

Abstract
Impulse control suppresses actions that are inappropriate in one context, but may be beneficial in others. The medial prefrontal cortex (mPFC) mediates this process by providing a top-down signal to inhibit competing responses, although the mechanism by which the mPFC acquires this ability is unknown. To that end, we examined synaptic changes in the mPFC associated with learning to inhibit an incorrect response. Rats were trained in a simple response inhibition task to withhold responding until a signal was presented. We then measured synaptic plasticity of excitatory synapses in the mPFC, using whole-cell patch-clamp recordings, in brain slices prepared from trained rats. Response inhibition training significantly increased the relative contribution of AMPA receptors to the overall EPSC in prelimbic, but not infralimbic, neurons of the mPFC. This potentiation of synaptic transmission closely paralleled the acquisition and extinction of response inhibition. Using a retrograde fluorescent tracer, we observed that these plastic changes were selective for efferents projecting to the ventral striatum, but not the dorsal striatum or amygdala. Therefore, we suggest that response inhibition is encoded by a selective strengthening of a subset of corticostriatal projections, uncovering a synaptic mechanism of impulse control. This information could be exploited in therapeutic interventions for disorders of impulse control, such as addiction, attention deficit–hyperactivity disorder, and schizophrenia.
Introduction
A deficit in impulse control, described as the tendency toward unplanned actions without due consideration of their consequences (Moeller et al., 2001), is a primary characteristic of many psychiatric disorders. This includes, but is not limited to, drug addiction, attention deficit–hyperactivity disorder, schizophrenia, obsessive–compulsive disorder, pathological gambling, binge eating, and antisocial personality disorders. These disorders also share a common element of altered frontal cortical functioning, fitting with evidence that frontal lobe damage disrupts inhibition of inappropriate actions (Miller, 1992).
Extensive animal studies propose a central role for the medial prefrontal cortex (mPFC) in impulse control. First, defined anatomical lesions in rodents have isolated behavioral disinhibition to the mPFC (Chudasama et al., 2003; Dalley et al., 2008). Second, blockade of glutamatergic transmission (Carli et al., 2006) or inactivation of the mPFC by muscimol infusion (Narayanan and Laubach, 2008) increases premature responses in tasks that require subjects to inhibit responding before a signal. Third, activity of mPFC neurons increases during periods that subjects must inhibit a response (Niki and Watanabe, 1976, 1979). Persistent firing of mPFC neurons, therefore, may signal subcortical targets to inhibit a motor response (Narayanan and Laubach, 2006).
The mPFC is not a unitary structure, but is divided into subregions based on connectivity and functionality (Heidbreder and Groenewegen, 2003). The efferent projections of the mPFC are heterogeneous (Vertes, 2004; Gabbott et al., 2005; Wang et al., 2006), with the prelimbic and infralimbic regions making denser projections to the striatum and amygdala, respectively. Projections from the mPFC to the striatum are of particular interest to the current study, as this region is implicated in impulse control (Dalley et al., 2007).
Despite extensive investigation into the neuropharmacological substrates of impulse control (see Hayton and Olmstead, 2009, for a review), few studies have examined the synaptic mechanisms that underlie this process. In fact, the neuronal changes associated with the acquisition of inhibitory control remain understudied. We addressed this gap in the literature by examining plasticity of excitatory synaptic transmission within the mPFC. More specifically, we combined behavioral testing with patch-clamp recordings in the whole-cell configuration to determine how the mPFC encodes impulse control.
Rats were trained in a simple response inhibition (RI) task; we then prepared brain slices containing the mPFC and examined changes in glutamatergic transmission in these subjects. We hypothesized that learning to withhold a response would increase the contribution of the AMPA relative to the NMDA component of EPSCs, a widely used indicator of plasticity at excitatory synapses (Nicoll and Malenka, 1999; Ungless et al., 2001).
In a second and third set of experiments, we recorded from slices prepared from rat brains midway through acquisition of the task, and following extinction of response inhibition. In a final experiment, we microinjected a retrograde fluorescent tracer into three subcortical targets of the mPFC: the ventral striatum, dorsal striatum, and basolateral amygdala. We then recorded from neurons projecting to these specific targets to determine whether changes in synaptic transmission were selective to a specific cortical efferent.
Materials and Methods
Subjects.
One hundred eight male Long–Evans rats (Charles River), aged 21 postnatal days (PND) at the start of the experiment, were singly housed in standard polycarbonate cages on a reverse light–dark cycle (lights on at 7:00 P.M.). All testing was conducted during the dark cycle. During a 10 d acclimatization period, rats had ad libitum access to food (Lab Diet; PMI Nutrition International) and water. Three days before training, food was restricted to 120 min of daily ad libitum access, such that animals gained 10–15 g per week.
Subjects in the retrograde labeling experiments (n = 53) had fluorescent microspheres injected into several prelimbic target regions. At age 35–46 PND, rats were anesthetized with isoflurane (2–5% at a rate of 2 L of O2/min) and the skull was exposed with 1.5 cm dermal incision running caudally along the midline of the skull. The skin was retracted and a small hole was drilled using a dental drill. A 500 nl Hamilton Syringe (Hamilton) was then lowered into the ventral striatum [+1.3 anteroposterior (AP), ±1.5 lateral (L), −6.5 dorsoventral (DV)], dorsal striatum (lateral: 0.0 AP, +3.7 L, −5.2 V; medial: 0.0 AP, +1.9 L, −5.0 V), or basolateral amygdala (−2.5 AP, ±4.5 L, −9.0 V), and 75 nl of fluorescent microbeads (0.04 μm far red-fluorescent TransFluoSpheres beads; Invitrogen) was injected over 60 s. The rats received 1 mg/kg Anafen both preoperatively and postoperatively for 3 d. Subjects were then food restricted for 3 d, and training began as described below. Following slice preparation (discussed below), the remaining brains were fixed in paraformaldehyde and the location of microbead infusions was verified postmortem. Data from subjects with injections beyond the anatomical boundaries of the intended targets were discarded.
All experiments were conducted in accordance with the guidelines provided by the Canadian Council on Animal Care, and were approved by the Queen's University Animal Care Committee.
Apparatus.
Behavioral testing was conducted in operant boxes (26.5 × 22.0 × 20.0 cm), each housed in a sound-attenuating chamber (built in house). Each box was fitted with two retractable levers positioned on one wall. A food magazine was located between the two levers, which dispensed 45 mg dustless food pellets (BioServ). Signal lights were located 4 cm above each lever and the food magazine, and an indirect house light illuminated the entire chamber. A tone generator produced a sine-wave 12–16 kHz, 80–90 db tone. A standard PC in an adjacent room controlled the equipment and was used for data collection (software written in-house using BASIC).
Behavioral procedures.
Behavioral inhibition was assessed in the RI task, which is a modified version of the signaled nose-poke task for mice (Bowers and Wehner, 2001). Initially, rats were magazine trained for 1 d, receiving 20 sucrose pellets on a variable-time 90 s schedule. Rats were then trained to lever press for food on a continuous reinforcement schedule. Only one lever was inserted into the chamber, with the lever assignment (left vs right) counterbalanced across animals. This lever remained consistent for future stages of the experiment. A signal (lever light) was turned on throughout these sessions, except during delivery of the reward (1 s). Training continued until the rat earned a minimum of 80 pellets in a 60 min session for 2 consecutive days.
Rats then progressed to the full RI task. Trials progressed through an intertrial interval (ITI), premature phase, and response phase (see Fig. 1 A). During the 10 s ITI, all lights were extinguished and the lever was retracted. During the premature phase (variable duration, see below), the lever was extended, the tone activated, and the house light illuminated. Lever presses during this period reinstated the ITI with no delivery of a sucrose pellet. If the rats did not respond during the premature phase, the trial progressed to the response phase (10 s), which was signaled by illumination of the lever light. A lever press during the response phase delivered a sucrose pellet, illuminated the magazine light for 1 s and initiated the next trial. If the response phase elapsed with no lever press, the lever retracted and the next trial was initiated. In each trial, responses were classified as “premature,” “correct,” or “omission.” Sessions were terminated once rats obtained 100 pellets or completed 200 trials, unless otherwise indicated.
Figure 1.

Visual schematic of behavioral tasks with arrows indicating possible outcomes. A, The RI task requires subjects to withhold responding until the correct phase. Responses during the correct phase result in a sucrose pellet reward and reinstate the intertrial interval (ITI). Responses during the premature phase restore the ITI with no reward. Failure to respond during the correct phase results in an omission and reinstates the ITI. B, The operant control is identical to the RI task, but the lever is withdrawn during the premature phase, preventing any premature responses. C, The yoked control has identical stimuli to the RI task, but lever presses have no programmed consequence. A sucrose pellet is delivered automatically 1 s after the initiation of the correct phase. D, In the extinction task, sucrose pellets are delivered following a response in either the premature or correct phase.
Rats were initially trained without a delay for two sessions. Subsequently, animals were trained on a 2 s delay, and advanced to a 4 s delay after achieving >80% accuracy for two consecutive sessions. One rat was advanced to the 4 s delay after failing to reach this criterion after 15 sessions. Electrophysiological measurements began after rats achieved >80% accuracy at the 4 s delay for two consecutive sessions.
Operant control rats underwent an identical training program, but the lever was withdrawn during the premature phase (Fig. 1 B), preventing premature responses. Yoked controls received identical cues as the RI task, such that all lights, sounds, lever extensions/retractions were identical to the experimental group, but reward deliveries were automatic and independent of lever presses. Reward delivery was set at 1 s to match mean response time in the RI group (Fig. 1 C). Yoked and operant controls were matched to rats in the RI task such that they underwent an identical number of sessions at each delay period. Food restricted controls were given an identical feeding schedule as other subjects, but did not receive any behavioral training. Age controls were left in their home cage with ad libitum access to food for the duration of the experiment. During extinction training (Fig. 1 D) any response after lever extension (correct or premature) resulted in reward delivery. That is, subjects continued to receive a sucrose pellet for each lever press, but no longer had to withhold responding until the discriminative stimulus was presented.
In a separate experiment, we recorded from brain slices prepared from rats as they acquired the ability to withhold responding. Specifically, animals were trained on only a 4 s delay with 100 trials per session, and we calculated AMPA/NMDA in groups of rats on the first session of the response inhibition task in which they achieved 40% (n = 5), 60% (n = 4), or 80% (n = 4) accuracy.
To further probe the plasticity of mPFC synapses, we examined whether AMPA/NMDA would be retained after extinction of the response inhibition. Three groups of rats were trained on the 4 s delay, until all subjects had achieved >80% accuracy. Next, one group underwent extinction by reinforcing any responses, either premature or correct, until accuracy was reduced to <20% for 2 consecutive days (Extinct group; n = 5). A second group received a matched number of days without any training (Pause group; n = 5), and a third group performed additional sessions of the standard response inhibition task for an equal number of days (Train group; n = 4).
Preparation of brain slices.
Within 2 h of the final training session, rats were anesthetized with isoflurane and killed, and the brains were extracted for slice preparation. Coronal slices (250 μm) were prepared on a vibrating blade microtome in an ice-cold, oxygenated physiological solution containing (in mm) 126 NaCl, 2.5 KCl, 1.2 MgCl2, 2.5 CaCl2, 1.2 NaH2PO4, 25 NaHCO3 and, 11 d-glucose. Slices were incubated in oxygenated physiological solution at 34°C for 60 min then transferred to a holding bath for patch-clamp electrophysiology. During patch clamping, slices were constantly perfused (1.5 ml/min) with physiological solution maintained at 34°C and equilibrated with 95% O2/5% CO2.
Electrophysiology recordings.
The mPFC was visualized and Layer V pyramidal neurons identified by shape, and located using white matter landmarks. Whole-cell voltage-clamp recordings were obtained from layer V pyramids with borosilicate glass pipettes (1.5–2.5 MΩ tip resistance). Recordings were obtained with a Multiclamp 700B amplifier connected to a Digidata 1440A digitizer (Molecular Devices). Data were collected and analyzed using Axograph X for windows (V 1.2, AxographX.com).
Neurons were voltage clamped at −60 mV, and EPSCs were evoked by stimulating (0.05–0.5 mA; 0.1 Hz) layer II/III with a biphasic stimulus isolator (World Precision Instruments) through a decapolar matrix electrode (FHC). Polysynaptic EPSCs were pharmacologically isolated by bath application of picrotoxin (100 μm; Sigma-Aldrich). Baseline recordings were made for 5 min at +40 mV. The AMPA receptor contribution to the EPSC determined by bath application (2–4 min) of d-AP5 (50 μm; 2-amino-5-phosphono-pentanoate; Sigma-Aldrich). NMDA receptor contribution was calculated offline by subtracting AMPA current from total EPSC. Focal recordings were obtained by an identical process, but used a theta glass pipette (Harvard Apparatus), placed in close proximity (∼50 μm) to the apical dendrite of recorded neurons.
Statistical analyses.
Accuracy was assessed as the percentage of trials in which animals successfully inhibited lever pressing in the premature phase. This dependent measure was calculated as a percentage of correct response out of total trials with a response [100 × (correct responses)/(premature + correct responses)]. Behavioral data were analyzed using repeated measures ANOVA with group as a between-subjects variable.
Electrophysiological data were analyzed using a two-way (region × group) ANOVA for the initial experiments, and a one-way (group) ANOVA for extinction, partial training, focal stimulation, and target-specific experiments using SPSS (V14.0). Post hoc tests (Fischer's LSD) were conducted where appropriate.
Results
Response inhibition training
Rats (n = 6) were initially trained in the RI task with a 2 s premature phase (Fig. 2 A). Despite poor performance during the initial session, the ability to inhibit premature responses improved rapidly from the first to the final session (t (5) = 4.21, p = 0.008). Five of the six subjects reached criterion (>80% accuracy for two consecutive sessions) within 15 sessions and one subject was advanced without reaching criterion (Fig. 1 B, inset). Performance on the first session of the 4 s premature phase was significantly worse than the final session of the 2 s phase (t (5) = 6.56, p = 0.001) and again, accuracy improved from the first to the final session at the 4 s delay (t (5) = 9.47, p < 0.001). All rats completed at least two sessions with >80% accuracy at the 4 s delay. Once this criterion was reached, rats were killed and mPFC brain slices were prepared for patch-clamp recordings.
Figure 2.
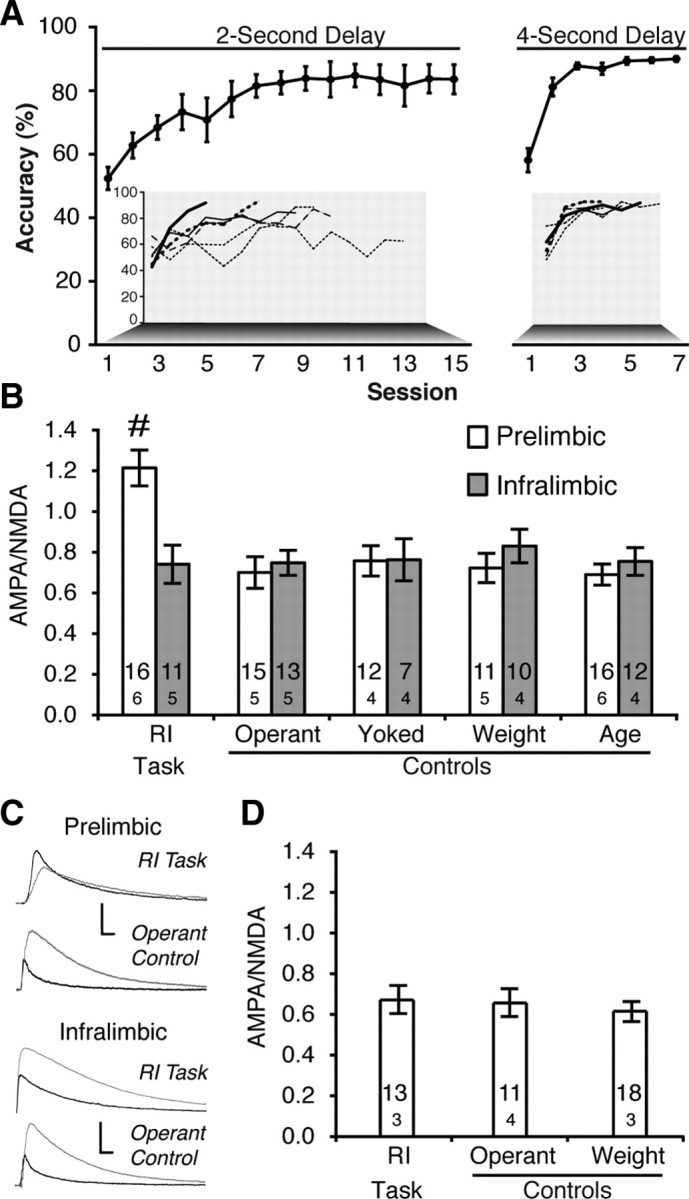
Training on an RI task strengthened AMPA/NMDA in prelimbic mPFC neurons. A, Accuracy improves after training in the RI task with both 2 s (left) and 4 s (right) premature phases. Individual subject data inset. B, Learning the RI task produced larger AMPA/NMDA in the prelimbic (white bars), but not the infralimbic (black bars), region of the mPFC (# p < 0.005 vs all groups). Sample sizes for cells (above) and subjects (below) are indicated within the bars. C, Representative AMPA (black) and NMDA (gray) EPSCs from RI task and operant control neurons in the prelimbic (top) and infralimbic (bottom) cortex (calibration: 500 nA/0.1 s). D, Training on the RI task failed to enhance AMPA/NMDA when measured by focal stimulation.
Synaptic changes underlying response inhibition
The ratio of AMPA to NMDA receptor currents (AMPA/NMDA) in layer V pyramidal cells was measured by stimulating layers II/II of the mPFC in the presence and absence of the NMDA antagonist d-AP5 (50 μm). Neurons in the prelimbic cortex, but not in the adjacent infralimbic cortex, substantially increased AMPA/NMDA after training in the RI task (Fig. 2 B,C) (group × region: F (4,105) = 3.58, p = 0.009; groupPL: F (4,63) = 10.27, p < 0.001; groupIL: F (4,42) = 0.58, p = 0.99). Post hoc tests revealed that increased AMPA/NMDA was restricted to impulse control (p < 0.005), and demonstrated that learning to lever press with identical cues, but no response inhibition (operant control; n = 5), failed to modify AMPA/NMDA. Likewise, passive food delivery (yoked control; n = 4), food restriction (weight control; n = 5), or development (age control; n = 6) failed to alter synaptic plasticity in the prelimbic or infralimbic cortices. These changes in AMPA/NMDA cannot be attributed to different cellular properties, as input resistance, series resistance, membrane capacitance, and holding current showed no differences between training conditions (Table 1).
Table 1.
Cell characteristics of layer V pyramidal neurons of rats trained in the RI task and behavioral controls
| Training group | Number of subjects | Number of cells | AMPA/NMDA | Series resistance (MΩ) | Input resistance (MΩ) | Membrane capacitance (pF) | Holding current (pA) |
|---|---|---|---|---|---|---|---|
| Prelimbic | |||||||
| RI task | 6 | 16 | 1.21 ± 0.09 | 7.1 ± 0.6 | 53.0 ± 3.5 | 45.2 ± 2.4 | 655.0 ± 38.8 |
| Operant control | 5 | 15 | 0.70 ± 0.08 | 6.3 ± 0.7 | 52.5 ± 6.0 | 53.4 ± 3.6 | 638.8 ± 85.9 |
| Yoked control | 4 | 12 | 0.76 ± 0.07 | 6.9 ± 0.8 | 55.4 ± 5.9 | 49.8 ± 4.2 | 637.1 ± 68.1 |
| Weight control | 5 | 11 | 0.72 ± 0.07 | 5.4 ± 0.5 | 48.2 ± 3.9 | 59.9 ± 8.3 | 595.1 ± 52.1 |
| Age control | 6 | 16 | 0.69 ± 0.05 | 5.4 ± 0.2 | 55.0 ± 6.0 | 54.5 ± 2.4 | 605.1 ± 67.0 |
| Infralimbic | |||||||
| RI task | 5 | 11 | 0.74 ± 0.09 | 6.6 ± 0.7 | 72.1 ± 11.0 | 48.1 ± 6.3 | 537.9 ± 70.8 |
| Operant control | 5 | 13 | 0.75 ± 0.06 | 8.3 ± 1.3 | 73.1 ± 7.6 | 41.3 ± 3.6 | 449.8 ± 39.7 |
| Yoked control | 4 | 7 | 0.76 ± 0.10 | 8.2 ± 1.1 | 101.5 ± 7.7 | 43.7 ± 5.5 | 366.4 ± 89.2 |
| Weight control | 4 | 10 | 0.83 ± 0.08 | 6.6 ± 0.6 | 61.6 ± 6.6 | 45.1 ± 2.4 | 550.9 ± 117.9 |
| Age control | 4 | 12 | 0.75 ± 0.07 | 5.5 ± 0.2 | 48.1 ± 3.5 | 48.1 ± 3.5 | 579.3 ± 90.2 |
Values were obtained with a 10 ms, +10 mV pulse while holding neurons at +40 mV. No significant differences between experimental groups were observed for any variable, with the exception of AMPA/NMDA (bolded).
Because of differing resistances, absolute values for AMPA and NMDA currents are intrinsically more variable than the AMPA/NMDA ratio. Thus, it is not surprising that peak AMPA and NMDA currents showed no significant changes with training (Table 2) (AMPA: F (4,63) = 1.21, p = 0.31; NMDA: F (4,63) = 0.50, p = 0.73). However, examining cumulative EPSC evoked in the 500 ms after stimulation (i.e., area under the curve) revealed increased AMPA current (F (4,63) = 2.87, p = 0.03), but no changes in cumulative evoked NMDA current (F (4,63) = 0.08, p = 0.99), suggesting that the shift in AMPA/NMDA is driven by increased AMPA, not decreased NMDA, current in the prelimbic cortex.
Table 2.
Absolute contribution of AMPA and NMDA receptors to EPSCs in the prelimbic cortex
| RI task | Operant control | Yoked control | Weight control | Age control | F | p | |
|---|---|---|---|---|---|---|---|
| AMPA | |||||||
| EPSC peak (pA) a | 1145.34 ± 106.5 | 813.73 ± 122.8 | 891.65 ± 163.3 | 864.46 ± 118.37 | 871.75 ± 114.59 | 1.21 | 0.31 |
| EPSC area (nA · ms) b | 232.18 ± 37.28 | 133.31 ± 23.76 | 145.83 ± 30.54 | 113.07 ± 18.06 | 129.50 ± 24.24 | 2.87 | 0.03 |
| NMDA | |||||||
| EPSC peak (pA) a | 1005.31 ± 113.92 | 1138.75 ± 94.51 | 1159.51 ± 146.84 | 1169.08 ± 123.58 | 1207.24 ± 95.56 | 0.50 | 0.73 |
| EPSC area (nA · ms) b | 341.5 ± 36.49 | 336.07 ± 34.09 | 358.06 ± 57.50 | 324.16 ± 41.59 | 336.93 ± 37.66 | 0.08 | 0.99 |
AMPA EPSC area showed a significant enhancement after training in the RI task.
aMaximum current observed following stimulation, subtracted from baseline holding current.
bCalculated as area under the curve for 500 ms following stimulation, subtracted from baseline holding current.
Focal stimulation was examined by stimulating proximal to the apical dendrite, within layer V, using a theta-glass stimulator. When AMPA/NMDA was measured using only focal stimulation (Fig. 2 D), learning the RI task failed to produce a shift in AMPA/NMDA (F (2,42) = 0.117, p = 0.89). This suggests the shift in glutamatergic transmission is selective to synapses between the external (II/III) and deep layers (V) of the cortex.
Partial training
Subjects were trained on a 4 s delay only, and we calculated AMPA/NMDA in groups of rats on the first session of the response inhibition task in which they achieved 40%, 60%, or 80% accuracy. As shown in Figure 3, AMPA/NMDA in the PL cortex increased progressively as the ability to inhibit a response improved (F (2,48) = 6.237, p = 0.004), and post hoc tests revealed a significant difference between subjects trained to 40% and 80% accuracy (p < 0.05).
Figure 3.

Partial training produces proportional increases in AMPA/NMDA ratios. Data points represent AMPA/NMDA of neurons from subjects trained to 40% accuracy (n = 5), 60% accuracy (n = 4), or 80% accuracy (n = 4) in the RI task. Representative AMPA (black) and NMDA (gray) EPSCs for each group at right (calibration: 500 nA/0.1 s; *p < 0.05).
Extinction
After subjects reached criterion in the RI task, response inhibition was extinguished by reinforcing all lever presses, even those that occurred before the signal (Fig. 4 A). AMPA/NMDA in the Extinct group was compared to subjects who continued to respond at criterion levels in the RI task (Train) and to subjects who had equivalent days without any training (Pause). Subjects in the Extinct group showed a rapid decline in accuracy relative to subjects in the Train group (session: F (5,35) = 14.74, p < 0.001; group: F (1,7) = 359.13, p < 0.001; session × group: F (5,35) = 26.95, p < 0.001). Extinguishing response inhibition reduced AMPA/NMDA in the PL cortex (Fig. 4 B) (F (2,76) = 8.88, p < 0.001), restoring it to near-baseline levels. Cessation of training (Pause group) also reduced AMPA/NMDA (albeit to a lesser degree than extinction), suggesting that maintenance of task performance is necessary to preserve these plastic changes.
Figure 4.

Extinguishing response inhibition returned AMPA/NMDA to baseline. A, Accuracy was rapidly reduced from baseline levels (day 0) in RI-task when all lever presses were reinforced (extinction). The Train group continued to perform the task without any changes in accuracy and the Pause group received equivalent days off. B, Extinction of response inhibition reduced AMPA/NMDA (Extinct group). Equivalent days without training reduced AMPA/NMDA in the Pause group, but to a lesser degree than in the Extinct group (*p < 0.05).
Target specificity
Fluorescent microbeads infused into the ventral striatum, medial dorsal striatum, lateral dorsal striatum, or basolateral amygdala were retrogradely transported to the PL and neurons containing these beads could be identified for patch-clamp electrophysiology after training was completed (Fig. 5).
Figure 5.

Retrograde transport of fluorescent beads permits identification of neurons projecting to target areas. A, Representative image of prelimbic region under fluorescence (medial at left; dorsal at top). Lines mark approximate boundaries of layer V, box indicates magnified area of interest (scale bar: 100 μm). B, C, Prelimbic pyramidal neuron under dark-field (B) and fluorescent (C) light (scale bars: 10 μm).
AMPA/NMDA increased in PL neurons projecting to the ventral striatum after training in the response inhibition task (Fig. 6) (F (2,45) = 6.74, p = 0.003), whereas neurons projecting to the basolateral amygdala showed no effect of training (Fig. 6) (F (2,44) = 0.27, p = 0.77). AMPA/NMDA was enhanced in neurons projecting to the dorsal striatum following training in the operant control version of the task. The enhancement of AMPA/NMDA in operant controls was observed equally in projections to lateral and medial regions of the dorsal striatum (training: F (2,85) = 8.57, p < 0.001; training × projection: F (2,85) = 0.61, p = 0.94; trainingMedial: F (2,33) = 4.64, p = 0.02; trainingLateral: F (2,52) = 4.75, p = 0.01).
Figure 6.

Target-specific enhancement of AMPA/NMDA after learning the RI task. A, Injections sites for subjects trained in the RI task (•), as well as operant (■) and weight (▴) controls. B, AMPA/NMDA increased in projections to the ventral striatum (left) after training in the RI task, and in projections to the dorsal striatum (center) following training in the operant control task. Projections to the basolateral amygdala (right) showed no effect of training. Sample sizes for cells (above) and subjects (below) are indicated within the bars (*p < 0.001 vs all groups).
Discussion
Our study demonstrates that learning to inhibit a simple response enhanced the relative contribution of AMPA to NMDA currents in prelimbic, but not infralimbic, synapses of layer V pyramidal neurons. We also observed a tight association between performance in the response inhibition task and changes in glutamatergic transmission in that performance during extinction of the response inhibition and partial training sessions closely tracked changes in AMPA/NMDA. This effect was selective to neurons making projections to the ventral striatum, but not the dorsal striatum or basolateral amygdala. Although we recognize the difficulty in establishing causation with this approach, the close relationship between alterations in AMPA/NMDA and successful performance during acquisition, maintenance, and extinction of response inhibition suggests that we uncovered a synaptic substrate for inhibiting inappropriate responses. Together, our data reveal a synaptic substrate of response inhibition via a target-specific enhancement of excitatory synaptic transmission in the mPFC. We propose that synapses within the prelimbic cortex change their relative contribution of AMPA and NMDA glutamate receptor components to encode response inhibition, a mechanism reminiscent of long-term potentiation (LTP) at excitatory synapses (Nicoll, 2003).
In this investigation, we designed a task that would be acquired rapidly with low demands on cognitive processes such as discrimination, timing, and attention. The simplicity of the task is an advantage in that we can minimize the possibility that changes in synaptic plasticity are due to confounding factors. A disadvantage is that it may be difficult to generalize our findings to other measures of response inhibition, particularly as many of these tasks require subjects to choose between several actions, time a delay without signals, inhibit an action already in progress, or sustain a response until a release signal is presented. Given the distinct pharmacological and anatomical systems that mediate these different tasks (see Hayton and Olmstead, 2009, for a review), it would be surprising if they were encoded by a mechanism that is identical to that of our simple response inhibition task. Indeed, lesion studies have confirmed a role for other mPFC regions, including the infralimbic and anterior cingulate cortices (Chudasama et al., 2003), in response inhibition. Moreover, antagonism of NMDA receptors in the infralimbic, but not the prelimbic, cortex disrupts response inhibition in the five-choice serial reaction time task (Murphy et al., 2005), suggesting that subregions of the mPFC make distinct contributions to response inhibition, depending on the task.
Learning-induced changes in AMPA/NMDA occur in a variety of paradigms and brain regions. For example, AMPA/NMDA increases in the ventral tegmental area following cocaine sensitization (Ungless et al., 2001; Borgland et al., 2004), in the amygdala following fear conditioning (Lin et al., 2010), and in the bed nucleus of the stria terminalis following lever pressing for sucrose or cocaine (Dumont et al., 2005). Most importantly, exposure to drug-paired cues, after responding for heroin has been extinguished, reduces AMPA/NMDA in the mPFC (Van den Oever et al., 2008), which in turn promotes relapse to drug seeking. This reduction in AMPA currents in the mPFC provides an exciting parallel to our findings, in that both relapse to drug seeking and premature responding can be defined as failures of impulse control. In line with these studies, inactivation of the dorsal mPFC blocks cue-, drug-, and stress-induced reinstatement of cocaine seeking (McFarland and Kalivas, 2001; McLaughlin and See, 2003; McFarland et al., 2004). Changes in glutamatergic transmission within the mPFC, therefore, may be a mechanism that mediates the withholding of responses for both drugs and natural rewards. Enhancement of AMPA/NMDA after learning the response inhibition task was restricted to ventral striatum projections, sparing dorsal striatum or basolateral amygdala projections. Given evidence that the ventral striatum is a substrate of impulse control (Pattij et al., 2007, Dalley et al., 2008), our results point to this structure as an intermediary in the mPFC-motor cortex circuit. In fact, neural recordings revealed that the mPFC indirectly inhibits the motor cortex during periods of response inhibition (Narayanan and Laubach, 2006). The fact that synaptic transmission was enhanced in a subpopulation of PL neurons demonstrates that increases in AMPA/NMDA were not an artifact of global changes in mPFC function or connectivity. Therefore, we identified an anatomical substrate for the behavioral/physiological relationship we observed originally. Explicitly, response inhibition is encoded as increased synaptic transmission in a specific prelimbic-ventral striatum projection. Although we did not examine it specifically, it is likely that this plasticity is confined to the core region of the nucleus accumbens as prelimbic neurons project, preferentially, to the core whereas infralimbic neurons project, primarily to the shell (Berendse et al., 1992; Vertes, 2004). Moreover, deep brain stimulation of the core enhances impulse control, whereas stimulation of the shell disrupts it (Sesia et al., 2008). Antagonism of dopamine receptors in the core and shell subregions also produce differential effects on impulsivity (Pattij et al., 2007; Besson et al., 2010), supporting the idea that the neural circuit of impulse control involves specific striatal targets from prelimbic afferents.
To date, the best-described example of PFC plasticity is fear extinction, which provides a fascinating parallel to impulse control (Morgan et al., 1993; Morgan and LeDoux, 1995). In fear extinction paradigms, subjects initially learn a simple association (shock–tone pairing), which is then extinguished by presenting the tone alone. Fear extinction, or inhibition of a previously learned association, is encoded by LTP-like changes in the infralimbic cortex (Santini et al., 2004; Quirk and Mueller, 2008). Thus, there appears to be a functional dissociation between the two adjoining mPFC regions: inhibition of conditioned fear is encoded by infralimbic projections to the amygdala (Quirk and Mueller, 2008), whereas inhibition of an operant response for food is encoded by prelimbic projections to the ventral striatum. The similarities between fear extinction and our response inhibition paradigm, as well as the previously discussed cue-induced relapse (Van den Oever et al., 2008), suggest a common mechanism for top-down control of subcortical processes by the mPFC.
Rats trained in the operant control version of the RI task showed a mild elevation in AMPA/NMDA of prelimbic cortex projections to the dorsal striatum. We did not observe AMPA/NMDA changes in the operant control group of the original experiment (Fig. 2 A), but less than one-fifth of prelimbic neurons project to the dorsal striatum (Gabbott et al., 2005). Thus, it is likely that we sampled only a few dorsal striatum-projecting neurons in the original experiment, which would have minimized the effect on AMPA/NMDA in the overall population. The functional significance of altered synaptic transmission in the prelimbic–dorsal striatal pathway may be revealed by comparing performance in the RI and operant control tasks. In the latter, the lever is withdrawn (i.e., unavailable) during the premature phase so rats learn a predictive association between environmental cues (sound plus light) and the opportunity to respond for reward. The dorsal striatum is likely activated under these conditions, given its prominent role in stimulus-response learning (McDonald and White, 1993; Packard and Knowlton, 2002). Prelimbic neurons may control dorsal striatum activity by directing attention to cues predicting reward availability: mPFC neurons fire when attending to stimuli (Totah et al., 2009), preparing to act (Cowen and McNaughton, 2007), or anticipating a reward (Mulder et al., 2003), and mPFC inactivation impairs cued responding in rats (Ishikawa et al., 2008). Our data, therefore, support the contention that prelimbic–dorsal striatal connections encode the relationship between operant contingencies (Balleine et al., 2009; Balleine and O'Doherty, 2010), and suggest that synaptic changes in this circuit may facilitate attention toward stimuli associated with responding for reward.
Our data fit within broader theories of mPFC function, including the idea that dorsal regions of the mPFC contribute to cognitive processing whereby animals make choices between a range of behavioral options at any given time (Seamans et al., 2008). We extend this theory by showing that strengthening of synapses in the mPFC may bias these processes toward specific actions, such as inhibiting or initiating a response. Our data also support the idea that the mPFC instructs “lower” motor systems, such as the ventral striatum, to wait for a stimulus through top-down control (Narayanan and Laubach, 2006), and identify prelimbic cortex synapses as a site for this learning. This concurs to previous findings showing that blockade of glutamatergic transmission (Carli et al., 2006) or inactivation of the mPFC (Narayanan and Laubach, 2008) impairs response inhibition. We observed changes in glutamatergic transmission between surface (II/III) and deep (V) layer neurons of the mPFC, but additional modifications to this area's functioning are likely to occur following the acquisition of response inhibition. For example, experience-dependent changes in neuronal excitability (Santini et al., 2008), metabotropic glutamate receptors (Melendez et al., 2004), and NMDA receptors (Turnock-Jones et al., 2009) have all been observed in the mPFC. In particular, persistent firing of mPFC neurons, which supports working memory and attention, is dependent on both AMPA and NMDA receptors (Wang, 1999; Durstewitz, 2009). In light of these findings, our results do not preclude the involvement of other mPFC mechanisms in response inhibition, but suggest that memories for impulse control are encoded by changes in AMPA/NMDA. Therefore, tests of impulse control are likely subserved by dissociable neural systems as each task involves a unique set of cognitive processes (Hayton and Olmstead, 2009).
Over the past two decades, extensive information has emerged regarding the mediation of behavioral disinhibition, and more broadly, impulse control. In rodents, lesions studies identified where the neural substrates of impulse control are localized (Chudasama et al., 2003; Dalley et al., 2008), while unit recordings showed what these neurons signal (Narayanan and Laubach, 2006). Our study provides the first evidence for how the mPFC encodes impulse control—by enhancing glutamatergic transmission within the prelimbic cortex. The idea that behavioral inhibition is represented in the CNS as a memory of a learned response may provide new directions for investigating and treating psychiatric disorders of impulse control.
Footnotes
This work was supported by National Science and Engineering Research Council (NSERC) discovery grants (E.C.D.: 327350-2008; M.C.O.: 203707-2006) and an NSERC postgraduate scholarship (S.J.H.). We gratefully acknowledge Drs. Denis Paré (Rutgers State University), Yogita Chudasama (McGill University), and Doug Munoz (Queen's University) for their comments on an earlier draft of this manuscript.
References
- Balleine BW, O'Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35:48–69. doi: 10.1038/npp.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, Liljeholm M, Ostlund SB. The integrative function of the basal ganglia in instrumental conditioning. Behav Brain Res. 2009;199:43–52. doi: 10.1016/j.bbr.2008.10.034. [DOI] [PubMed] [Google Scholar]
- Berendse HW, Galis-de Graaf Y, Groenewegen HJ. Topographical organization and relationship with ventral striatal compartments of prefrontal corticostriatal projections in the rat. J Comp Neurol. 1992;316:314–347. doi: 10.1002/cne.903160305. [DOI] [PubMed] [Google Scholar]
- Besson M, Belin D, McNamara R, Theobald DE, Castel A, Beckett VL, Crittenden BM, Newman AH, Everitt BJ, Robbins TW, Dalley JW. Dissociable control of impulsivity in rats by dopamine d2/3 receptors in the core and shell subregions of the nucleus accumbens. Neuropsychopharmacology. 2010;35:560–569. doi: 10.1038/npp.2009.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgland SL, Malenka RC, Bonci A. Acute and chronic cocaine-induced potentiation of synaptic strength in the ventral tegmental area: electrophysiological and behavioral correlates in individual rats. J Neurosci. 2004;24:7482–7490. doi: 10.1523/JNEUROSCI.1312-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers BJ, Wehner JM. Ethanol consumption and behavioral impulsivity are increased in protein kinase Cgamma null mutant mice. J Neurosci. 2001;21(RC180) doi: 10.1523/JNEUROSCI.21-21-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carli M, Baviera M, Invernizzi RW, Balducci C. Dissociable contribution of 5-HT1A and 5-HT2A receptors in the medial prefrontal cortex to different aspects of executive control such as impulsivity and compulsive perseveration in rats. Neuropsychopharmacology. 2006;31:757–767. doi: 10.1038/sj.npp.1300893. [DOI] [PubMed] [Google Scholar]
- Chudasama Y, Passetti F, Rhodes SE, Lopian D, Desai A, Robbins TW. Dissociable aspects of performance on the 5-choice serial reaction time task following lesions of the dorsal anterior cingulate, infralimbic and orbitofrontal cortex in the rat: differential effects on selectivity, impulsivity and compulsivity. Behav Brain Res. 2003;146:105–119. doi: 10.1016/j.bbr.2003.09.020. [DOI] [PubMed] [Google Scholar]
- Cowen SL, McNaughton BL. Selective delay activity in the medial prefrontal cortex of the rat: contribution of sensorimotor information and contingency. J Neurophysiol. 2007;98:303–316. doi: 10.1152/jn.00150.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Fryer TD, Brichard L, Robinson ES, Theobald DE, Lääne K, Peña Y, Murphy ER, Shah Y, Probst K, Abakumova I, Aigbirhio FI, Richards HK, Hong Y, Baron JC, Everitt BJ, Robbins TW. Nucleus accumbens D2/3 receptors predict trait impulsivity and cocaine reinforcement. Science. 2007;315:1267–1270. doi: 10.1126/science.1137073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalley JW, Mar AC, Economidou D, Robbins TW. Neurobehavioral mechanisms of impulsivity: fronto-striatal systems and functional neurochemistry. Pharmacol Biochem Behav. 2008;90:250–260. doi: 10.1016/j.pbb.2007.12.021. [DOI] [PubMed] [Google Scholar]
- Dumont EC, Mark GP, Mader S, Williams JT. Self-administration enhances excitatory synaptic transmission in the bed nucleus of the stria terminalis. Nat Neurosci. 2005;8:413–414. doi: 10.1038/nn1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durstewitz D. Implications of synaptic biophysics for recurrent network dynamics and active memory. Neural Netw. 2009;22:1189–1200. doi: 10.1016/j.neunet.2009.07.016. [DOI] [PubMed] [Google Scholar]
- Gabbott PL, Warner TA, Jays PR, Salway P, Busby SJ. Prefrontal cortex in the rat: projections to subcortical autonomic, motor, and limbic centers. J Comp Neurol. 2005;492:145–177. doi: 10.1002/cne.20738. [DOI] [PubMed] [Google Scholar]
- Hayton SJ, Olmstead MC. Fractionating animal models of motor impulsivity: reconciling the neurochemistry of disinhibition. In: Granon S, editor. Endophenotypes of psychiatric and neurodegenerative disorders in rodent models. Trivandrum, India: Transworld Research Network; 2009. pp. 135–158. [Google Scholar]
- Heidbreder CA, Groenewegen HJ. The medial prefrontal cortex in the rat: evidence for a dorso-ventral distinction based upon functional and anatomical characteristics. Neurosci Biobehav Rev. 2003;27:555–579. doi: 10.1016/j.neubiorev.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Ambroggi F, Nicola SM, Fields HL. Contributions of the amygdala and medial prefrontal cortex to incentive cue responding. Neuroscience. 2008;155:573–584. doi: 10.1016/j.neuroscience.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HC, Mao SC, Su CL, Gean PW. Alterations of excitatory transmission in the lateral amygdala during expression and extinction of fear memory. Int J Neuropsychopharmacol. 2010;13:335–345. doi: 10.1017/S1461145709990678. [DOI] [PubMed] [Google Scholar]
- McDonald RJ, White NM. A triple dissociation of memory systems: hippocampus, amygdala, and dorsal striatum. Behav Neurosci. 1993;107:3–22. doi: 10.1037//0735-7044.107.1.3. [DOI] [PubMed] [Google Scholar]
- McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21:8655–8663. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin J, See RE. Selective inactivation of the dorsomedial prefrontal cortex and the basolateral amygdala attenuates conditioned-cued reinstatement of extinguished cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2003;168:57–65. doi: 10.1007/s00213-002-1196-x. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Gregory ML, Bardo MT, Kalivas PW. Impoverished rearing environment alters metabotropic glutamate receptor expression and function in the prefrontal cortex. Neuropsychopharmacology. 2004;29:1980–1987. doi: 10.1038/sj.npp.1300507. [DOI] [PubMed] [Google Scholar]
- Miller LA. Impulsivity, risk-taking, and the ability to synthesize fragmented information after frontal lobectomy. Neuropsychologia. 1992;30:69–79. doi: 10.1016/0028-3932(92)90015-e. [DOI] [PubMed] [Google Scholar]
- Moeller FG, Barratt ES, Dougherty DM, Schmitz JM, Swann AC. Psychiatric aspects of impulsivity. Am J Psychiatry. 2001;158:1783–1793. doi: 10.1176/appi.ajp.158.11.1783. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE. Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci. 1995;109:681–688. doi: 10.1037//0735-7044.109.4.681. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE. Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett. 1993;163:109–113. doi: 10.1016/0304-3940(93)90241-c. [DOI] [PubMed] [Google Scholar]
- Mulder AB, Nordquist RE, Orgüt O, Pennartz CM. Learning-related changes in response patterns of prefrontal neurons during instrumental conditioning. Behav Brain Res. 2003;146:77–88. doi: 10.1016/j.bbr.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Murphy ER, Dalley JW, Robbins TW. Local glutamate receptor antagonism in the rat prefrontal cortex disrupts response inhibition in a visuospatial attentional task. Psychopharmacology (Berl) 2005;179:99–107. doi: 10.1007/s00213-004-2068-3. [DOI] [PubMed] [Google Scholar]
- Narayanan NS, Laubach M. Top-down control of motor cortex ensembles by dorsomedial prefrontal cortex. Neuron. 2006;52:921–931. doi: 10.1016/j.neuron.2006.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan NS, Laubach M. Neuronal correlates of post-error slowing in the rat dorsomedial prefrontal cortex. J Neurophysiol. 2008;100:520–525. doi: 10.1152/jn.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. Expression mechanisms underlying long-term potentiation: A postsynaptic view. Philos Trans R Soc Lond B Biol Sci. 2003;358:721–726. doi: 10.1098/rstb.2002.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Niki H, Watanabe M. Cingulate unit activity and delayed response. Brain Res. 1976;110:381–386. doi: 10.1016/0006-8993(76)90412-1. [DOI] [PubMed] [Google Scholar]
- Niki H, Watanabe M. Prefrontal and cingulate unit activity during timing behavior in the monkey. Brain Res. 1979;171:213–224. doi: 10.1016/0006-8993(79)90328-7. [DOI] [PubMed] [Google Scholar]
- Packard MG, Knowlton BJ. Learning and memory functions of the basal ganglia. Annu Rev Neurosci. 2002;25:563–593. doi: 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- Pattij T, Janssen MC, Vanderschuren LJ, Schoffelmeer AN, van Gaalen MM. Involvement of dopamine D1 and D2 receptors in the nucleus accumbens core and shell in inhibitory response control. Psychopharmacology (Berl) 2007;191:587–598. doi: 10.1007/s00213-006-0533-x. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Mueller D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology. 2008;33:56–72. doi: 10.1038/sj.npp.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Ge H, Ren K, Peña de Ortiz S, Quirk GJ. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J Neurosci. 2004;24:5704–5710. doi: 10.1523/JNEUROSCI.0786-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Quirk GJ, Porter JT. Fear conditioning and extinction differentially modify the intrinsic excitability of infralimbic neurons. J Neurosci. 2008;28:4028–4036. doi: 10.1523/JNEUROSCI.2623-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Lapish CC, Durstewitz D. Comparing the prefrontal cortex of rats and primates: insights from electrophysiology. Neurotox Res. 2008;14:249–262. doi: 10.1007/BF03033814. [DOI] [PubMed] [Google Scholar]
- Sesia T, Temel Y, Lim LW, Blokland A, Steinbusch HW, Visser-Vandewalle V. Deep brain stimulation of the nucleus accumbens core and shell: opposite effects on impulsive action. Exp Neurol. 2008;214:135–139. doi: 10.1016/j.expneurol.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Totah NK, Kim YB, Homayoun H, Moghaddam B. Anterior cingulate neurons represent errors and preparatory attention within the same behavioral sequence. J Neurosci. 2009;29:6418–6426. doi: 10.1523/JNEUROSCI.1142-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnock-Jones JJ, Jennings CA, Robbins MJ, Cluderay JE, Cilia J, Reid JL, Taylor A, Jones DN, Emson PC, Southam E. Increased expression of the NR2A NMDA receptor subunit in the prefrontal cortex of rats reared in isolation. Synapse. 2009;63:836–846. doi: 10.1002/syn.20665. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, De Vries TJ. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Vertes RP. Differential projections of the infralimbic and prelimbic cortex in the rat. Synapse. 2004;51:32–58. doi: 10.1002/syn.10279. [DOI] [PubMed] [Google Scholar]
- Wang XJ. Synaptic basis of cortical persistent activity: the importance of NMDA receptors to working memory. J Neurosci. 1999;19:9587–9603. doi: 10.1523/JNEUROSCI.19-21-09587.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Markram H, Goodman PH, Berger TK, Ma J, Goldman-Rakic PS. Heterogeneity in the pyramidal network of the medial prefrontal cortex. Nat Neurosci. 2006;9:534–542. doi: 10.1038/nn1670. [DOI] [PubMed] [Google Scholar]