET | Unfilled copolymer | 114 ± 19 (4) | 125 ± 35 (4) |
| composite | 61 ± 6 (12) | 59 ± 7 (15) | |
| EHmH | Unfilled copolymer | 122 ± 13 (3) | 120 ± 27 (4) |
| composite | 58 ± 9 (12) | 51 ± 9 (15) | |
| ETH | Unfilled copolymer | 133 ± 38 (3) | 128 ± 49 (4) |
| composite | 57 ± 11 (12) | 49 ± 8 (17) | |
| ETHM1.00 | Unfilled copolymer | 110 ± 21 (4) | 125 ± 8 (3) |
| composite | 69 ± 10 (29) | 49 ± 8 (22) | |
| ETHM0.67 | Unfilled copolymer | 128 ± 24 (4) | 114 ± 27 (3) |
| composite | 72 ± 14 (14) | 48 ± 9 (13) | |
| ETHM0.50 | Unfilled copolymer | 130 ± 16 (4) | 151 ± 38 (3) |
| composite | 76 ± 10 (17) | 4 ± 6 (19) | |
| ETHM0.25 | Unfilled copolymer | 135 ± 19 (4) | 160 ± 37 (3) |
| composite | 65 ± 9 (19) | 46 ± 10 (26) | |
| c. UDMA-based resins | |||
|---|---|---|---|
|
| |||
| BFS (MPa) | |||
| Resin | Filler | Dry | Wet |
| U | Unfilled copolymer | 206 ± 15 (4) | 159 ± 18 (4) |
| composite | 76 ± 6 (12) | 53 ± 8 (12) | |
| UH14.00 | Unfilled copolymer | 196 ± 6 (4) | 155 ± 24 (4) |
| composite | 70 ± 8 (9) | 61 ± 4 (12) | |
| UH6.47 | Unfilled copolymer | 191 ± 29 (4) | 151 ± 22 (4) |
| composite | 74 ± 3 (10) | 53 ± 4 (12) | |
| UHm | Unfilled copolymer | 183 ± 22 (4) | 117 ± 38 (3) |
| composite | 65 ± 8 (11) | 60 ± 13 (14) | |
| UT | Unfilled copolymer | 192 ± 46 (4) | 93 ± 30 (4) |
| composite | 61 ± 11 (12) | 57 ± 10 (11) | |
| UHmH | Unfilled copolymer | 170 ± 32 (4) | 123 ± 14 (4) |
| composite | 63 ± 7 (12) | 37 ± 12(13) | |
| UTH | Unfilled copolymer | 124 ± 30 (4) | 74 ± 27 (4) |
| composite | 54 ± 11 (12) | 40 ± 11 (12) | |
| d. Other | |||
|---|---|---|---|
|
| |||
| BFS (MPa) | |||
| Resin | Filler | Dry | Wet |
| PT | Unfilled copolymer | 164 ± 31 (5) | 101 ± 29 (10) |
| composite | 72 ± 9 (40) | 30 ± 6 (36) | |
Studies conducted on the water sorption of dental materials indicate that excessive water uptake may cause a decrease in mechanical strength, distortion, and depression of the glass transition temperature [25] due to plasticization, solvation, reversible rupture of weak inter-chain bonds and irreversible disruption of the polymer matrix [26]. In the case of ACP/methacrylate composites, not only water-polymer but also water-ACP interactions occur and both contribute significantly to the overall water sorption profiles. Besides affecting the strength (BFS usually decreases upon soaking), water sorption/diffusion influences the mineral ion release kinetics and consequently the remineralizing ability of these bioactive materials. Kinetic WS data for unfilled and ACP-filled (u-, Si- and Zr-ACP) XT, XHm, XTH and XHmH (X = Bis-GMA, EBPADMA or UDMA, T = TEGDMA, H = HEMA) resins (data not shown) indicate that plateau values are reached within a week for XTH and within 2 weeks for XT, XHm and XHmH systems. Generally, XTH composites exhibited the highest and XHm composites exhibited the lowest WS (plateau values are compared in Fig. 12 a-c). Observed differences in WS are primarily due to whether hydrophilic (TEGDMA and HEMA) or hydrophobic (HmDMA) monomers are the dominant components of the resin matrices. No clear-cut conclusion could be made on the effect of the filler type on WS, although certain trends were evident: more water was absorbed by u-ACP composites compared to hybrid ACP composites in Bis-GMA and EBPADMA systems, Zr-ACP composites seemingly have the lowest WS regardless of the resin composition.
Fig. 12.
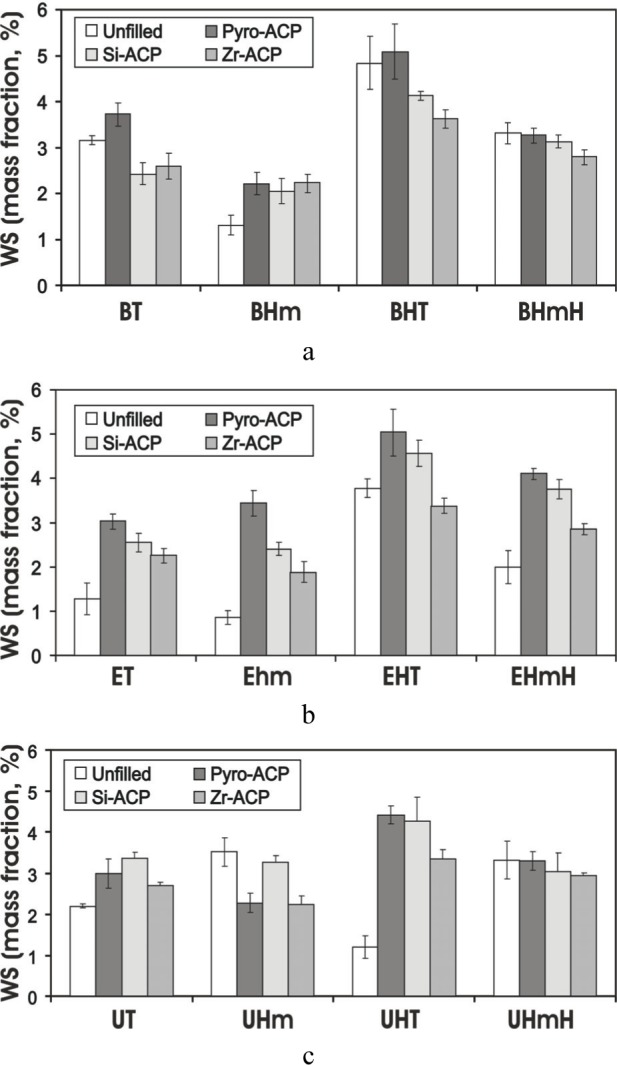
Maximum water sorption (WS) values [mean ± standard deviation (indicated by bars)] for unfilled (a) and ACP-filled XT, XHm, XTH and XHmH resins (X = Bis-GMA (part a) EBPADMA (part b) or UDMA (part c) T = TEGDMA, H = HEMA) after 5 weeks of exposure to 75 % relative humidity at 23 °C. Number of specimens in each group n ≥ 5. The standard deviation is taken as a measure of the standard uncertainty.
It has also been demonstrated that the filler's loading level has a significant effect on the WS of ACP composites [27]. As seen in Fig. 13, Si-ACP filled BT, BH and BG composites adsorbed more water than unfilled resin samples and the sorption was proportional to the mass fraction of Si-ACP in the composite. The differences may have resulted from increased stresses at the ACP/resin boundaries that, in turn, promoted water diffusion and enhanced the hydration of ACP surfaces. Also, the presence of silanol (≡Si-OH) groups in the Si-ACP could enhance WS. The possible relevance of this finding is that increased ACP levels increased WS which, in turn, increased ion release and faster ACP to HAP conversion. The resulting disruption in the integrity of the filler/resin interface decreased BFS.
Fig. 13.
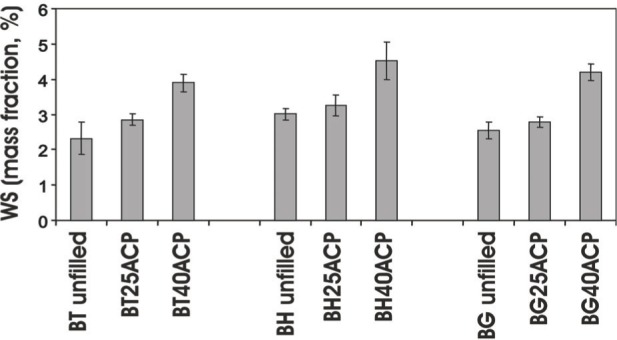
Maximum water sorption (WS) values [mean ± SD (indicated by bars)] of unfilled BT, BH and BG resins and corresponding composites with a mass fraction of 25 % or 40 % of Si-ACP after 5 weeks of exposure to 75 % relative humidity at 23 °C. Number of specimens in each group n ≥ 5. The standard deviation is taken as a measure of the standard uncertainty.
These results exemplify the fact that water plays a very significant if not a major role in filler-matrix interactions. It may leach out filler elements, induce filler failures, cause filler-matrix de-bonding and reduce the strength of matrix material. Since the effect of water on most dental composites is irreversible [28], a true degeneration must have occurred either within the BT, BH or BG matrices or within TEOS/ACP “interphase” region of our experimental composites. Which region, the outer filler layer, the intra-silane coating or the silane-ACP bonds should be regarded as “critical” remains an open question. Through the future evaluation of hybrid ACP composites with reformulated resins, spectrum of their potential applications should extend to prevention of demineralization in orthodontically treated teeth and/or promotion remineralization of white spots in addition to initially envisioned applications as sealants and/or base/liner (Table 10).
Table 10.
Potential benefits of bioactive ACP-based composites
| Field | Foreseen benefit |
|---|---|
| Preventive dentistry | Remineralizing material that can counteract recurrent decay, known to develop near the surfaces of teeth in contact with conventional fillings (50 % of all dental fillings require replacement because of recurrent caries) • Particularly useful for patients that are especially susceptible to cavities as a result of radiation therapy and diseases or medications that cause dry mouth • Ameliorate the development and promote healing of root caries • Desensitizing agents for patients with tooth sensitivity |
| Orthodontics | Remineralizing adhesive cement that can minimize demineralization that frequently occurs under orthodontic brackets |
| Endodontics | Remineralizing root canal sealers or filling materials |
| Orthopedics | Biodegradable, potentially osteoconductive composites for healing defective bony tissues |
4. Conclusions
In conclusion, results of this study demonstrate that it is possible to improve the remineralizing potential of ACP composites by introducing Si or Zr elements during the low-temperature synthesis of the filler. Si- and Zr- ACPs enhanced the duration of mineral ion release through their ability to slow down the intra-composite ACP to HAP conversion. Additionally, when compounded with BTHZr resins, hybrid ACPs showed improved mechanical properties compared to composites that utilized unmodified ACP. Also, the chemical structure and composition of the monomer system used to form the matrix phase significantly affected ion release, water sorption and the DC of the composites. Utilizing EBPADMA in addition to Bis-GMA as a base monomer and adding moderate amounts of hydrophilic HEMA may be the best route to maximize remineralizing ability of the filler while maintaining low leachability of unreacted monomeric species, i.e., high DC. However, additional adjustments in resin formulations will be necessary to improve the PS of current experimental ACP composites. Finally, Bis-GMA and EBPADMA have proven to be essential in maintaining the mechanical integrity of the composites. As currently formulated, resins that included surface-active adhesive monomers PMGDMA or GDMA did not meet basic physicochemical requirements for ACP composites.
Acknowledgments
This investigation was supported, in part, by USPHS Research Grant 13169 to the American Dental Association Health Foundation from the National Institutes of Health—National Institute of Dental and Craniofacial Research and is part of the dental research program conducted by the National Institute of Standards and Technology in cooperation with the American Dental Association Health Foundation.
Biography
About the authors: Drago Skrtic is a physical chemist in the Cariology Division of the Paffenbarger Research Center, American Dental Association Health Foundation at NIST. Joseph M. Antonucci is a research chemist and David E. Eanes is a guest scientist in the Biomaterials Group of the Polymers Division, NIST Materials Science and Engineering Laboratory. The National Institute of Standards and Technology is an agency of the Technology Administration, U.S. Department of Commerce.
Footnotes
Certain commercial equipment, instruments, or materials are identified in this paper to foster understaning. Such identification does not imply recommendation or endorsement by the American Dental Association Health Foundation or the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
Contributor Information
D. Skrtic, Email: drago.skrtic@nist.gov.
J. M. Antonucci, Email: joseph.antonucci@nist.gov.
E. D. Eanes, Email: edward.eanes@nist.gov.
5. References
- 1.LeGeros RZ. Calcium Phosphates in Oral Biology and Medicine. Karger; Basel: 1991. pp. 1–201. [PubMed] [Google Scholar]
- 2.LeGeros RZ. Calcium Phosphate Materials in Restorative Dentistry: A Review. Adv Dent Res. 1988;2(1):164–180. doi: 10.1177/08959374880020011101. [DOI] [PubMed] [Google Scholar]
- 3.Eanes ED. Amorphous Calcium Phosphate: Thermodynamic and Kinetic Considerations. In: Amjad Z, editor. Calcium Phosphates in Biological and Industrial Systems. Kluwer Academic Publ; Boston: 1998. pp. 21–40. [Google Scholar]
- 4.Meyer JL, Eanes ED, Thermodynamic A. Analysis of the Amorphous to Crystalline Calcium Phosphate Transformation. Calcif Tissue Res. 1978;25:59–68. doi: 10.1007/BF02010752. [DOI] [PubMed] [Google Scholar]
- 5.Meyer JL, Eanes ED, Thermodynamic A. Analysis of the Secondary Transition in the Spontaneous Precipitation of Calcium Phosphate. Calcif Tissue Res. 1978;25:209–216. doi: 10.1007/BF02010771. [DOI] [PubMed] [Google Scholar]
- 6.Tung MS. Calcium Phosphates: Structures, Composition, Solubility and Stability. In: Amjad Z, editor. Calcium Phosphates in Biological and Industrial Systems. Kluwer Academic Publ; Boston: 1998. pp. 1–20. [Google Scholar]
- 7.Antonucci JM, Skrtic D, Eanes ED. Remineralizing Dental Composites Based on Amorphous Calcium Phosphate. Polymer Preprints. 1995;36(1):779–780. [Google Scholar]
- 8.Antonucci JM, Skrtic D, Eanes ED. Bioactive Dental Materials Based on Amorphous Calcium Phosphate—Effect of Coupling Agents. In: Ottenbrite R, Huang S, Park K, editors. Hydrogels and Biodegradable Polymers for Bioapplications. ACS; Washington DC: 1996. 1996. pp. 243–254. [Google Scholar]
- 9.Skrtic D, Hailer AW, Takagi S, Antonucci JM, Eanes ED. Quantitative Assessment of the Efficacy of Amorphous Calcium Phosphate/methacrylate Composites in Remineralizing Caries-like Lesions Artificially Produced in Bovine Enamel. J Dent Res. 1996;75(9):1679–1686. doi: 10.1177/00220345960750091001. [DOI] [PubMed] [Google Scholar]
- 10.Eanes ED, Gillessen IH, Posner AS. Intermediate States in the Precipitation of Hydroxyapatite. Nature. 1965;208:365–367. doi: 10.1038/208365a0. [DOI] [PubMed] [Google Scholar]
- 11.Murphy J, Riley JP. Single Solution Method for the Determination of Phosphate in Natural Waters. Anal Chim Acta. 1962;27:31–36. [Google Scholar]
- 12.Reed B, Dickens B, Dickens S, Parry E. Volumetric Contraction Measured by a Computer-controlled Mercury Dilatometer. J Dent Res. 1996;75:290. [Google Scholar]
- 13.Stansbury JW, Dickens SH. Determination of Double Bond Conversion in Dental Resins by Near Infrared Spectroscopy. Dent Mater. 2001;17:71–79. doi: 10.1016/s0109-5641(00)00062-2. [DOI] [PubMed] [Google Scholar]
- 14.ASTM F394-78, Standard Test Method for Biaxial Strength (Modulus of Rapture) of Ceramic Substrates, re-approved 1991
- 15.Skrtic D, Antonucci JM, Eanes ED, Brunworth RT. Silica- and Zirconia-hybridized Amorphous Calcium Phosphate. Effect on Transformation to Hydroxyapatite. J Biomater Res. 2002;59(4):597–604. doi: 10.1002/jbm.10017. [DOI] [PubMed] [Google Scholar]
- 16.Venhoven BAM, de Gee AJ, Davidson CL. Polymerization Contraction and Conversion of Light-curing BisGMA-based Methacrylate Resins. Biomaterials. 1993;14(11):871–875. doi: 10.1016/0142-9612(93)90010-y. [DOI] [PubMed] [Google Scholar]
- 17.Stansbury JW, Dickens SH. Network Formation and Compositional Drift During Photo-initiated Copolymerization of Dimethacrylate Monomers. Polymer. 2001;42:6363–6369. [Google Scholar]
- 18.Labella R, Lambrechts P, Van Meerbeek B, Vanherle G. Polymerization Shrinkage and Elasticity of Flowable Composites and Filled Adhesives. Dent Mater. 1999;15:128–137. doi: 10.1016/s0109-5641(99)00022-6. [DOI] [PubMed] [Google Scholar]
- 19.Price RB, Rizkalla AS, Hall GC. Effect of Stepped Light Exposure on the Volumetric Polymerization Shrinkage and Bulk Modulus of Dental Composites and an Unfilled Resin. Am J Dent. 2000;13:176–180. [PubMed] [Google Scholar]
- 20.Guggenberger R, Weinmann W. Exploring Beyond Methacrylates. Am J Dent. 2002;13:82D–84D. [PubMed] [Google Scholar]
- 21.Tilbrook DA, Clarke RL, Howle NE, Braden M. Photocurable Epoxy-polyol Matrices for Use in Dental Composites. Biomaterials. 2000;21:1743–1753. doi: 10.1016/s0142-9612(00)00059-4. [DOI] [PubMed] [Google Scholar]
- 22.Skrtic D, Antonucci JM, Eanes ED. Effect of the Monomer and Filler systems on the Remineralizing Potential of Bioactive Dental Composites Based on Amorphous Calcium Phosphate. Polym Adv Technol. 2001;12:369–379. [Google Scholar]
- 23.Skrtic D, Antonucci JM, Eanes ED, Eidelman N. Dental Composites Based on Hybrid and Surface-modified Amorphous Calcium Phosphates—A FTIR Microscopic Study. in preparation. [DOI] [PubMed] [Google Scholar]
- 24.Schumacher GE, Eichmiller FC, Antonucci JM. Effects of Surface-active Resins on Dentin/composite Bonds. Dent Mater. 1992;8:278–282. doi: 10.1016/0109-5641(92)90100-q. [DOI] [PubMed] [Google Scholar]
- 25.Arima T, Hamada T, McCabe JF. The Effects of Cross-linking Agents on Some Properties of HEMA-based Resins. J Dent Res. 1995;74(9):1597–1601. doi: 10.1177/00220345950740091501. [DOI] [PubMed] [Google Scholar]
- 26.Garcia-Fiero JL, Aleman JV. Sorption of Water by Epoxide Prepolymers. Macromolecules. 1982;15:1145–1149. [Google Scholar]
- 27.Skrtic D, Antonucci JM. Effect of Bifunctional Co-monomers on Mechanical Strength and Water Sorption of Amorphous Calcium Phosphate- and Silanized Glass-filled Bis-GMA-based Composites. Biomaterials. 2003 doi: 10.1016/s0142-9612(03)00119-4. in press. [DOI] [PubMed] [Google Scholar]
- 28.Soderholm KJM, Roberts MJ. Influence of Water Exposure on the Tensile Strength of Composites. J Dent Res. 1990;69:1812–1816. doi: 10.1177/00220345900690120501. [DOI] [PubMed] [Google Scholar]