

Abstract
USP14 is a major regulator of the proteasome and one of three proteasome-associated deubiquitinating enzymes1–9. Its effects on protein turnover are substrate specific, for unknown reasons. We report that USP14 shows a dramatic preference for ubiquitin-cyclin B conjugates that carry more than one ubiquitin modification or chain. This specificity is conserved from yeast to humans and is independent of chain linkage type. USP14 has been thought to cleave single ubiquitin groups from the distal tip of a chain but we find that it removes chains from cyclin B en bloc, proceeding until a single chain remains. The suppression of degradation by USP14’s catalytic activity reflects its capacity to act on a millisecond time scale, before the proteasome can initiate degradation of the substrate. In addition, single-molecule studies showed that the dwell time of ubiquitin conjugates at the proteasome was reduced by USP14-dependent deubiquitination. In summary, the specificity of the proteasome can be regulated by rapid ubiquitin chain removal, which resolves substrates based on a novel aspect of ubiquitin chain architecture.
The proteasome, a ~2.5-MDa molecular machine, is the focal point for global regulation of ubiquitin pathway output. Among its major regulators is a deubiquitinating enzyme known in mammals as USP14 (in yeast, Ubp6). USP14 negatively regulates proteasome activity by ubiquitin chain disassembly as well as by a noncatalytic mechanism1–6. USP14 inhibitors produce selective effects on the turnover of proteasome substrates3,8, suggesting that the rate at which USP14 disassembles proteasome-bound ubiquitin chains may depend on the nature of the substrate. We test this hypothesis in the present work.
USP14 is thought to progressively trim monoubiquitin groups from the substrate-distal tip of a chain9 (Fig. 1a). To assess this model we employed a canonical substrate of USP14, ubiquitinated cyclin B11,3. When cyclin B1 is modified by the ubiquitin ligase Anaphase-Promoting-Complex/Cyclosome (APC/C) in the presence of E2 enzyme UbcH10, the resulting conjugates typically carry multiple short ubiquitin chains of varied length10,11. We refer to these as supernumerary chains, insofar as only one chain is in principle required for substrate degradation. The flexible, 88-residue N-terminal element of cyclin B1 (NCB1) is enriched in lysine residues, nine of which are competent for ubiquitination10. The N-terminal element is necessary for cyclin B1 degradation, and contains an APC/C recognition motif, the destruction box10–12.
Figure 1. USP14 cleaves supernumerary ubiquitin chains from substrates.

a, Canonical model of USP14 activity. USP14 is thought to shorten ubiquitin chains progressively from their distal tip. b–f, In vitro degradation and deconjugation assays with polyubiquitinated N-terminal fragment of cyclin B1 (Ubn-NCB1; HA tagged). Cartoons show idealized representations of the expected distribution of ubiquitin moieties throughout. b, Human proteasome (Ptsm; 4 nM) was incubated with Ubn-NCB1 (~110 nM final) generated by UbcH10. Where indicated, USP14 (80 nM) was added. c, As in b, except using polyubiquitinated NCB1 generated with lysine-less Ub. d, As in b, except using polyubiquitinated conjugates on K64-only NCB1 (1KNCB1, unless otherwise noted), generated by UbcH10 and Ube2S. e, Ubn-NCB1 (~110 nM) was incubated with hexokinase-treated ADP-proteasome (ADP-Ptsm) in the presence or absence of USP14 (80 nM). f, As in e, except using polyubiquitinated K64-only NCB1. Samples were analysed by SDS–PAGE/immunoblotting for HA. For gel source data, see Supplementary Information.
Ubiquitinated NCB1 was rapidly degraded by proteasomes lacking USP14, but in the presence of USP14 deubiquitinated species were observed. These were not fully deubiquitinated, but rather carried 2–4 ubiquitin groups and were resistant to both degradation and further deubiquitination (Fig. 1b). This strong stop to USP14-dependent processing might be assumed to reflect that these conjugates retain too few ubiquitin moieties for efficient proteasome binding. However, studies below show that USP14 activity depends on the architecture of ubiquitin chains on the substrate and not simply substrate binding to the proteasome. The behavior of NCB1 was comparable to that of full-length cyclin B1 (data not shown)3,11.
We used this experimental system to test whether the architecture of ubiquitin chains on NCB1 was relevant to its rapid deubiquitination. Cyclin B1 is modified by APC/C and UbcH10 to form mixed-linkage chains10 primarily via residues K11, K48, and K63. Thus, a lysine-free mutant of ubiquitin11 will modify NCB1 via multiple mono-ubiquitination events (Fig. 1c). USP14 deubiquitinated this form of NCB1 at a rate comparable to that of wild-type conjugates, indicating that USP14 does not act obligatorily on ubiquitin-ubiquitin linkages (Fig. 1c).
In a complementary experiment, we eliminated lysines from NCB1 rather than ubiquitin, leaving behind only K64 of NCB1 to allow for the synthesis of a single ubiquitin chain on NCB1. This mutant is competent for degradation (Fig. 1d)11. Although single-chain conjugates are canonical proteasome substrates13, USP14 showed no detectable activity on this substrate (Fig. 1d). The lack of deubiquitinating activity on the single-chain substrate was accompanied by a failure to inhibit its degradation. Similar results were obtained with NCB1 ubiquitinated with Ubc4 in place of UbcH10 (Extended Data Fig. 1a, b).
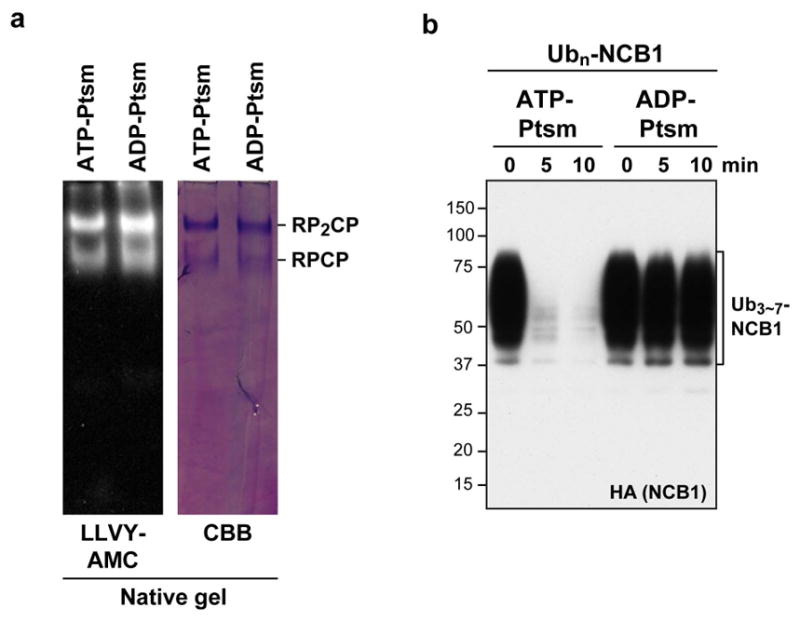
A caveat to the experiment above is that, because NCB1 is degraded quickly by the proteasome, USP14 has little time to act. However, the proteasome must be present in the assay because it is required to activate USP14. When we quenched substrate degradation by replacing ATP with ADP (Extended Data Fig. 2), USP14-dependent deubiquitination of WT NCB1 conjugates was preserved (Fig. 1e). In contrast, no deubiquitination was detected with single-chain conjugates (Fig. 1f). Thus, USP14 shows dramatic specificity for NCB1 conjugates bearing multiple ubiquitin chains, whereas the proteasome does not effectively distinguish between these two classes of substrate in the absence of USP14.
We next considered whether the resistance of single-chain conjugates to USP14 was idiosyncratic to the particular modified lysine in NCB1. On the contrary, conjugates generated using a K36-only form of NCB1 behaved equivalently to the K64-anchored conjugates (Extended Data Fig. 1c). The supernumerary chain effect was seen with the substrate PY-Sic1 as well as NCB1 (Extended Data Fig. 1d, e). We identified a third preferred substrate of USP14, and it is likewise modified at multiple lysines (Extended Data Fig. 1f)12,14. Ubp6, the S. cerevisiae ortholog of USP14, showed the same preference for supernumerary chains, arguing for the biological significance of this property (Extended Data Fig. 3).
It has been hypothesized that USP14 or Ubp6 may preferentially cleave K63-linked chains15,16. This model could account for our data if single-chain NCB1 conjugates were devoid of K63 linkages. To test this model, we prepared single-chain NCB1 with homogeneous K63 linkages. These conjugates were also cleaved slowly if at all by USP14 (Extended Data Fig. 4), regardless of length.
One explanation for the specificity of USP14 for substrates modified at multiple sites is that cleavage of one chain is promoted allosterically by binding of a second chain to USP14 or another site on the proteasome. However, adding free chains over a wide concentration range failed to stimulate deubiquitination of single-chain NCB1 (Extended Data Fig. 5). Another measure that failed to stimulate removal of singleton chains was phosphomimetic substitution of Ser432, a site subject to modification by Akt17 (Extended Data Fig. 6). If singleton chains are as a rule poor substrates for USP14, then all free ubiquitin chains–those not attached to an acceptor protein–must be poor USP14 substrates, regardless of length or linkage type. Extended Data Fig. 7 shows that free chains of diverse length and linkage type are all cleaved minimally by proteasome-activated USP14, even upon long incubation.
By ubiquitinating NCB1 with preformed tetraubiquitin chains, we prepared conjugates carrying multiple chains of homogeneous linkage type and length. When these conjugates were incubated with USP14, proteasomes, and either ATP or ADP, a limit digest product was formed consisting of NCB1 carrying four ubiquitin groups (Fig. 2a, b). Thus, USP14 appeared to completely remove each supernumerary chain, and stop when a single chain remained. Accordingly, ubiquitin was released from this substrate in the form of tetraubiquitin chains (Fig. 2c). Using mass spectrometry, we confirmed that K48 linkages were not consumed in this reaction (Fig. 2d). Studies with other linkage types (Extended Data Fig. 8) further indicated that USP14 removes chains preferentially en bloc (Fig. 2e), rather than trimming them from the distal tip, as thought.
Figure 2. USP14 cleaves arrayed tetraubiquitin chains proximally, stopping at the last chain.

a, K48-linked Ub4 conjugates of NCB1 were prepared, resulting in one or more tetraubiquitin modifications. ADP-Ptsm (3 nM) was incubated with multi-K48Ub4-NCB1 (~90 nM) in the presence or absence of USP14 (60 nM). Arrow indicates K48Ub4-NCB1 species modified with a single chain and derived by USP14 cleavage. b, Assays as in a, except in the presence of ATP. c, K48Ub4-multichain conjugate of NCB1 was purified, and assays were performed as in a, except that a control was added in which the pan-specific deubiquitinating enzyme USP21 (0.5 μM) was used (10 min incubation). Arrowheads indicate free Ub4 chains released by USP14 (K48Ub4) and products of USP21 activity such as free Ub. Samples were analysed by SDS–PAGE/immunoblotting for HA (a and b) or Ub (c). d, Tandem Mass Tag (TMT) labeling and quantitative mass spectrometry. Purified K48Ub4-multi-chain NCB1 conjugates were assayed in biological duplicates for each condition as in a. The reaction was then quenched and subjected to trypsin digestion, TMT labeling, and tMS2 analysis in a Q-Exactive for quantification of K48-linked Ub peptides. e, Proposed en bloc mode of deubiquitination by USP14. f, Modeling of free ubiquitin chain cleavage by proteasome-activated Ubp6. Access of a K11-linked di-ubiquitin chain to the active site of Ubp6 appears occluded. Structures were obtained from the PDB database: Ubp6 and proteasome (5A5B)5 and Ub (1UBQ). C118 contains the active site nucleophile.
Why does USP14 discriminate against cleavage within chains? The presumptive active form of Ubp6 sits with its catalytic domain directly contacting the OB domain of subunit Rpt15,6. Modeling studies with di-ubiquitin as substrate indicated that, regardless of chain linkage type, the proximal ubiquitin may have restricted access to the active site of the enzyme (Fig. 2f, Extended Data Fig. 9). The coiled-coils of Rpt1 and Rpt2 are responsible for this occlusion (Fig. 2f). This effect may account for the dominant en bloc cleavage mode of Ubp6 and USP14. Cleavage within ubiquitin chains by free forms of USP14 and Ubp6, as observed by many researchers, may reflect that free USP14 is not subject to occlusion from proteasome subunits.
In a recent study, we established a single-molecule (SM) TIRF microscopy method to capture deubiquitination events mediated by the en bloc zinc-dependent activity of RPN1118,19, using USP14-free proteasomes14. Deubiquitination was identified as a stepwise reduction of signals from fluorescently-labeled ubiquitin on substrates (Fig. 3a, Extended Data Fig. 10). To study USP14-mediated deubiquitination, we inhibited RPN11 activity (as well as substrate degradation) using ADP and a zinc chelator14. ‘Stepped’ events representing deubiquitination increased seven-fold upon USP14 addition (Fig. 3a). SM traces with USP14-reconstituted proteasome in the presence of ADP showed remarkable similarity to those produced by RPN11 (Fig. 3b, Extended Data Fig. 10). The step size of ubiquitin removal by RPN11 and USP14 followed a similar distribution, supporting en bloc cleavage by USP14 (Fig. 3c).
Figure 3. Single-molecule analysis of deubiquitination by USP14.

Single molecule (SM) assays were performed on immobilized human proteasome from which USP14 had been removed. Securin conjugates were generated using fluorescently labeled ubiquitin and added to the proteasome under conditions as indicated. a, Percent of stepped traces under conditions measuring RPN11 (ATP, no USP14) or USP14 (ADP, USP14-WT) activities. N(ATP, no USP14)=1574; N(ADP, no USP14)=578; N(ADP, USP14-WT)=733. b, Examples of step-trace segmentation graphs from an SM analysis on polyubiquitinated securin conjugates with USP14-reconstituted human proteasome in the presence of ADP (See Extended Data Fig. 10 for RPN11 step traces). c, Step size distribution of RPN11-mediated (ATP, no USP14, N=356) or USP14-mediated (ADP, USP14-WT, N=129) deubiquitination events as a function of the number of ubiquitins. d, Average dwell time of securin conjugates on the proteasome in the presence or absence of USP14. ATP was added for the no-USP14 condition. Dwell time was plotted as a function of the multiplicity of ubiquitin groups on securin. Error bars indicate SD of the mean (n=3). ADP denotes the condition of ADP plus o-PA to suppress RPN11 activity.
USP14-mediated deubiquitination reduced the dwell time of ubiquitin conjugates on the proteasome, suggesting that rapid deubiquitination can suppress degradation of the substrate by contracting the duration of its encounter with the proteasome (Fig. 3d). This dwell time effect suggests that, to suppress degradation, USP14 must remove chains at least as rapidly as the substrate is degraded in the absence of USP14. To test this scenario we used quench-flow methods, which allow reactions to be followed on a millisecond time scale. An excess of proteasome over substrate was employed to approximate single-turnover conditions.
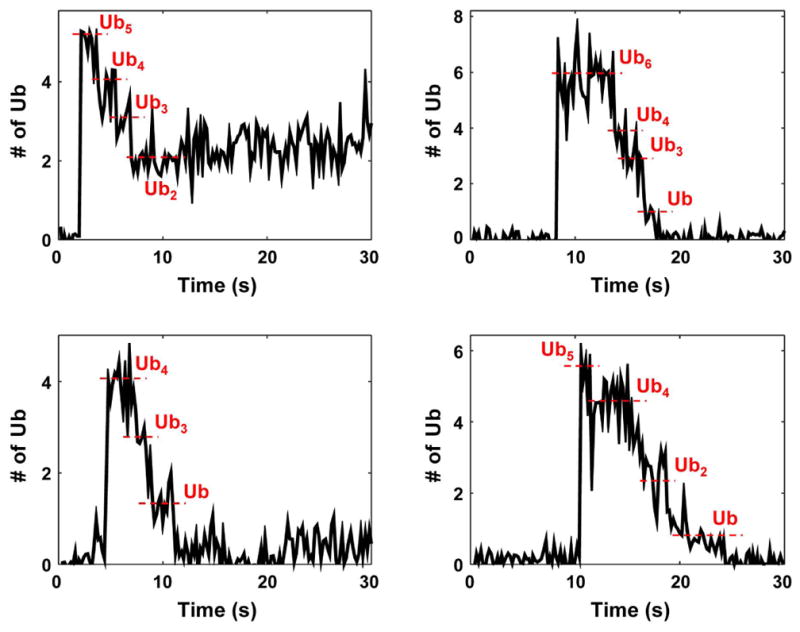
USP14-free proteasomes degraded NCB1 conjugates efficiently, with most degradation occurring between 9 and 30 seconds (Fig. 4a, b). USP14 delayed degradation (Fig. 4b), as high molecular weight conjugate bands were instead converted into partially deubiquitinated species. The production of lower molecular weight species through deubiquitination was initiated before degradation was evident with USP14-free proteasomes (Fig. 4a–c). Thus, USP14 is sufficiently fast that deubiquitination can be accomplished before the proteasome would otherwise degrade NCB1. Similar results were obtained using full length cyclinB1 (data not shown), and we confirmed that deubiquitination was mediated by proteasome-bound rather than free USP14 (Extended Data Fig. 6b).
Figure 4. Kinetic competition between USP14 and the proteasome.

a, Proteasome (1.2 μM) in the presence or absence of USP14 (17 μM) was mixed with Ubn-NCB1 (~0.47 μM) in a quench flow device. (Top) Degradation of NCB1 conjugates is detectable by approximately 15 s. (Bottom) Deubiquitination of Ub4~6-NCB1 can be observed by 100 ms or earlier as Ub2~3-NCB1 species are generated. b, Quantification of Ubn-NCB1 degradation from a. c, Quantification of Ub3- or Ub2-NCB1 species generated by USP14-mediated deubiquitination. USP14-derived reaction products can appear as early as hundreds of ms. Collected samples were analysed by SDS–PAGE/immunoblotting, using antibody to HA. Note that, for b, axes are logarithmic in scale.
The ability of USP14 to prevail over the proteasome in a kinetic competition may explain the suppression of protein degradation by USP14’s deubiquitinating activity3. Thus, USP14-loaded proteasomes may discriminate against substrates bearing multiple ubiquitin chains whereas USP14-free proteasomes do not. Because USP14 and Ubp6 are modulated by growth factors and stress conditions2,17, the ability of the proteasome to discriminate between single-chain and multi-chain substrates is likely under biological control.
We expect that the supernumerary chain rule should bias USP14 action towards the products of ubiquitin ligases that are weak in chain-extending activity, thus adding ubiquitin to multiple sites on a target protein. Many ubiquitinated proteins can be modified at multiple lysines10,11,20,21, as originally described for cell surface receptors22. In most cases however, it is unclear whether simultaneous modification of lysines occurs on endogenous substrates, because proteomics methods generally rely on protein fragmentation. Therefore, the development of new methodologies may be critical for a better understanding of how USP14 regulates proteasome function.
We suggest that the last ubiquitin chain resists cleavage by USP14 because its site of attachment to substrate is not docked in proximity to USP14, regardless of the ubiquitin receptor to which the chain is bound. If the chain that is spared by USP14 is short, degradation will be suppressed; if long, then proteasome-substrate interaction will be preserved and substrate degradation will proceed. This chain will be removed by RPN11 once substrate translocation is initiated. Because RPN11 also cleaves chains proximally18,19, our data reveal an unexpected point of similarity between these enzymes, which may act sequentially. RPN11 activity differs from that of USP14 however, in requiring ATP hydrolysis18,19, presumably to drive translocation of the conjugate to its active site23,24. That RPN11 promotes substrate degradation whereas USP14 suppresses degradation may reflect that RPN11 fails to remove chains prior to the commitment step of the proteasome, which by hypothesis negates the ubiquitin requirement for degradation.
It will be interesting to determine whether supernumerary chain removal is the principal role of USP14 in cells, or whether other specificity rules exist for this enzyme. Substrate flexibility, a feature of the cyclin B and securin degrons25, but not of ubiquitin itself, may be important for USP14 to act on these substrates. Indeed, the occlusion that we hypothesize to impair docking of proximal ubiquitin in a free chain near the USP14 active site may apply as well to many folded globular domains that are ubiquitinated, which would be required to access the same site. This substrate docking constraint may further limit the substrate range of USP14. In contrast to USP14, RPN11 is expected to have a robust activity against chains on domains that are normally folded, because such chains will typically be presented to RPN11 only after the domain has been unfolded by mechanical force applied by the proteasomal ATPases.
Online Content
Methods, along with any additional Extended Data display items, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Methods
Recombinant proteins and biochemical reagents
Wild-type or single lysine N-terminal cyclin B1 (NCB1; N-t HA tag and C-t His tag), MBP-human E1, UbcH10, Ubc4, and Ube2S proteins were expressed in BL21 (DE3) or Rosetta2 (DE3) cells (Millipore), and purified essentially as previously described11. The baculoviruses of full length cyclin B1, CDK1, and Cak1 (generous gifts from RP. Fisher and E. Winter) were re-amplified in Sf21 cells (Life Technologies) and used to infect High Five cells (Life Technologies). Expressed proteins were purified on Ni-NTA beads (Qiagen) and by gel filtration as previously described26.
N-terminal His-tagged USP14 in pET-15b (Novagen), tags-cleaved USP14 wild-type and S432E in pET-32M17 (generous gifts from J. Yuan), non-tagged USP14 and non-tagged Ubp6 in pTXB1 (NEB) were expressed in Rosetta2 (DE3), and bound and eluted from Ni-NTA beads and chitin beads (NEB), respectively, according to the manufacturers’ instructions. The eluates from the resins was further applied to HiTrap Q HP5 anionic exchange chromatography (GE Healthcare) followed by gel filtration with Hi Load 16/600 Superdex 200 PG column (GE Healthcare) using AKTA Purifier FPLC system (GE Healthcare).
Sources of some commercial reagents not described elsewhere are as follows: anti-HA-HRP (3F10, Roche); anti-cyclin B1 (Ab-2, Thermo Scientific); anti-T7-HRP (Novagen); anti-Cdc27 (AF3.1, custom-made); anti-Ub (P4D1, Santa Cruz and Dako); anti-securin (MBL International); anti-Ubp6 (custom-made); anti-UbcH10 (Millipore); anti-20S (MCP21, custom-made); Ub, Ub mutants, and free Ub chains (R&D systems and Life Sensors); IU1 (custom-synthesized); o-phenanthroline (o-PA, Sigma); ATPγS (Santa Cruz); PS-341 (Apex Bio); MG-262 (R&D Systems); USP21 (a gift from Y. Ye).
Proteasome purification and ATP-depletion
Human and yeast proteasomes were affinity-purified as previously described1,3. For native human proteasomes, HEK293T cells stably expressing epitope-tagged RPN11 (a generous gift from L. Huang; mycoplasma negative) were lysed by Dounce-homogenizer in buffer A (50 mM NaH2PO4 [pH 7.5], 100 mM NaCl, 10% glycerol, 5 mM MgCl2, 0.5% NP-40, 5 mM ATP and 1 mM DTT) containing protease inhibitors. The cleared lysates were incubated with NeutrAvidin resin (Thermo Scientific) for at least 2 hr at 4°C. The beads were extensively washed with buffer A to remove endogenous USP14 associated with the proteasome, followed by buffer B (50 mM Tris-HCl [pH 7.5], 1 mM MgCl2, 1 mM ATP, and 10% glycerol), then equilibrated in cleavage buffer C (50 mM Tris-HCl [pH 7.5], 1 mM MgCl2, and 1 mM ATP). Human proteasomes were eluted from the beads by cleavage, using TEV protease (Life Technologies) for 1 hr at 30°C. For yeast proteasomes, each strain was cultured to an OD600 of about 0.7, and lysed in buffer D (50 mM Tris-HCl [pH 8.0], 1 mM EDTA, 5 mM MgCl2, and 1 mM ATP) containing protease inhibitors by a French press device. The cleared lysates were incubated with rabbit IgG resin (MP Biomedicals) for 1 hr at 4°C. The proteasome-bound beads were washed with an excess of wash buffer E (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, and 1 mM ATP), then equilibrated in elution buffer F (50 mM Tris-HCl [pH 7.5], 1 mM EDTA, 5 mM MgCl2, 1 mM ATP, and 10% glycerol). TEV cleavage was performed at 30°C for 1 hr, and the protease was removed from the eluate using Ni-NTA resin.
Proteasomes were generally purified in the presence of ATP to preserve their integrity. To deplete the ATP from the proteasome prep where needed, purified proteasomes were incubated in reaction buffer G (50 mM Tris-HCl [pH 7.5] and 5 mM MgCl2) containing 20 mM glucose and 0.2 U/μl hexokinase (Sigma) for 1 hr at 30°C, and then final 10% glycerol was added for low temperature storage. The hexokinase-treated ‘ADP-proteasome’ was tested by native gel, peptidase assay, and in vitro degradation assay for structural integrity and loss of activity in degradation assays.
Ubiquitin conjugate preparation and purification
Ubiquitination reactions were carried out as previously described3,10,11. In summary, for APC/C-mediated reaction, APC/C was immunopurified from X. laevis egg extract with anti-Cdc27 antibodies bound to protein A-Sepharose 4B fast flow (Sigma). The conjugation mixture generally contained 2.5 μM MBP-E1, 3.8 μM UbcH10, 133 μM ubiquitin, 2.3 μM NCB1 or 0.55 μM full length cyclin B1-CDK1 complex, and APC/C on the beads in the reaction buffer H (20 mM Tris-HCl [pH 7.5], 100 mM KCl, 2 mM ATP, and 2.5 mM MgCl2). Where indicated, 3.8 μM of Ubc4, 2 μM of Ube2S (along with 0.7 μM of UbcH10), or 2 μM of securin was used. For conjugation with pre-formed ubiquitin chains, 18.4 μM (K48- or K63-linked Ub4) or 10.5 μM (K63-linked Ub8) of free chains was incubated with 0.7 ~ 1 μM of NCB1. Reactions were typically performed at least for 90 min at room temperature with agitation at 2000 rpm. The reaction mixture was collected and then combined with one bed volume of the first wash of the APC/C-beads with reaction buffer lacking salt. Sic1PY ubiquitin conjugates were generated essentially as previously described3.
For TMT mass spec-coupled Ub linkage analysis and SM assays, ubiquitin conjugates were further purified to remove excess unreacted free ubiquitin or ubiquitin chains and auto-ubiquitinated enzymes. The conjugation reaction was incubated with magnetic Co2+-beads (Life Technologies) for 30 min on ice. The conjugate-bound beads were washed two times with buffer I (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.01% Tween-20, and 1 mg/ml BSA), then washed twice with the same buffer lacking detergent (for securin) or lacking detergent and BSA (for NCB1). Dylight550Ubn-securin was eluted in the buffer J (50 mM Tris-HCl [pH 7.5], 50 mM NaCl, 250 mM imidazole, and 1 mg/ml BSA). Polyubiquitinated NCB1 was eluted first in buffer K (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, and 250 mM imidazole), and combined with the next two washes of the beads with buffer K lacking NaCl and imidazole to reduce the concentration of salt. The eluted conjugate was not further subjected to desalting, buffer exchange, or ultrafiltration, since all these steps resulted in a significant loss of yield and also aggregation.
Yeast strains
Yeast proteasomes with intact ubiquitin receptors were purified from strain SY409 (MATa rpn11-TEV-ProA::HIS3 ubp6::URA3), which derived from SDL133 (MATa RPN11-TEV-ProA)7 and SJH14 (MATa ubp6::URA3), the latter being prepared from a direct transformation of SUB61 with the ubp6::URA3 cassette previously used to develop similar strains1.
In vitro degradation and deubiquitination assays
Purified human proteasomes (3 to 5 nM) or yeast proteasomes (40 to 50 nM) were incubated with polyubiquitinated NCB1 (~110 nM) or full length cyclin B1 (~30 nM) in buffer G supplemented with 5 mM ATP at room temperature. Where indicated, purified recombinant USP14 or Ubp6 was pre-incubated with proteasome for at least 5 min before initiating the reaction. The reaction was terminated by adding 5x SDS-PAGE sample buffer and heating for 10 min at 70°C. Samples were subsequently subjected to SDS–PAGE/immunoblot analysis. Conjugates were treated with pan-specific ubiquitin isopeptidase USP21 (0.4 μM) for 10 min. Assays with polyubiquitinated Sic1PY (~200 nM) or securin (~100 nM) were performed similarly.
For USP14 deubiquitination assays uncoupled to degradation, ADP-proteasome (3 to 4 nM) was used for assays in buffer G supplemented with 3 to 5 mM ‘ATP-free’ ADP. To remove the residual ATP contamination, commercial ADP (Calbiochem) was first dissolved in water at 0.5 to 1 M at pH 7.0, and then treated with hexokinase and glucose in the reaction buffer as described above. The final ADP concentration in solution was adjusted to 0.25 to 0.5 M (pH 7.5). Degradation-suppressed deconjugation assays were also alternatively performed with ATP-proteasome in the assay buffer G supplemented with 3 to 5 mM ADP, 6 mM o-PA, 0.75 mM ATPγS, 1.5 μM PS-341, and 7.5 μM MG-262, with equivalent results.
To perform deubiquitination assays with pre-formed unanchored chains, human proteasomes (5 to 12 nM) were incubated at 30°C with free chains (80 to 300 nM) in the presence or absence of recombinant USP14 (at 20-fold excess over proteasome) in buffer G supplemented with 5 mM ATP. As a control, chains were treated with USP21 (0.5 μM) for 20 min.
Tandem mass tagging mass spectrometry for Ub linkage analysis
To quantify ubiquitin conjugate cleavage by USP14, purified multi-chain NCB1 conjugates (Ubn-NCB1 or multi-K48Ub4-NCB1) were subjected to in vitro deubiquitination assays as described above. Deubiquitination reactions with ADP-proteasome in the presence or absence of USP14 were performed in duplicate and quenched by adding the inhibitor mixture of USP14 inhibitor related to IU1 (70 μM), o-PA (6 mM), ATPγS (0.5 mM), PS-341 (1.5 μM), and MG-262 (7.5 μM). As a pan-specific deubiquitination control, USP21 (0.5 μM) was also incubated with purified conjugates for 10 min in duplicate. Each quenched reaction was subjected to overnight trypsin digestion at 37°C. Peptides obtained were desalted and labeled with TMT 10plex isobaric reagents (Thermo Scientific) following the manufacturer’s protocol. Finally, all 10 samples containing TMT-labeled peptides were combined and desalted before LC-MS analysis. The peptide mixture was fractionated using a solid-phase C18 column equipped in an Accela 600 liquid chromatography (LC) pump coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific). To detect and quantify all possible linkage-specific ubiquitin peptides we designed a TMT-PRM method consisting in one full MS scan acquired in the Orbitrap (70k resolution and automatic gain control [AGC] of 1 × 106) followed by 11 tMS2 (covering all detectable linkage-specific ubiquitin peptides) for each data collection cycle. Fragment ions generated by HCD (normalized collision energy was set to 27%) were acquired in the Orbitrap (70k resolution, AGC of 1 × 106, 200 ms of maximum injection time and 1 m/z isolation window). RAW files obtained were processed using an in-house software pipeline based on Sequest27,28. Signal to noise ratio of the reporter ions was used to calculate the abundance of each linkage across all samples.
Single-molecule deubiquitination assays
Coverslip passivation, fluorescently labeled ubiquitin-securin conjugates, TIRF microscope configuration, and image analysis were as previously described14. For single-molecule assays, 20 nM USP14-free human proteasome was incubated with 10 nM biotinylated anti-20S (MCP21) antibody for 20 min in buffer L (50 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 3 mM ATP, 20 mM imidazole, and 3 mg/ml BSA). After immobilizing antibody-bound proteasome on streptavidin-functionalized coverslip, unbound proteasome was washed off and securin (ubiquitinated with dylight550-labeled ubiquitin) was added at a final concentration of ~10 nM. For studying USP14-mediated deubiquitination, hexokinase-treated ADP was used, together with 6 mM o-PA. Deubiquitination kinetics was monitored under a TIRF microscope (Ti, Nikon) at 5 frames/sec for 2 min.
Quench-flow analysis
To resolve rapid single-encounter events for USP14-catalyzed deubiquitination, millisecond reactions were performed on a quench-flow device (Kintek RQF-3). The quench-flow machine was first calibrated with known amount of BSA, ovalbumin, and UbcH10 to achieve maximum recovery of samples according to the manufacturer’s instructions. Generally, the recovery ranged between 75 to 85% of the input and was relatively even among the reaction loops (3.5 milliseconds to minutes). For these reactions, human proteasomes were purified on a large scale as described above and concentrated to 2 μM or higher in buffer M (50 mM Tris-HCl [pH 7.5], 1 mM MgCl2, 1 mM ATP, and 5% glycerol). Recombinant USP14 was purified and concentrated to 250 μM or higher in buffer N (35 mM Tris-HCl [pH 7.5], 50 mM NaCl, 1 mM DTT, and 5% glycerol). Before the quench-flow reaction, proteasomes were pre-incubated with USP14, and the solution was adjusted to 50 mM Tris-HCl [pH 7.5], 3.5 mM NaCl, 3 mM MgCl2, 3 mM ATP, 0.74 mM DTT, and 3.3% glycerol. For the proteasome-only control (i.e., no USP14), only the buffer N was added as a mock to achieve the same final composition of buffer ingredients as above. Ubiquitinated NCB1 was generated on a large scale, and before the reaction, adjusted to about 0.47 μM in buffer O (20 mM Tris-HCl [pH 7.5], 36 mM KCl, 3 mM ATP, and 3 mM MgCl2). To enhance the recovery, prevent aggregation, and reduce non-specific binding to the tubing, 1 mg/ml of BSA and 0.0025% of NP-40 (each final concentration after mixing with enzymes) were added to the conjugate solution. Ubiquitinated full length cyclin B1 was essentially prepared as above except adjusting the substrate at ~0.17 μM.
Millisecond single-encounter reactions were carried out from 5 milliseconds to minutes by mixing the two reactants in the corresponding reaction loops and then quenched in the device with 3.3x quench solution P (300 mM Tris-HCl [pH 6.8], 3% SDS, and 10% glycerol) with freshly added 330 mM DTT. A small amount of SDS-PAGE loading dye was then added to each collected sample. For the 0 sec sample, enzymes or ubiquitin conjugates alone were separately quenched inside the machine with each corresponding mock buffer by utilizing reaction loop 1 at 3.5 msec. The reactions were resolved in SDS-PAGE/immunoblotting.
For quantification, the total ubiquitin conjugate or each conjugate species was analysed by Amersham 600 RGB system (GE Healthcare) or Odyssey CLx Infrared Imaging System (LI-COR Biosciences). The conjugate signal was normalized by UbcH10 for each time point to correct for recovery, and then further normalized to the 0 time control. For Ubn-NCB1, proteasome alone showed some non-specific signals detected by HA antibody. This number was subtracted before normalization. The data were curve-fitted by non-linear regression with the best fit model using GraphPad Prism (GraphPad Software).
Extended Data
Extended Data Figure 1. USP14 and Ubp6 are highly specific for multi-chain ubiquitin-protein conjugates.
In vitro degradation and deconjugation assays with polyubiquitinated conjugates. a, b, In vitro deubiquitination assays with Ubn-NCB1 or Ubn-1KNCB1 generated by Ubc4 in parallel. Each conjugate sample (~110 nM) was incubated with 4 nM of human proteasome in the presence or absence of USP14 (80 nM). c, This experiment tests whether the failure of USP14 to deubiquitinate a single-chain conjugate formed using K64-only NCB1 (Figs. 1d and 1f) is predictive of the behavior of USP14 on other single-chain conjugates. Conjugates (~110 nM) were incubated with human proteasome (4 nM) in the presence or absence of USP14 (80 nM). d, e, In vitro degradation and deubiquitination assays with polyubiquitinated Sic1PY or polyubiquitinated K36-only Sic1PY (1KSic1PY). d, Ubn-Sic1PY (~200 nM) was incubated with human proteasome (5 nM) in the presence or absence of USP14 (100 nM). e, Assays were performed similarly as in d, except using a single-lysine variant of Sic1 (1KSic1PY) modified with wild-type Ub. IU1 (75 μM)3 was used to inhibit USP14 deubiquitinating activity. f, In vitro degradation and deconjugation assays with polyubiquitinated securin. Human proteasome (4 nM) was incubated with Ubn-securin (~100 nM) generated by UbcH10 and APC/C. Where indicated, USP14 (80 nM) was added. Samples were resolved by SDS–PAGE and immunoblotted using an antibody to the HA (a–c) or T7 (d, e) epitopes, and securin (f).
Extended Data Figure 2. Preparation of ATP-depleted proteasome for uncoupling degradation from deubiquitination.

a, For ATP-depletion, purified human proteasomes were treated with hexokinase or mock-treated with vehicle for hexokinase (i.e. water). Native gel analysis of untreated proteasome (ATP-Ptsm) or hexokinase-treated ADP-proteasome (ADP-Ptsm) with suc-LLVY-AMC and Coomassie Brilliant Blue (CBB) staining. Each lane was loaded with 5 μg of protein. Note that there was essentially no change in proteasome integrity upon hexokinase treatment. b, In vitro Ubn-NCB1 degradation assays with ATP-Ptsm or ADP-Ptsm (4 nM each). Assays were performed and analysed as in Fig. 1b or 1e.
Extended Data Figure 3. The substrate specificity of USP14 is evolutionarily conserved.

In vitro degradation and deconjugation assays by yeast proteasome and Ubp6 with NCB1 conjugate. a, Purified ubp6Δ yeast proteasome (50 nM) was incubated with Ubn-NCB1 or Ubn-1KNCB1 (~110 nM) in the presence or absence of Ubp6 (200 nM). b, Assays as in a, except yeast proteasome was pre-incubated with ADP and other inhibitors of the proteasome (ATPγS, o-phenanthroline, PS-341, and MG-262) to suppress the degradation. c, Assays with human proteasome, USP14, and polyubiquitinated NCB1 or 1KNCB1 conjugates were carried out as in Figs. 1b and 1d, and compared side-by-side in one gel. These data serve as a control for Extended Data Fig. 3a, allowing Ubp6 and USP14 to be compared directly.
Extended Data Figure 4. USP14 deubiquitinates singleton chains poorly, regardless of length or linkage type.

In vitro deubiquitination assays are shown. a, b, ADP-Ptsm (4 nM) was incubated with K63-linked octa-Ub conjugates of K64-only NCB1 (~110 nM) in the presence or absence of USP14 (80 nM). c, Single chain conjugates were generated with Ube2S/UbcH10/APC and K11-only Ub to achieve a homogeneously K11-linked singleton species. Assays were done as in Fig. 1f. d, e, UbcH10/APC-mediated conjugation reactions were performed on the K64-only substrate 1KNCB1 using K48-only Ub (d) or K63-only Ub (e). The conjugation reaction formed rather short chains containing no more than three ubiquitin groups. In vitro degradation and deubiquitination assays were performed as described above. All panels show samples resolved by SDS–PAGE and immunoblotted with HA antibody (a, c–e) or Ub antibody (b).
Extended Data Figure 5. Free chains in trans do not stimulate USP14 activity for singleton conjugates.

In vitro deubiquitination assays with single chain conjugates in the presence of free chains. ADP-Ptsm (4 nM) was incubated with Ubn-1KNCB1 (~110 nM) in the presence or absence of USP14 (80 nM), and with K48Ub4 (a), K48Ub2 (b), or free ubiquitin (c) at the indicated concentrations. Samples were analysed by SDS-PAGE and immunoblotted with an antibody to the HA epitope.
Extended Data Figure 6. A phosphomimetic mutant of USP14 that mimics AKT-phosphorylated enzyme does not promote cleavage of singleton ubiquitin chains.

a, Free USP14 wild-type (400 nM) does not cleave multi-chain cyclin B1 conjugates (~110 nM). b, High concentration of free USP14 does not deubiquitinate Ubn-NCB1. USP14 (15 μM) activity on Ubn-NCB1 (~0.45 μM) in the absence of proteasome was tested by incubating at room temperature for time periods as indicated. Note that there is no obvious deubiquitination of the conjugate by free USP14, although high levels of USP14 were tested (>1000-fold over that of activated, proteasome-associated USP14 in our standard assay). This experiment confirms that deubiquitination of Ubn-NCB1 by USP14 occurs on and requires the proteasome. c, Free USP14-S432E (400 nM) does not cleave single chain cyclin B1 conjugates (~110 nM). d, Proteasome-associated USP14-S432E (4 nM proteasome and 80 nM USP14) does not cleave single chain cyclin B1 conjugates (~110 nM). Assay in d was performed in the presence of ATP under conditions permissive for degradation. Samples were resolved by SDS-PAGE and immunoblotted using an antibody to the HA epitope.
Extended Data Figure 7. USP14 deubiquitinates free chains poorly, regardless of linkage type.

In vitro deubiquitination assays with free chains. a–d, Each linkage-specific free chain (200 nM) was incubated with human proteasome (12 nM) in the presence or absence of USP14 (240 nM). The sample was treated with pan-specific Ub isopeptidase USP21 (400 nM) for 20 min as a positive control showing that chain disassembly is readily detected. e, f, K48-linked Ub3-7 free chains (125 nM) or K63-linked octa-Ub free chains (300 nM) were incubated with human proteasomes (5 nM for e and 10 nM for f) in the presence or absence of USP14 (20-fold excess over proteasome). Samples were resolved by SDS–PAGE and immunoblotted using an antibody to ubiquitin.
Extended Data Figure 8. USP14 cleaves ubiquitin chains at the proximal site.

In vitro deubiquitination assays with multi-chain conjugates. a, Multi-chain K63Ub4 conjugates on NCB1 were prepared using pre-formed K63Ub4 free chains as described in Methods. Assays were performed as in Fig. 2a. Arrow indicates accumulated K63Ub4-NCB1 as major deubiquitinated species generated by USP14 cleavage. Asterisks indicate presumptive deconjugated K63Ub5- or other multi-chain K63Ubn-NCB1 species originating from other K63Ubn contaminants in the commercial preparation of K63Ub4. b, Ubn-NCB1 was generated using UbcH10, APC/C, and wild type ubiquitin, and purified as described in Methods. Note that this conjugate may be enriched with short multi-ubiquitin chains with different linkages, based on previous reports11. Deubiquitination assays were performed as in Fig. 2c. Arrowhead indicates di-Ub chains as major products of USP14 activity, suggesting that Ubn-NCB1 in this sample was rich in di-ubiquitin chains. Samples were resolved by SDS–PAGE and immunoblotted with antibody against the HA epitope (a) or Ub (b). c, TMT labelling and quantitative mass spectrometry. Purified Ubn-NCB1 was assayed in biological duplicates for each condition, and the resulting samples were analysed for specific ubiquitin chain linkages by mass spectrometry as in Fig. 2d. The experiment was repeated in biological duplicates at a 4-min point with equivalent results (data not shown).
Extended Data Figure 9. Modeling of free ubiquitin chain cleavage by proteasome-activated Ubp6 or USP14.
Possible steric conflict between the proximal ubiquitin and proteasome-associated Ubp6 or USP14 was assessed by molecular modeling. a, As in Fig. 2f, except employing K48- or K63-linked proximal ubiquitin. b, Proximal ubiquitin with each different linkage was fitted in USP14 structure with minimal steric hindrance, and then docked onto the proteasome EM structure. Structures were obtained from the PDB database: Ubp6 and proteasome (5A5B), USP14 (2AYO), K63 di-Ub (2RR9), and ubiquitin (1UBQ). Note that for K6, 27, 29, and 33 linkages, modeling on the proteasome was not attempted because of extensive steric occlusion between proximal ubiquitin and USP14.
Extended Data Figure 10. Single-molecule studies of RPN11 show patterns of chain removal similar to those of USP14.

These experiments serve as controls for Fig. 3. Since RPN11 is known to work as a proximal-ubiquitin-specific DUB, its similarity to USP14 in these particular chain removal assays supports the hypothesis that USP14 is a proximal-specific deubiquitinating enzyme. SM assays were performed and analysed as described in Fig. 3, and were carried out using the same samples of proteasomes and ubiquitinated securin. The data shown are examples of step-trace segmentation graphs from an SM analysis of purified Dylight550-labeled Ubn-securin conjugates incubated with human proteasome lacking USP14 in the presence of ATP. These deubiquitination events represent RPN11-mediated activity as previously reported14.
Supplementary Material
Acknowledgments
We thank J. Hanna and Y. Ye for comments on the manuscript. We thank N. Dimova for NCB1 constructs, Y. Ye for USP21 protein, Y. Saeki for PY-Sic1 clones, J. Yuan for USP14 plasmids, and R.P. Fisher and E. Winter for full-length cyclin B1 baculoviruses. This study was supported by NIH grant R01GM5660052 (to D.F.), NIH grant R37-GM043601 (to D.F.), NIH grant R01GM66492-9 (to R.W.K.), NIH grant 5R01GM039023-26 (to M.W. Kirschner), and a grant from the Rainwater Foundation to D.F.
Footnotes
Author Contributions
B.-H.L. performed and analyzed most of biochemical experiments. Y.L. performed single molecule analysis. M.A.P. performed mass spectrometric analysis. Y.S., S.S., G.T., and S.E. generated biochemical reagents. G.T. also performed structural modeling. S.P.G. guided the mass spectrometry experiments. B.-H.L., R.W.K., and D.F. supervised the study.
The authors declare competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Hanna J, et al. Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell. 2006;127:99–111. doi: 10.1016/j.cell.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 2.Hanna J, Meides A, Zhang D, Finley D. A ubiquitin stress response induces altered proteasome composition. Cell. 2007;129:747–759. doi: 10.1016/j.cell.2007.03.042. [DOI] [PubMed] [Google Scholar]
- 3.Lee B, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peth A, Besche HC, Goldberg AL. Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol Cell. 2009;36:794–804. doi: 10.1016/j.molcel.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aufderheide A, et al. Structural characterization of the interaction of Ubp6 with the 26S proteasome. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8626–8631. doi: 10.1073/pnas.1510449112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bashore C, et al. Ubp6 deubiquitinase controls conformational dynamics and substrate degradation of the 26S proteasome. Nature structural & molecular biology. 2015;22:712–719. doi: 10.1038/nsmb.3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leggett D, et al. Multiple associated proteins regulate proteasome structure and function. Mol Cell. 2002;10:495–507. doi: 10.1016/s1097-2765(02)00638-x. [DOI] [PubMed] [Google Scholar]
- 8.Homma T, et al. Ubiquitin-specific protease 14 modulates degradation of cellular prion protein. Scientific reports. 2015;5:11028. doi: 10.1038/srep11028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu M, et al. Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 2005;24:3747–3756. doi: 10.1038/sj.emboj.7600832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirkpatrick DS, et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nature cell biology. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 11.Dimova NV, et al. APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nature cell biology. 2012;14:168–176. doi: 10.1038/ncb2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin L, Williamson A, Banerjee S, Philipp I, Rape M. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell. 2008;133:653–665. doi: 10.1016/j.cell.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. The EMBO journal. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Lee BH, King RW, Finley D, Kirschner MW. Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science. 2015;348:1250834. doi: 10.1126/science.1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mansour W, et al. Disassembly of Lys11 and mixed linkage polyubiquitin conjugates provides insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J Biol Chem. 2015;290:4688–4704. doi: 10.1074/jbc.M114.568295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vaden JH, et al. Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Molecular neurodegeneration. 2015;10:3. doi: 10.1186/1750-1326-10-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu D, et al. Phosphorylation and activation of ubiquitin-specific protease-14 by Akt regulates the ubiquitin-proteasome system. eLife. 2015;4 doi: 10.7554/eLife.10510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verma R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 19.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 20.Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Molecular cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kravtsova-Ivantsiv Y, Cohen S, Ciechanover A. Modification by single ubiquitin moieties rather than polyubiquitination is sufficient for proteasomal processing of the p105 NF-kappaB precursor. Advances in experimental medicine and biology. 2011;691:95–106. doi: 10.1007/978-1-4419-6612-4_10. [DOI] [PubMed] [Google Scholar]
- 22.Haglund K, et al. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol. 2003;5:461–466. doi: 10.1038/ncb983. [DOI] [PubMed] [Google Scholar]
- 23.Pathare GR, et al. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc Natl Acad Sci U S A. 2014;111:2984–2989. doi: 10.1073/pnas.1400546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Worden EJ, Padovani C, Martin A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat Struct Mol Biol. 2014;21:220–227. doi: 10.1038/nsmb.2771. [DOI] [PubMed] [Google Scholar]
- 25.Cox CJ, et al. The regions of securin and cyclin B proteins recognized by the ubiquitination machinery are natively unfolded. FEBS Lett. 2002;527:303–308. doi: 10.1016/s0014-5793(02)03246-5. [DOI] [PubMed] [Google Scholar]
- 26.Krude T, Jackman M, Pines J, Laskey RA. Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell. 1997;88:109–119. doi: 10.1016/s0092-8674(00)81863-2. [DOI] [PubMed] [Google Scholar]
- 27.Huttlin EL, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulo JA, Gaun A, Gygi SP. Global Analysis of Protein Expression and Phosphorylation Levels in Nicotine-Treated Pancreatic Stellate Cells. Journal of proteome research. 2015;14:4246–4256. doi: 10.1021/acs.jproteome.5b00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.