

Abstract
Smooth muscle sphincters exhibit basal tone and control passage of contents through organs such as the gastrointestinal tract; loss of this tone leads to disorders such as faecal incontinence. However, the molecular mechanisms underlying this tone remain unknown. Here, we show that deletion of myosin light-chain kinases (MLCK) in the smooth muscle cells from internal anal sphincter (IAS-SMCs) abolishes basal tone, impairing defecation. Pharmacological regulation of ryanodine receptors (RyRs), L-type voltage-dependent Ca2+ channels (VDCCs) or TMEM16A Ca2+-activated Cl− channels significantly changes global cytosolic Ca2+ concentration ([Ca2+]i) and the tone. TMEM16A deletion in IAS-SMCs abolishes the effects of modulators for TMEM16A or VDCCs on a RyR-mediated rise in global [Ca2+]i and impairs the tone and defecation. Hence, MLCK activation in IAS-SMCs caused by a global rise in [Ca2+]i via a RyR-TMEM16A-VDCC signalling module sets the basal tone. Targeting this module may lead to new treatments for diseases like faecal incontinence.
 The molecular basis of the basal tone generated by internal anal sphincters (IAS) is largely unknown. Here, the authors show that the tone arises from a global rise in intracellular Ca2+ in smooth muscle cells via a Ryanodine receptor-TMEM16A-L-type Ca2+ channel-MLC kinase pathway, suggesting a potential therapy for IAS motility disorders.
The molecular basis of the basal tone generated by internal anal sphincters (IAS) is largely unknown. Here, the authors show that the tone arises from a global rise in intracellular Ca2+ in smooth muscle cells via a Ryanodine receptor-TMEM16A-L-type Ca2+ channel-MLC kinase pathway, suggesting a potential therapy for IAS motility disorders.
The human body, and those of other mammals, contains up to 50 sphincters, ring-shaped structures encircling an opening or passage in hollow organs such as the intestine and the bladder. These sphincters control the entrance of material into, or the release of contents from, these organs, and participate in a variety of biological functions essential for homeostasis1. Dysfunction in the sphincters, either structurally or functionally, can have severe consequences leading to diseases/disorders including gastroesophageal reflux disease, achalasia, gastroparesis, dysphagia, recurrent episodes of pancreatitis or biliary pain, faecal incontinence and urinary incontinence1. Healthy sphincters open transiently but, in the basal state, remain closed and therefore require constant force generation from the smooth muscle cells that make up sphincters. It is thus of fundamental importance to determine the molecular and cellular mechanisms that dictate sphincter smooth muscle contraction at rest (basal tone formation).
The internal anal sphincter (IAS) located at the end of the gastrointestinal tract, has served as a prototypical model to understand basal tone genesis in sphincters. A significant number of in vitro and in vivo experiments have indicated that the basal tone of IAS is independent of extrinsic nerve and hormone stimulation2, but instead is an intrinsic property of the sphincter smooth muscle itself. Smooth muscle force generation results from the cross-bridge movement of myosin and actin filaments on 20-kDa myosin regulatory light-chain phosphorylation (p-RLC)3. The amount of p-RLC is controlled by the balanced activation of Ca2+/calmodulin-dependent MLC kinase (MLCK) and Ca2+-independent MLC phosphatase (MLCP). MLCP consists of three subunits including a regulatory 110–130 kDa subunit, called the myosin-targeting subunit of MLCP (MYPT1), which anchors MLCP to p-RLC. MLCP can be phosphorylated by activation of small GTPase RhoA and Rho-associated, coiled-coil containing serine/threonine kinase (ROCK)4. Based on pharmacological and biochemical evidence, it has been suggested that a lower activity of MLCP as a result of ROCK-mediated phosphorylation of pThr696-MYPT1 may be responsible for the basal tone in IAS (refs 5, 6, 7).
In this study, we use smooth-muscle-specific MYPT1 knockout mice to directly test this hypothesis. We find that the basal tone of IAS from the knockout mice is the same as that from wild-type mice. We, therefore, also test a new hypothesis that Ca2+-mediated MLCK activation is required for the IAS basal tone. We find that the basal tone in IAS from MLCK knockout mice is essentially abolished and these mice give rise to larger faeces, a sign of impaired faecal continence. By directly examining Ca2+ signals and ion channel activity, we further find that Ca2+-releasing ryanodine receptors/channels (RyRs), TMEM16A Ca2+-activated Cl− (ClCa) channels and L-type voltage-dependent Ca2+ channels (VDCCs) form a module which generates a global rise in Ca2+, and that pharmacologically altering any one of the three channels can severely impair IAS basal tone (to the same degree as MLCK deletion). Moreover, genetic deletion of TMEM16A in IAS smooth muscle cells (IAS-SMCs) severely impairs both the RyR-mediated Ca2+ rise and the basal tone, and results in wider and longer faeces. Our results hence demonstrate that MLCK activation by a RyR-TMEM16A ClCa channel-L-type VDCC signalling cascade in the IAS-SMCs is required for basal tone formation and maintenance, and is essential for faecal continence.
Results
MLCK is required for basal tone and evoked contraction in IAS
IAS is a phenotypical sphincter consisting mainly of circular smooth muscle. To assay the basal tone, we employed a standard protocol8,9 in which excised mouse IAS strips had a 0.5-g ‘load' applied to them. In response to this load, the IAS gradually generated force (Fig. 1a, black trace; Supplementary Fig. 1 for the tone measurement). The slow development of the tone may reflect the unique smooth muscle arrangement in IAS, that is, it is divided into ‘minibundles' separated by connective tissue septa without electrical couplings10,11, so that the IAS is not readily synchronized as a whole. To directly examine the role of MYPT1 in the IAS basal tone5,6,7, we assessed the effect of MYPT1 deletion on this tone using MYPT1 knockout mice12. The development (time to 50% of the plateau) of the IAS basal tone was no different between MYPT1 knockout mice (8.4±0.62 min, n=12) and their littermate controls (10±0.97 min, n=10; P>0.05, by two-tailed Student's t-test), neither was the amplitude of the tone (MYPT1 knockout: 0.24±0.03 g, n=15; control: 0.22±0.02 g, n=13, P>0.05 by two-tailed Student's t-test) (Supplementary Fig. 2). Our genetic evidence thus indicates that p-RLC regulated by MYPT1-mediated MLCP activity may be not necessary for basal tone generation. This implies that p-RLC, regulated by MLCK, may be a determinant of IAS basal tone.
Figure 1. MLCK and RLC phosphorylation are required for the basal tone in IAS.

(a) Time courses of changes in force after application of 0.5 g tension in CTR and MlckSMKO mice. (b) Summarized data showing much smaller IAS tone in MlckSMKO mice than in control (mean±s.e.m., CTR n=7, MlckSMKO n=5, ****P<0.0001 by two-tailed Student's t-test). (c) Faeces from CTR mice and MlckSMKO mice after 20 days of tamoxifen treatment. (d) The length (left) and width (right) of faeces were increased in MlckSMKO mice compared with the controls. Bars represent mean±s.e.m., n=20, ****P<0.0001 by two-tailed Student's t-test. (e) Examples of RLC phosphorylation during the process of spontaneous tone generation in IAS from CTR and MlckSMKO mice. (f) Quantification of p-RLC during basal tone generation in CTR and MlckSMKO IAS. Bars represent mean±s.e.m., n=3, *P<0.05, **P<0.01, ***P<0.001 by two-tailed Student's t-test.
To directly test whether MLCK is required for IAS basal tone formation, we analysed the tone generation and contractile properties of IAS from tamoxifen inducible MLCK-deficient mice (MlckSMKO)13. We found that 20 days after tamoxifen injection14, MLCK in IAS from MlckSMKO mice was reduced by 70–98%. For the phenotypic analysis, other than where noted, only the IAS tissue in which MLCK was decreased by >95% was used (Supplementary Fig. 3a). But, as shown in Fig. 1a,b, MLCK deletion completely inhibited the basal tone (MlckSMKO: 0.07±0.01 g; control (CTR): 0.29 ±0.02 g, P<0.0001 by two-tailed Student's t-test; Fig. 1b). Moreover, after the 18th day post tamoxifen induction, mouse faeces became softer and larger. At the 20th day, the length of the faeces from MlckSMKO mice increased from 5.4±0.1 to 11.3±0.8 mm (P<0.0001 by two-tailed Student's t-test, n=20), and the diameter increased from 2.1±0.1 to 2.7±0.1 mm (P<0.0001 by two-tailed Student's t-test, n=20) (Fig. 1c,d). These results suggest that MLCK deletion abolishes the basal tone, likely resulting in weaker compacting capability and slight rectoanal incontinence15,16.
To determine whether the suppression of the IAS basal tone in MlckSMKO mice was due to a decrease in regulatory light-chain (RLC) phosphorylation, we measured p-RLC in MlckSMKO mice and their controls. As shown in Fig. 1e,f, IAS muscle strips from the controls generated phosphorylated myosin light chain during tone development. Although the time course of p-RLC level was not strictly correlated with the time course of basal tone, p-RLC level was always above its basal level during tone development and maintenance. Significantly p-RLC in IAS from MlckSMKO was not detectable during the same time frame.
It is known that MLCK is required for phasic smooth muscle contraction induced by contractile agonists13. To determine whether this is also the case in sphincter smooth muscle, we examined the dependency of agonist-induced contraction on MLCK and RLC phosphorylation. On stimulation with KCl and bethanechol , IAS smooth muscle from the control mice displayed a robust contraction with prolonged tension (Supplementary Fig. 3b), whereas IAS from MlckSMKO mice developed much weaker contraction (26 and 28% of the controls with KCl and bethanechol, respectively) (Supplementary Fig. 3b,c). Moreover, the IAS from the MlckSMKO mice showed a significantly lower level of RLC phosphorylation at different time points on stimulation with KCl or bethanechol (Supplementary Fig. 3d,e).
The decreases in both the basal tone and the evoked contraction in the MLCK-deficient IAS cells were not due to a structural change in IAS because with standard H&E staining, no apparent changes in structure or cell morphology in IAS from these MlckSMKO mice were detected (Supplementary Fig. 4). Neither could they be explained by compensatory changes in the cyclic guanosine monophosphate (cGMP)/cGMP-dependent kinase (PKG), protein kinase C (PKC) and RhoA/ROCK signalling pathways because, as shown in Supplementary Fig. 5, the expressions of soluble guanylate cyclase (sGC), PKG, integrin-linked kinase (ILK), PKC and CPI-17 were not changed significantly in IAS between MlckSMKO mice and control mice. Together, the above results demonstrate that both the generation of basal tone and evoked contraction in IAS requires MLCK activation and RLC phosphorylation.
Ion channel activation is required for basal tone in IAS
The requirement of MLCK activation in the basal tone formation in IAS prompted us to search for the Ca2+ signalling mechanism essential for this tone. Given the myogenic nature of the basal tone and the autonomic activation of ryanodine receptors (RyRs) at resting Ca2+ concentration ([Ca2+]i), we hypothesized that RyRs could be a critical component of this essential Ca2+ signal. To examine this possibility, we investigated the effects of (RyR inhibitor) ryanodine and (RyR agonist) caffeine on IAS basal tone. As shown in Fig. 2, ryanodine (100 μM) fully blocked the tone (Fig. 2a,c). On the other hand, caffeine (3 mM) increased the maximal level of the basal tone ∼68% over the control (P<0.05 by paired two-tailed Student's t-test; Fig. 2b,c). These results indicate that RyRs alone, or in conjunction with other molecules, can generate the calcium signal essential for basal tone formation in IAS. L-type VDCCs blockers reverse the IAS basal tone of various animals9,17. We confirmed this in mice since (1) mouse IAS-SMCs exhibit typical L-type Ca2+ currents, which can be blocked by nifedipine, a specific L-type VDCC blocker (Supplementary Fig. 6); (2) nifedipine fully inhibited the basal tone, and FPL64176, a specific L-type VDCC agonist,18,19 enhanced the tone (Fig. 2d; Supplementary Fig. 7e,g). The above results concerning RyRs and VDCCs raise a possibility that RyR-mediated Ca2+ events functionally couple with L-type VDCCs to control the basal tone. In several smooth muscles, ClCa channels act as the mediator coupling RyR and L-type VDCC activity, that is, a local or global rise in Ca2+ due to RyR opening activates ClCa channels, which depolarizes the membrane and turns on L-type VDCCs (refs 20, 21, 22). To investigate whether this mechanism underlies the IAS tone formation, we examined changes in the tone by 16Ainh-A01 (a specific blocker of TMEM16A ClCa channels23) and niflumic acid (a non-specific blocker of ClCa channels24). We found that niflumic acid and 16Ainh-A01 dose-dependently reversed the basal tone (Supplementary Fig. 7a–d). Niflumic acid at 100 μM and 16Ainh-A01 at 10 μM fully reversed the basal tone (Fig. 2d; Supplementary Fig. 7a–d), similar to the effect of MLCK deletion. Moreover, Eact, a newly developed activator of the TMEM16A ClCa channel25, potentiated the basal tone (Fig. 2d; Supplementary Fig. 7f). Since the inhibitors for RyRs, the ClCa channels and L-type VDCCs can all fully reverse the basal tone while their agonists can potentiate the tone, we propose that these three channels form a signalling module which sets this tone in IAS.
Figure 2. RyRs, ClCachannels and VDCCs are essential for the basal tone in IAS.

(a) Ryanodine (100 μM) significantly decreased the spontaneous tone in IAS. Bars represent mean±s.e.m., control n=5, ryanodine n=6, ****P<0.0001 by analysis of variance (ANOVA) comparing the sustained phases. (b) Caffeine (3 mM) increased the tone in IAS. Bars represent mean±s.e.m., n=7, ****P<0.0001 by ANOVA comparing the sustained phases. (c) Summarized results on the IAS tone affected by 100μM ryanodine (n=6), 3 mM caffeine (n=8). Bars depict mean±s.e.m., *P<0.05, **P<0.01 by paired two-tailed Student's t-test. (d) Summarized results on the IAS tone affected by 100 μM niflumic acid (NFA; n=8), 10 μM 16Ainh-A01 (n=4), 30 μM Eact (n=5), 1 μM nifedipine (n=8) and 0.1 μM FPL64176 (n=5). Bars depict mean±s.e.m., *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 by paired two-tailed Student's t-test.
A RyR-mediated rise in global [Ca2+]i contracts IAS-SMCs
Having established that the RyR-ClCa channel-VDCC module controls the basal tone of IAS, we next studied the nature of the Ca2+ signalling generated by activation of these channels in IAS-SMCs. Since RyRs generate spontaneous Ca2+ sparks in a variety of smooth muscle cells26,27,28, these Ca2+ events may directly regulate IAS smooth muscle contraction. To test this possibility, we examined the relationship between Ca2+ sparks and cell length using isolated single IAS-SMCs. We found that freshly isolated IAS-SMCs generated spontaneous localized Ca2+ events (Fig. 3a) at a frequency of 1.4±0.2 Hz (n=36; Fig. 3b). These events can be classified as Ca2+ sparks because caffeine (180 μM) increased their activity by almost eightfold (n=10) while ryanodine at 100 μM suppressed them by 62%±22 (n=4) (Fig. 3b). Interestingly, as shown in Fig. 3c, there was an inverse relationship between the spark frequency and cell length, that is, the higher the frequency, the shorter the cell length. This inverse relationship raises a possibility that Ca2+ sparks may be a causal signal for cell shortening. If this is the case, we should observe that an increase in Ca2+ spark activity by other means shortens IAS-SMCs. To assess this possibility, we used caffeine to evoke Ca2+ sparks. In IAS-SMCs, caffeine elicited Ca2+ sparks at a concentration as low as 10 μM, which is about one-tenth the concentration needed in other types of smooth muscle cells27 (Supplementary Fig. 8). Interestingly, caffeine at concentrations that only increase Ca2+ sparks did not induce the shortening of cells with different original lengths (Fig. 3d). Only at the level which caused both a burst of Ca2+ sparks and a rise in global [Ca2+] did caffeine cause cell shortening (Fig. 3e). This caffeine-induced rise in global [Ca2+]i was mediated by RyR since ryanodine could essentially block the Ca2+ response evoked by caffeine (Fig. 3f). These results indicate that a RyR-mediated rise in global [Ca2+]i, and not the RyR-mediated Ca2+ sparks, regulate the shortening of IAS-SMCs.
Figure 3. RyR-mediated global [Ca2+]i, but not Ca2+ sparks, contracts IAS-SMCs.

(a) Top panels show the spatiotemporal evolution of a single Ca2+ spark in an IAS-SMC, and the Roman numerals correspond to the time marked on the trace of time course of ΔF/F0 (%) (a proxy for the change in [Ca2+]i at the epicentre pixel (333 × 333 nm) of this spark. (b) Effects of caffeine (180 μM, red bars) and ryanodine (100 μM, blue bars) on Ca2+ sparks. Bars represent mean±s.e.m., n=20 cells for caffeine, and n=4 cells for ryanodine. *P<0.05; **P<0.01 by two-tailed Student's t-test. (c) Calcium spark frequency was inversely correlated with cell length (Pearson correlation coefficient: −0.8398, P<0.0001 by two-tailed Student's t-test; n=36). (d) Ca2+ spark activity was increased by caffeine (30, 200 or 500 μM) but did not associate with any detectable shortening (upper panel) of cells with different initial lengths (lower panel; Pearson correlation coefficient: 0.3409, P>0.05 by two-tailed Student's t-test; n=19). (e) An increase in global [Ca2+]i by 1 or 10 mM caffeine did cause cell shortening by 27.0±3.4% on average (P<0.01 by the Student's t-test; n=15) although no correlation exists between the magnitude of the caffeine-induced global [Ca2+]i and amount of cell shortening (Pearson correlation coefficient: 0.1596, P>0.05 by the Student's t-test; n=15). (f) Ryanodine (100 μM) blocked the caffeine-induced calcium increase. The bar chart on the right is the mean±s.e.m. Per cent inhibition by ryanodine for six cells.
ClCa channels and VDCCs contribute to the RyR-mediated global [Ca2+]i
Since RyRs, ClCa channels and L-type VDCCs form a signalling module to control the basal tone (Fig. 2), and a RyR-mediated rise in global [Ca2+]i is required to induce shortening of IAS-SMCs (Fig. 3), we asked whether ClCa channels and L-type VDCCs contribute to the RyR-mediated rise in global [Ca2+]i. In the absence of extracellular Ca2+, caffeine increased [Ca2+]i to a smaller extent (ΔF/F0: 108±23%, n=10) than in the presence of extracellular Ca2+ ((ΔF/F0: 159±28%, n=12)), indicating that in addition to Ca2+ release from ryanodine-sensitive Ca2+ stores, caffeine also induced Ca2+ influx. To determine the potential roles of the ClCa channel and L-type VDCCs in this Ca2+ influx we compared the caffeine-induced Ca2+ rise in control cells with the rise produced when modulators of ClCa channels or L-type VDCCs were present. Figure 4a shows that niflumic acid (100 μM), 16Ainh-A01 (10 μM) and nifedipine (1 μM) inhibited the caffeine-induced increase in global [Ca2+]i by 33, 35 and 74.0%, respectively. Eact (1μM) and FPL64176 (1 μM) enhanced the caffeine-induced rise in [Ca2+]i by 28 and 20%, respectively. These results demonstrated that activation of RyRs can turn on ClCa channels and L-type VDCCs, resulting in a global rise in [Ca2+]i.
Figure 4. RyRs, ClCa channels and VDCCs form a signaling module in IAS-SMCs.

(a) Niflumic acid (100 μM), 16Ainh-A01 (10 μM) and nifedipine (1 μM) partially inhibited, while Eact (30 μM) and FPL64176 (0.1 μM) potentiated, a caffeine (1 mM)-induced calcium increase. Data were calculated as the ratio of increase in Ca2+ by a second caffeine pulse over the first caffeine pulse. For each group, cells were pretreated with the modulator for ∼3 min before the application of the second caffeine pulse. Bars represent mean±s.e.m., n=3–8, *P<0.05; **P<0.01 versus caffeine alone group by two-tailed Student's t-test. (b) Individual sparks do not activate any Cl− currents. Top panel shows the spatiotemporal evolution of two Ca2+ sparks (sites marked 1 and 3) in an IAS-SMC, below it is the associated time course for site 3 calculated as ΔF/F0 (%), that is, the change in [Ca2+]i from the spark's epicentre pixel, 330 × 330 nm, divided by its value from time 0. Images are displaying ΔF/F0 (%). Roman numerals on the time course are the time points of the four images on the top panel from site 3. Sites 2 and 4 displayed sparks at times not shown in this analysis. The lowest panel is the membrane current associated with these Ca2+ sparks. Note that none of the events is associated with membrane current changes, that is, no STICs (that is, spontaneous transient inward currents) were evoked by Ca2+ sparks. (c) Caffeine (10 mM) increased global Ca2+ and ClCa currents. The currents reversed at ECl (∼0 mV), which indicates that the currents detected at −70 and +40 mV are due to the opening of ClCa channels. (d) Summarized results for the 10 mM caffeine-induced global Ca2+ increases and their corresponding ClCa currents at −70, 0 and +40 mV. Bars represent mean±s.e.m., n=4 cells, one from each of four mice.
A global rise in [Ca2+]i activates ClCa currents in IAS-SMCs
To directly examine whether an increase in [Ca2+]i can activate ClCa channels, we examined the relationship between Ca2+ signals and ClCa currents by simultaneously measuring [Ca2+]i with high- speed imaging and membrane currents with the patch-clamp technique28. In several types of smooth muscle cells, Ca2+ sparks activate ClCa channels to generate spontaneous transient inward currents (STICs)29. To our surprise, in mouse IAS-SMCs, Ca2+ sparks did not associate with any detectable currents when the membrane was clamped at voltages more negative than the reversal potential for Cl− (Fig. 4b, n=5), indicating they do not activate STICs. We then assessed whether an increase in global [Ca2+]i could elicit ClCa currents. To raise global [Ca2+]i, we stimulated the cells with caffeine (10 mM) and recorded the membrane current at different voltages. As shown in Fig. 4c, on stimulation with caffeine, the IAS-SMC generated an inward current when held at −70 mV. This inward current was determined to result from the opening of ClCa channels, because at the reversal potential for Cl− (that is, 0 mV), the same cell failed to generate current in response to the same caffeine stimulation. At +40 mV, the cell produced a markedly outward current, as would be predicted if this current was a Cl− current. Finally, caffeine raised the global [Ca2+]i to the same level at three different holding potentials (Fig. 4c,d), indicating the difference in Cl− currents at different holding potentials is not due to the variation in [Ca2+]i. The cells that generated these Cl− currents are authentic smooth muscle cells as the IAS cells with positive green fluorescent protein (GFP) from α-smooth muscle actin-GFP mice produced similar currents in response to caffeine (Supplementary Fig. 9). In conclusion, these results argue that a rise in global [Ca2+]i via RyR activation can activate ClCa channels in IAS-SMCs.
Ion channel expression in IAS-SMCs
It is well established that RyRs underlie Ca2+ sparks, and Cav1 encodes L-type VDCCs in smooth muscle. PCR with reverse transcription (RT–PCR) detected three types of Ryrs with the dominant expression of Ryr1 (Supplementary Fig. 10a), and of Cav1 (that is, Cav1.1, Cav1.2 and Cav1.3; Supplementary Fig. 10b) in IAS-SMCs. The TMEM16 family (that is, TMEM16A and TMEM16B) was recently found to function as ClCa channels in several cell types30,31,32,33,34,35. We therefore decided to determine whether TMEM16A and/or TMEM16B are also expressed in IAS cells. With RT–PCR, we detected the transcripts of Tmem16a (Supplementary Fig. 10c). Interestingly, we failed to detect Tmem16b in IAS tissue (Supplementary Fig. 10d). An immunohistochemistry assay showed the colocalization between TMEM16A and Myh11, a specific smooth muscle marker, in IAS-SMCs, particularly in its inner layer (Supplementary Fig. 10e).
Knockout of TMEM16A impairs the basal tone in IAS
If a RyR-TMEM16A-Cav1 module is critical for the basal tone formation, genetic interruption of one of the members in this module could impair, or even abolish, the tone. Since IAS tissues express multiple RyRs and Cav1s, but only TMEM16A ClCa channels, we tested this prediction by generating smooth-muscle-specific TMEM16A knockout mice. A conditional knockout of this gene is necessary because global deletion of TMEM16A causes post-natal lethality36, which makes force measurement in IAS quite a challenge. Supplementary Fig. 11a depicts the schematic of the strategy used to produce smooth-muscle-specific knockout mice with Tmem16a gene deletion, in which exon 12 was floxed with two loxP sites. That homologous recombination occurred in the floxed mice was confirmed with Southern blot analysis (Supplementary Fig. 11b). To delete TMEM16A specifically in smooth muscle, we crossed Tmem16a floxed mice with SMA-Cre transgenic mice12. The resultant mice Tmem16aflox/flox, SMA-Cre (that is, Tmem16aSMKO) were used as the KO mice, while Tmem16aflox/+, SMA-Cre littermates were used as control mice (CTR). Birth of Tmem16aSMKO and CTR pups occurred in the expected Mendelian ratio. Tmem16aSMKO mice were fertile, viable and lacked apparent developmental defects. Western blot demonstrated that TMEM16A in the KO IAS tissues was significantly decreased to ∼20% of the control levels (Supplementary Fig. 11c). The residual TMEM16A is most likely from other types of cells (for example, interstitial cells of Cajal (ICCs); Supplementary Fig. 12) in the IAS. To determine the changes in ClCa currents due to TMEM16A deletion in IAS-SMCs, we compared these currents in cells from CTR and Tmem16aSMKO mice. In the CTR cells, caffeine at 10 mM generated a ClCa current of −1.67±0.37 pA pF−1 at the holding potential of −70 mV (n=12), while in the Tmem16a−/− IAS-SMCs, the same concentration of caffeine yielded essentially no ClCa current (−0.05±0.01 pA pF−1; n=14; Fig. 5a). To further characterize ClCa currents in these cells, the IAS cells from both the KO and CTR were dialyzed with 600 nM [Ca2+]i via the patch pipette. Out of 29 control cells, 6 cells produced a current of 36.54±6.18 pA pF−1 (n=6) at +100 mV and showed an outward rectification and time-dependent activation at positive potentials (Supplementary Fig. 13); In the Tmem16aSMKO cells, one cell out of 29 had about one-half that current (16.54 pA pF−1) at +100 mV, and the other 28 cells had markedly reduced currents with 6.31±0.30 pA pF−1 (n=28) at the same potential (Supplementary Fig. 13). These results confirm that Tmem16a encodes ClCa channels in smooth muscle cells34,37,38,39,40. They further reveal that some of cytosolic factors that bind with TMEM16A (ref. 41) may be required to prevent TMEM16A from its rundown, a characteristic often observed in ClCa currents from native smooth muscle cells and other cell types42,43.
Figure 5. TMEM16A deletion impairs IAS smooth muscle function and faecal defecation.

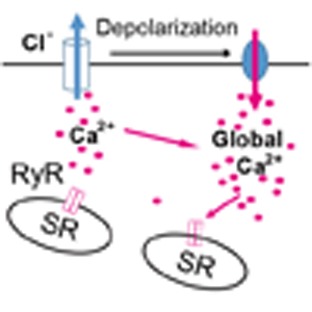
(a) Caffeine (10 mM)-induced ClCa currents at the holding potential of −70 mV in IAS-SMCs from control and Tmem16aSMKO mice. ****P<0.0001 KO versus CTR by two-tailed Student's t-test, n=14 for CTR and n=12 for KO (An additional five cells did not generated detectable ClCa currents and were excluded in this calculation). (b) Effects of ClCa channel and L-type Ca2+ channel modulators on caffeine-induced Ca2+ rise in IAS-SMCs from Tmem16aSMKO mice. Experiments were carried out with a protocol as described in Fig. 4a. n=7–10 cells. (c) Basal tone in Tmem16aSMKO mice is significantly reduced compared with CTR mice. Bars represent mean±s.e.m., CTR (n=5) versus KO (n=9), ****P<0.0001 by analysis of variance (ANOVA) comparing the sustained phases. (d) Faeces sizes in CTR and Tmem16aSMKO mice. *P<0.05; **P<0.01 by two-tailed Student's t-test, n=8 for each group. (e) A model of the molecular basis of basal tone generation in IAS. See text for the details. CaM/MLCK, camodulin/myosin light-chain kinase; p-RLC, phosphorylated 20-kDa myosin RLC; PM, plasma membrane; RyR, ryanodine receptor; SR, sarcoplasmic reticulum; VDCC, voltage-dependent calcium channel.
We further examined potential functional changes in IAS as a result of TMEM16A deletion in IAS-SMCs. We first studied the effect of TMEM16A modulators on caffeine-induced Ca2+ signals in the Tmem16aSMKO cells. We found that both 16Ainh-A01 and Eact did not decrease caffeine-induced Ca2+ release, nor did nifedipine and FPL64176 affect caffeine-induced Ca2+ release (Fig. 5b). We next compared the basal tone of IAS in the CTR and Tmem16aSMKO mice. As shown in Fig. 5c, TMEM16A deficient IAS tissue produced ∼50% of the tone compared with the CTR IAS. We finally determined whether TMEM16A deletion in IAS-SMCs affect faecal continence by comparing the size of faeces from CTR and Tmem16aSMKO mice (Fig. 5d). We found that faeces from Tmem16aSMKO were both longer and wider (length: 5.4±0.26 mm in CTR versus 7.2±0.3 mm in Tmem16aSMKO; n=7 for each group, P<0.01 by two-tailed Student's t-test; width: 1.9±0.1 mm in CTR versus 2.3±0.1 mm in Tmem16aSMKO, n=7 for each group, P<0.05 by the t-test). These results indicate that TMEM16A is required for ClCa currents in IAS-SMCs, and its deficiency impairs IAS basal tone formation and faecal continence.
Discussion
Our results support a model in which RyRs, TMEM16A ClCa channels and L-type VDCCs in IAS-SMCs form a signalling module to regulate global [Ca2+]i, which activates MLCK and sets IAS basal tone (Fig. 5e). This model is supported by several lines of evidence. First, genetic deletion of MLCK or TMEM16A specifically in IAS-SMCs abolishes or severely impairs the basal tone, leading to faecal impairment. Second, pharmacological activation of RyRs, TMEM16A or L-type Ca2+ channels increases the tone, while their blockage suppresses it. Third, the RyR-mediated increase in global [Ca2+]i induced by caffeine is enhanced by TMEM16A agonist Eact and L-type Ca2+ channel agonist FPL64176, and inhibited by TMEM16A antagonist 16Ainh-A01 and L-type Ca2+ channel blocker nifedipine. Fourth, the aforementioned effects by the modulators of TMEM16A and L-type Ca2+ channels are abolished when TMEM16A in IAS-SMCs is deleted. Finally, a global increase in [Ca2+]i activates ClCa channels, as directly recorded by patch clamp. Our model provides a molecular explanation for a long-standing notion that the basal tone in IAS is intrinsic to smooth muscle and independent of external stimuli1,2.
In our model, calcium release from the opening of RyRs in the sarcoplasmic reticulum of IAS-SMCs could be an initial signal for the basal tone generation and maintenance. The increased calcium then activates TMEM16A ClCa channels subsequently activating L-type Ca2+ channels, resulting in Ca2+ influx. Given that TMEM16A has a low-Ca2+ sensitivity with an EC50 at ∼3 μM at −70 mV (refs 31, 44), an unexpected finding is that although Ca2+ sparks, a phenotypical localized Ca2+ release event, are present in IAS-SMCs, they do not activate TMEM16A ClCa channels to generate STICs as in other smooth muscle cells26. Instead, a global rise in [Ca2+]i created by activating RyRs is required to activate TMEM16A ClCa channels, which in turn depolarizes the membrane and activates VDCCs, leading to the Ca2+-MLCK signalling cascade. Our results motivate three new questions. The first regards the reasons for the lack of STICs in IAS-SMCs. One possibility is that TMEM16A ClCa channels do not concentrate near enough to Ca2+ spark sites. Since the unitary conductance of TMEM16A ClCa channels is on the order of a few picosiemens, it requires ∼300 of them to localize near a spark site in order for a STIC to be generated. A direct visualization of TMEM16A ClCa channels and RyRs at high spatial resolution will help solve this puzzle. The second question concerns the endogenous signalling molecules that convert local calcium events like Ca2+ sparks to a global rise in [Ca2+]i. One possibility is that applied stretch may activate a diffusible messenger such as cyclic ADP-ribose (cADPR), a potential endogenous ligand for RyR, or a stretch-gated ion channel. In this regard, it is worth mentioning that stretch can activate the non-selective cation channel TRPV4, which is known to activate Ca2+ sparks in vascular smooth muscle45. And the third question raised is the mechanism which terminates RyR-TMEM16A-L-type VDCC signalling. Interestingly, in smooth muscle Ca2+ levels in both the cytosol and the internal Ca2+ store can auto-regulate RyR activity28,46,47, and high Ca2+ in the cytosol inhibits L-type VDCCs and ClCa channels48,49. One or more of these regulators may be able to terminate the RyR-TMEM16A-L-type VDCCs signalling process in IAS-SMCs. The activation and termination of this signalling module also imply that global [Ca2+]i in these cells could be oscillating during basal tone development and maintenance. It would be interesting to examine whether an oscillating [Ca2+]i leads to a low level of RLC phosphorylation during tone generation as revealed in Fig. 1e,f.
Out of two identified ClCa channel genes32,35, we detected the expression of TMEM16A but not TMEM16B in IAS, consistent with the report that TMEM16B is predominantly expressed in the nervous system35. Interestingly, TMEM16A is thought to be absent from smooth muscle cells in the gastrointestinal tract where they generate phasic contractions30. With evidence from electrophysiological recordings, functional assays, RT–PCR and immunofluorescence, we have unambiguously demonstrated that TMEM16A is present and functional in IAS-SMCs. More importantly, using TMEM16A smooth muscle conditional knockout mice, we established that this gene is required for the generation and maintenance of basal tone in IAS. The fact that TMEM16A is expressed in sphincter smooth muscle cells while it is absent in phasic GI smooth muscle cells suggests this gene may be a differentiator for the phenotype of sphincters and phasic muscles. This finding suggests that TMEM16A may be an attractive therapeutic target for IAS motility disorders.
In the TMEM16A deficient mice, the tone was decreased by 51% compared with littermate controls. What accounts for this remaining tone? One possibility is that Cre in a small subset of IAS-SMCs has an insufficient efficiency, resulting in an incomplete deletion of the TMEM16A in these cells. Another possibility could be that activation of RyRs alone (that is, without amplification due to TMEM16A and L-type VDCCs) is sufficient to generate a certain amount of IAS tone. This is supported by our finding that caffeine-induced Ca2+ release is intact in IAS-SMCs from TMEM16A knockout mice (Fig. 5b). Finally the remaining tone could be attributed to the TMEM16A in ICCs in IAS tissues (Supplementary Fig. 12). In some types of smooth muscle a depolarization in ICCs can propagate to smooth muscle cells via gap junctions50. Whether this is the case in IAS-ICCs is not clear as IAS tone and slow waves are similar in control and W/WV mice (that is, ICC depleted mice)51,52. Nevertheless, the presence of TMEM16A in the ICCs in IAS would require the application of an ICC cell specific knockout technique to uncover the role of ICC's TMEM16A in IAS function. Our study shows the power of this technique in addressing the role of TMEM16A in the IAS smooth muscle and so did by Heinze et al. 2014 who also elegantly demonstrated its role in the blood vessel smooth muscle38.
It is interesting that TMEM16A unevenly distributes across IAS smooth muscle, with the highest concentration apparently in the region adjacent to the submucosal space. This pattern of distribution implies that mechanical stretch by faeces may be able to preferentially activate TMEM16A in IAS smooth muscle, making this channel an attractive sensor of anal contents. This is in line with findings that membrane stretch can activate TMEM16A in vascular smooth muscle cells40, perhaps through the actin cytoskeleton53. This role for TMEM16A necessitates gap junctions among IAS-SMCs to propagate the electrical signals initiated in ClCa channels near the submucosal space to the entire tissue. This is certainly possible as gap junctions are expressed abundantly in IAS cells (Supplementary Fig. 14) and other smooth muscle sphincter tissue54,55.
In summary, with an integrative approach combining genetically modified mice, bioassays, molecular biology and electrophysiology, we have uncovered the molecular mechanism underlying the genesis of spontaneous basal tone in IAS. This understanding will facilitate further insight into the pathophysiology of IAS disorders and the therapeutic options for treating them, as well as many other diseases related to smooth muscle sphincters.
Methods
Preparation of IAS tissue
C57BL/6, MYPT1 CTR (Mypt1flox/+; SMA-Cre) and Mypt1SMKO (Mypt1flox/flox; SMA-Cre) mice, TMEM16A CTR (Tmem16aflox/+; SMA-Cre) and Tmem16aSMKO (Tmem16aflox/flox; SMA-Cre) mice at 6–10 weeks were decapitated. MLCK CTR (Mlckflox/+; SM22-CreERT2) and MlckSMKO (Mlckflox/flox; SM22-CreERT2) mice were decapitated 20 days after tamoxifen injection. MYPT1 CTR, Mypt1SMKO, MLCK CTR and MlckSMKO mice were in a C57BL/6 background. TMEM16A CTR and Tmem16aSMKO mutant mice were in a mixed C57BL/6 and Sv/129 background. Both genders of the mice were used equally. The anal canal and an adjacent region of the rectum were quickly removed and transferred to ice-cold and oxygenated Krebs physiological buffer (KPS) which was comprised of (in mM): 118.07 NaCl, 4.69 KCl, 2.52 CaCl2, 1.16 MgSO4, 1.01 NaH2PO4, 25 NaHCO3 and 11.10 glucose. Strained skeletal muscle fibres, mucosal layer and other extraneous tissues (for example, large blood vessels) were carefully dissected away, whereas the anal canal was left intact. The IAS was identified as a thickened circular smooth muscle situated at the lowermost part of anal canal, and the strips of IAS smooth muscle (∼1 × 6 mm) were prepared for experiments.
Animal experiments in this study were conducted in accordance with the guidelines of the Animal Care and Use Committee of Model Animal Research Center of Nanjing University (Nanjing, China), or University of Massachusetts Medical School, Massachusetts, USA.
Measurement of IAS basal tone and contractility
Isolated strips of IAS smooth muscle were transferred to 8 ml muscle baths containing ice-cold oxygenated KPS. One end of the smooth-muscle strip was anchored to the bottom of the muscle bath. The other end of the smooth-muscle strip was connected to a force transducer (MLT0202, AD Instruments) and isometric tension was measured by a PowerLab (AD Instruments) recording device. A wire myograph (610-M, Danish Myo Technology, Aarhus, Denmark) was also used for force measurements. Each smooth-muscle strip was equilibrated for 60 min followed by a 0.5-g load8,9. The basal tone measurement is described in Supplementary Fig. 1. For evoked contractility measurements (that is, Supplementary Fig. 3), the resting tension of the IAS was adjusted to ∼0.2 g after developing spontaneous tone. KPS containing (in mM) 124 KCl, 2.52 CaCl2, 1.16 MgSO4, 1.01 NaH2PO4, 25 NaHCO3 and 11.10 glucose was used to achieve membrane depolarization. Bethanechol (100 μM) was used to induce agonist-induced contraction in IAS. For RyR inhibitor ryanodine experiments, IAS strips were pretreated with 100 μM ryanodine for 5 min before giving the initial stretch. For RyR agonist caffeine experiments, 3 mM caffeine was added into the bath 8 min after the initial stretch. Both ryanodine and caffeine were present through the experiments after administration. For L-type VDCC modulators (nifedipine and FPL64176) and ClCa channel modulators (Eact, niflumic acid or 16Ainh-A01), experiments were performed in the presence of 1 μM atropine and 10 μM guanethidine to eliminate the possible effects of cholinergic and sympathetic nerves on the basal tone.
Generation of Tmem16a knockout mice (Tmem16a SMKO )
Bacterial artificial chromosome retrieval methods were used for constructing the TMEM16A targeting vector56. Briefly, the Tmem16a locus including Exon 12 was retrieved from a 129/sv bacterial artificial chromosome clone (bMQ 379h21, provided by the Sanger Institute) by a retrieval vector containing two homologous arms. Exon 12 which encodes a partial transcript in the second trans-member domain was floxed by 2 loxP sites, and an frt-Neo-frt cassette was inserted as a positive selection marker (Supplementary Fig. 11a). The deletion of this domain causes an out-of-frame reading shift and thereby generates a premature stop codon and a loss-of-function allele36. Embryonic stem W4 cells were electroporated with the linearized targeting vector, selected by long-fragment PCR and Southern blot analysis. Chimeric mice were generated by injecting homologous recombined embryonic stem cells into the blastocysts of C57BL/6 mice. Floxed Tmem16a mice with germ-line transmission were further confirmed by genotyping analysis and Southern blot analysis. To generate smooth-muscle-specific Tmem16a knockout mice, floxed Tmem16a mice were crossed with SMA-Cre mice.
Isolation of mouse IAS-SMCs
Mice as listed above from 6 to 10 weeks of age of both genders were decapitated or euthanized with CO2. IAS was quickly removed and placed in a pre-chilled dissociation solution consisting of (in mM): 135 NaCl, 6 KCl, 5 MgCl2, 0.1 CaCl2, 0.2 EDTA, 10 Hepes and 10 glucose (pH 7.3). The tissue was then incubated in the dissociation medium containing 30 unit per ml papain, 1 mM DTT and 0.5 mg ml−1 BSA, at 35 °C for 30 min. The tissue was then transferred to a dissociation medium containing 3 unit per ml collagenase F and 0.5 mg ml−1 BSA, and incubated at 35 °C for another 15 min to produce isolated IAS cells. Finally, the tissue was agitated with a fire polished wide-bore glass pipette to release the cells.
Western blot assay
IAS smooth muscle strips were homogenated in 120 μl lysis buffer (containing 20 mM Tris base, 137 mM NaCl, 2 mM EDTA, 1% NP-40, 10% glycerol, protease inhibitor cocktail (Roche), pH=8). Then the homogenization was incubated on ice for 15 min, and centrifugation at 3,000g, 4 °C to remove cell debris. Protein concentration was measured with a bicinchoinic acid protein assay kit (Pierce). The proteins were denatured at 95 °C for 5 min with sample buffer and reducing agent (5 × sample buffer contains 10% SDS, 20% glycerol, 0.05% bromophenol blue 10 mM β-mercapto-ethanol, 200 mM Tris-HCl and 8M urea). General SDS-page processes were followed. The blots were visualized by using the enhanced chemiluminescence method, the Super Signal West Dura substrate (PIERCE) and MaxiSignal Western Solution (SUDGEN) were used. All the primary antibodies used in western blot assay are listed in Supplementary Table 1, and the full scans of western blots are available in Supplementary Figs 15–17.
Immunohistochemical analyses
Anal tubes were isolated and fixed in ice-cold acetone for 10 min, then washed with PBS overnight at 4 °C and rewashed for 4 h with a change of PBS per hour. Cryosections with a 10-μm thickness were used. The non-specific binding of primary antibodies was blocked by incubation with PBS containing 1% BSA and 5% non-immune goat serum for 1 h. Incubation was carried out overnight at 4 °C with a rabbit polyclonal antibody to TMEM16A (ab53212, abcam) diluted 1:200, together with a rat anti-c-Kit antibody (ACK2, Chemicon, 1:100) or mouse monoclonal Myh11 antibody (ab683 clone 1G12, 1:200; abcam). After washing in PBST, sections were incubated with a Alexa Fluor 555-conjugated goat anti-Rabbit antibody (Sigma) diluted 1:250 and a FITC-conjugated goat anti-rat antibody (Invitrogen) diluted 1:250 or a Alexa Fluor 488–conjugated goat anti-mouse antibody (Cell signaling technology) for 1 h. 4,6-diamidino-2-phenylindole was used for nuclear staining. Immunoreactivity was evaluated using a FV1000 confocal laser scanning microscope system (Olympus).
Patch-clamp recording
Membrane currents were recorded using the perforated whole-cell patch recording configuration. The extracellular solution contained (in mM): 130 NaCl, 5.5 TEA-Cl, 2.2 CaCl2, 1 MgCl2, 10 Hepes and 5.6 glucose; pH adjusted to 7.4 with NaOH. The pipette solution contained (in mM): 137 CsCl, 1 MgCl2, 10 Hepes, 3 Na2ATP; pH adjusted to 7.3 with CsOH. Amphotericin B was freshly made and added to the pipette solution at a final concentration of 200 μg ml−1. Whole-cell currents were recorded when cells were held at the designated potentials and the currents were low-pass filtered using the Axopatch 1D amplifier (200 Hz cutoff) and then digitally sampled at 1 kHz and stored for analysis. Caffeine was applied to the cells via a puffing pipette placed ∼100 μm from the cells under the control of a picospritzer.
For recording of 600 nM Ca2+-induced currents, conventional whole-cell patch clamp was carried out. The extracellular solution contained (in mM) 144.5 NaCl, 5.5 TEA-Cl, 1 CaCl2, 1 MgCl2, 10 glucose, 10 mannitol and 10 Hepes; pH was adjusted to 7.4 with NaOH. The pipette solution contained (in mM) 130 CsCl, 10 EGTA, 1 MgCl2, 8 CaCl2, 10 Hepes, 1 MgATP; pH was adjusted to 7.3 with NaOH. Whole-cell currents were recorded in response to 1 s voltage pulses from −80 to +100 mV in 10 mV increments followed by 700 ms pulses to −60 mV, in freshly isolated IAS-SMCs. Holding potential was 0 mV. Currents were sampled at 20 kHz using the Axon MultiClamp 700B amplifier (Molecular Devices) and then low-pass filtered at 2 kHz.
For recording L-type VDCCs in IAS-SMCs, conventional whole-cell patch clamp was carried with extracellular solution containing (in mM) 130 TEA-Cl, 10 CaCl2, 1 MgCl2, 10 Hepes, 10 glucose; pH was adjusted to 7.4 with NaOH. The pipette solution contained (in mM) 130 CsCl, 4 MgCl2, 10 Hepes, 10 EGTA, 5 Na2ATP; pH was adjusted to 7.2 with CsOH. Cells were held at −80 mV, whole-cell currents were recorded in response to 200 ms voltage pulses from −80 to +50 mV in 10 mV increments. Freshly isolated IAS-SMCs were incubated in extracelluar solution with 1 μM Nifedipine or vehicle (0.1% EtOH) during recording. Currents were sampled at 20 kHz using the Axon MultiClamp 700B amplifier (Molecular Devices) and then low-pass filtered at 2 kHz.
Imaging and measurement of Ca2+ sparks
Fluorescent images were obtained using fluo-3 as the Ca2+ indicator and a custom-built wide- field, high-speed digital imaging system57. Rapid imaging was made possible by using a cooled high-sensitivity charge-coupled device camera (128 × 128 pixels) developed in conjunction with the Massachusetts Institute of Technology Lincoln Laboratory. The camera was interfaced to a custom-made inverted microscope equipped with a × 60 oil immersion lens (numerical aperture of 1.3), with each pixel covering a 333 × 333-nm area of the cell. The 488-nm line of a multiline argon laser provided fluorescence excitation for the indicator fluo-3, and a laser shutter controlled the exposure duration. Emission of the Ca2+ indicator was monitored at wavelengths >510 nm. Ca2+ sparks were measured as the conventional fluorescence ratio (ΔF/F0) within a restricted volume as described previously57.
Reverse transcription PCR detection of messenger RNA
The IAS and rectal smooth muscle (4 mm above IAS) from mice were carefully isolated, quickly removed from connective tissue and then frozen in liquid nitrogen. The total RNA was isolated using the TRIZOL (Invitrogen) method according to the manufacturer's guidelines, and cDNA was synthesized. Primers were synthesized by Invitrogen, and their sequences are available in Supplementary Table 2.
Statistical analysis
Data are presented as the mean±s.e.m. Differences between groups were determined by Student's t-test, or two-way analysis of variance (ANOVA) for significant differences. The significance levels were indicated as follows: NS P>0.05, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Additional information
How to cite this article: Zhang, C.-H. et al. The molecular basis of the genesis of basal tone in internal anal sphincter. Nat. Commun. 7:11358 doi: 10.1038/ncomms11358 (2016).
Supplementary Material
Supplementary Figures 1-17, Supplementary Tables 1-2 and Supplementary References
Acknowledgments
This work was supported by the National Natural Science Funding of China (31272311 and 31330034 to M.-S.Z.), National Key Scientific Research Program of China (2014CB964701 to M.-S.Z.) and US National Institute of Health grants NIDDK098586, and HL117104 (to R.Z.G.).
Footnotes
Author contributions C.-H.Z., R.Z.G. and M.-S.Z. designed research; C.-H.Z., P.W., W.Z., X.C., D.-H.L., P.L., K.Z., R.Z.G. and C.C. performed research; P.W. generated Tmem16a knockout mice; W.-Q.H. and Y.-J.P. generated MLCK and MYPT1 knockout mice; C.-P.C., Y.-N.Q., T.T., L.M.L., S.M.C., J.F.K., K.E.F. and J.S. contributed new reagents/analytic tools; C.-H.Z., P.W., D.-H.L., R.Z.G. and M.-S.Z. analysed data; C.-H.Z., M.-S.Z. and R.Z.G. wrote the paper.
References
- Vander A., Sherman J. & Luciano D. Human Physiology: The Mechanisms of Body Function 6th edn, International Edition McGraw-Hill international editions (1994) . [Google Scholar]
- Rattan S. The internal anal sphincter: regulation of smooth muscle tone and relaxation. Neurogastroenterol. Motil. 17, 50–59 (2005) . [DOI] [PubMed] [Google Scholar]
- Murthy K. S. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 68, 345–374 (2006) . [DOI] [PubMed] [Google Scholar]
- Somlyo A. P. & Somlyo A. V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 83, 1325–1358 (2003) . [DOI] [PubMed] [Google Scholar]
- Rattan S. & Singh J. RhoA/ROCK pathway is the major molecular determinant of basal tone in intact human internal anal sphincter. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G664–G675 (2012) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel C. A. & Rattan S. Cellular regulation of basal tone in internal anal sphincter smooth muscle by RhoA/ROCK. Am. J. Physiol. Gastrointest. Liver Physiol. 292, G1747–G1756 (2007) . [DOI] [PubMed] [Google Scholar]
- Patel C. A. & Rattan S. Spontaneously tonic smooth muscle has characteristically higher levels of RhoA/ROK compared with the phasic smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 291, G830–G837 (2006) . [DOI] [PubMed] [Google Scholar]
- Mcdonnell B., Hamilton R., Fong M., Ward S. M. & Keef K. D. Functional evidence for purinergic inhibitory neuromuscular transmission in the mouse internal anal sphincter. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G1041–G1051 (2008) . [DOI] [PubMed] [Google Scholar]
- Chakder S., Mchugh K. M. & Rattan S. Inhibitory neurotransmission in lethal spotted mutant mice: a model for Hirschsprung's disease. Gastroenterology 112, 1575–1585 (1997) . [DOI] [PubMed] [Google Scholar]
- Hall K. A., Ward S. M., Cobine C. A. & Keef K. D. Spatial organization and coordination of slow waves in the mouse anorectum. J. Physiol. 592, 3813–3829 (2014) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobine C. A. et al. Interstitial cells of Cajal in the cynomolgus monkey rectoanal region and their relationship to sympathetic and nitrergic nerves. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G643–G656 (2010) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. Q. et al. Altered contractile phenotypes of intestinal smooth muscle in mice deficient in myosin phosphatase target subunit 1. Gastroenterology 144, 1456–1465 (2013) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W. Q. et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology 135, 610–620 (2008) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. C. et al. Myosin light chain kinase is necessary for tonic airway smooth muscle contraction. J. Biol. Chem. 285, 17–19 (2010) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S., Kempf J. & Stessman M. Anal seepage: sphincter dysfunction or incomplete evaluation? Gastroenterology 114, A824 (1998) . [Google Scholar]
- Rao S. S. Pathophysiology of adult fecal incontinence. Gastroenterology 126, S14–S22 (2004) . [DOI] [PubMed] [Google Scholar]
- Cobine C. A., Fong M., Hamilton R. & Keef K. D. Species dependent differences in the actions of sympathetic nerves and noradrenaline in the internal anal sphincter. Neurogastroenterol. Motil. 19, 937–945 (2007) . [DOI] [PubMed] [Google Scholar]
- Zheng W., Rampe D. & Triggle D. J. Pharmacological, radioligand binding, and electrophysiological characteristics of FPL 64176, a novel nondihydropyridine Ca2+ channel activator, in cardiac and vascular preparations. Mol. Pharmacol. 40, 734–741 (1991) . [PubMed] [Google Scholar]
- McDonough S. I., Mori Y. & Bean B. P. FPL 64176 modification of CaV1.2L-type calcium channels: dissociation of effects on ionic current and gating current. Biophys. J. 88, 211–223 (2005) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuge R., Bao R., Fogarty K. E. & Lifshitz L. M. Ca2+ sparks act as potent regulators of excitation-contraction coupling in airway smooth muscle. J. Biol. Chem. 285, 2203–2210 (2010) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley S. & Jaggar J. H. Cl− channels in smooth muscle cells. Pflug. Arch. 466, 861–872 (2013) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha J. K., Sengupta J. N. & Goyal R. K. Role of chloride ions in lower esophageal sphincter tone and relaxation. Am. J. Physiol. Gastrointest. Liver Physiol. 263, G115–G126 (1992) . [DOI] [PubMed] [Google Scholar]
- Namkung W., Phuan P. W. & Verkman A. S. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J. Biol. Chem. 286, 2365–2374 (2011) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M. M. & Aylwin M. Niflumic and flufenamic acids are potent reversible blockers of Ca2+-activated Cl- channels in Xenopus oocytes. Mol. Pharmacol. 37, 720–724 (1990) . [PubMed] [Google Scholar]
- Namkung W., Yao Z., Finkbeiner W. E. & Verkman A. S. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 25, 4048–4062 (2011) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson M. T. et al. Relaxation of arterial smooth muscle by calcium sparks. Science 270, 633–637 (1995) . [DOI] [PubMed] [Google Scholar]
- Zhuge R. et al. Ca2+ spark sites in smooth muscle cells are numerous and differ in number of ryanodine receptors, large-conductance K+ channels, and coupling ratio between them. Am. J. Physiol. Cell Physiol. 287, C1577–C1588 (2004) . [DOI] [PubMed] [Google Scholar]
- Zhuge R. et al. The influence of sarcoplasmic reticulum Ca2+ concentration on Ca2+ sparks and spontaneous transient outward currents in single smooth muscle cells. J. Gen. Physiol. 113, 215–228 (1999) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuge R., Sims S. M., Tuft R. A., Fogarty K. E. & Walsh J. V. Jr Ca2+ sparks activate K+ and Cl- channels, resulting in spontaneous transient currents in guinea-pig tracheal myocytes. J. Physiol. 513, 711–718 (1998) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F. et al. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc. Natl Acad. Sci. USA 106, 21413–21418 (2009) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. D. et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 455, 1210–1215 (2008) . [DOI] [PubMed] [Google Scholar]
- Schroeder B. C., Cheng T., Jan Y. N. & Jan L. Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 134, 1019–1029 (2008) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A. et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 322, 590–594 (2008) . [DOI] [PubMed] [Google Scholar]
- Manoury B., Tamuleviciute A. & Tammaro P. TMEM16A/anoctamin 1 protein mediates calcium-activated chloride currents in pulmonary arterial smooth muscle cells. J. Physiol. 588, 2305–2314 (2010) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohr H. et al. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J. Neurosci. 29, 6809–6818 (2009) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock J. R., Futtner C. R. & Harfe B. D. The transmembrane protein TMEM16A is required for normal development of the murine trachea. Dev. Biol. 321, 141–149 (2008) . [DOI] [PubMed] [Google Scholar]
- Zhang C. H. et al. The transmembrane protein 16A Ca2+-activated Cl- channel in airway smooth muscle contributes to airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 187, 374–381 (2013) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinze C. et al. Disruption of vascular Ca2+-activated chloride currents lowers blood pressure. J. Clin. Invest. 124, 675–686 (2014) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dam V. S., Boedtkjer D. M., Nyvad J., Aalkjaer C. & Matchkov V. TMEM16A knockdown abrogates two different Ca2+-activated Cl− currents and contractility of smooth muscle in rat mesenteric small arteries. Pflug. Arch. 466, 1391–1409 (2014) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulley S. et al. TMEM16A/ANO1 channels contribute to the myogenic response in cerebral arteries. Circ. Res. 111, 1027–1036 (2012) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Cornejo P. et al. Anoctamin 1 (Tmem16A) Ca2+-activated chloride channel stoichiometrically interacts with an ezrin-radixin-moesin network. Proc. Natl Acad. Sci. USA 109, 10376–10381 (2012) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angermann J. E., Sanguinetti A. R., Kenyon J. L., Leblanc N. & Greenwood I. A. Mechanism of the inhibition of Ca2+-activated Cl- currents by phosphorylation in pulmonary arterial smooth muscle cells. J. Gen. Physiol. 128, 73–87 (2006) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris A. P. & Frizzell R. A. Ca2+-dependent Cl- channels in undifferentiated human colonic cells (HT-29). II. Regulation and rundown. Am. J. Physiol. Cell Physiol. 264, C977–C985 (1993) . [DOI] [PubMed] [Google Scholar]
- Bao R. et al. A close association of RyRs with highly dense clusters of Ca2+-activated Cl- channels underlies the activation of STICs by Ca2+ sparks in mouse airway smooth muscle. J. Gen. Physiol. 132, 145–160 (2008) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H. et al. Membrane stretch-induced activation of a TRPM4-like nonselective cation channel in cerebral artery myocytes. J. Pharmacol. Sci. 103, 417–426 (2007) . [DOI] [PubMed] [Google Scholar]
- Collier M., Ji G., Wang Y. X. & Kotlikoff M. Calcium-induced calcium release in smooth muscle loose coupling between the action potential and calcium release. J. Gen. Physiol. 115, 653–662 (2000) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essin K. et al. Indirect coupling between CaV1. 2 channels and ryanodine receptors to generate Ca2+ sparks in murine arterial smooth muscle cells. J. Physiol. 584, 205–219 (2007) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl A., Lee H. K. & Sanders K. M. Regulation of ion channels in smooth muscles by calcium. Am. J. Physiol. Cell Physiol. 271, C9–C34 (1996) . [DOI] [PubMed] [Google Scholar]
- Piper A. S. & Large W. A. Multiple conductance states of single Ca2+-activated Cl- channels in rabbit pulmonary artery smooth muscle cells. J. Physiol. 547, 181–196 (2003) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders K. M., Ward S. M. & Koh S. D. Interstitial cells: regulators of smooth muscle function. Physiol. Rev. 94, 859–907 (2014) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy A. M., Cobine C. A. & Keef K. D. Changes in neuromuscular transmission in the W/WV mouse internal anal sphincter. Neurogastroenterol. Motil. 24, e41–e55 (2012) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorijn F. et al. Interstitial cells of Cajal are involved in the afferent limb of the rectoanal inhibitory reflex. Gut 54, 1107–1113 (2005) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshiro J., Yamamura H., Suzuki Y. & Imaizumi Y. Modulation of TMEM16A-channel activity as Ca2+ activated Cl- conductance via the interaction with actin cytoskeleton in murine portal vein. J. Pharmacol. Sci. 125, 107–111 (2014) . [DOI] [PubMed] [Google Scholar]
- Wang Y. F. & Daniel E. E. Gap junctions in gastrointestinal muscle contain multiple connexins. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G533–G543 (2001) . [DOI] [PubMed] [Google Scholar]
- Samuel U., Lutjen-Drecoll E. & Tamm E. R. Gap junctions are found between iris sphincter smooth muscle cells but not in the ciliary muscle of human and monkey eyes. Exp. Eye Res. 63, 187–192 (1996) . [DOI] [PubMed] [Google Scholar]
- Liu P., Jenkins N. A. & Copeland N. G. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476–484 (2003) . [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZhuGe R. et al. Dynamics of signaling between Ca2+ sparks and Ca2+- activated K+ channels studied with a novel image-based method for direct intracellular measurement of ryanodine receptor Ca2+ current. J. Gen. Physiol. 116, 845–864 (2000) . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures 1-17, Supplementary Tables 1-2 and Supplementary References