

Abstract
The chirality, or ‘handedness’, of a biologically active molecule can alter its physiological properties. For this reason, it is routine procedure in the drug discovery and development process to prepare and fully characterize all possible stereoisomers of a drug candidate for biological evaluation1,2. Despite many recent advances in asymmetric synthesis, the development of general and practical strategies to obtain all possible stereoisomers of an organic compound bearing multiple contiguous stereocenters remains a significant challenge3. In this manuscript, we report a stereodivergent copper-based approach for the expeditious construction of amino alcohols with high levels of chemo-, regio-, diastereo- and enantioselectivity. Specifically, these amino alcohol products were synthesized using the sequential copper hydride-catalyzed hydrosilylation and hydroamination of readily available enals and enones. This strategy provides a route to all possible stereoisomers of the amino alcohol products, which contain up to three contiguous stereocenters. Catalyst control and stereospecificity were simultaneously leveraged to attain exceptional control of the product stereochemistry. Beyond the utility of this protocol, the strategy demonstrated here should inspire the development of methods providing complete sets of stereoisomers for other valuable synthetic targets.
Different stereoisomers of drugs can have distinct therapeutic properties or adverse effects due to the chiral environments of enzymes and receptors in biological systems. The most well known example is thalidomide, the R-enantiomer of which was an effective sedative, while the S-enantiomer caused severe teratogenic side effects and resulted in tragic birth defects during the 1950s. Stereoisomers of drugs can also have contrasting indications as in the case of quinine and quinidine or even opposing biological activities (Fig. 1a and 1b). For these reasons, regulatory agencies require the evaluation of the bioactivity of all stereoisomers of pharmaceutical candidates during the drug discovery and development process1,2. Furthermore, manufacturers must develop assays to determine stereochemical purity to ensure drug safety. Consequently, all stereoisomers of a molecule must be prepared for use in biological testing or as standard samples. Thus, the construction of complete stereoisomeric sets represents a practically important synthetic problem as well as a fundamentally significant research topic1,2.
Figure 1. Bioactive stereoisomers and strategies for construction of all stereoisomers of amino alcohols.
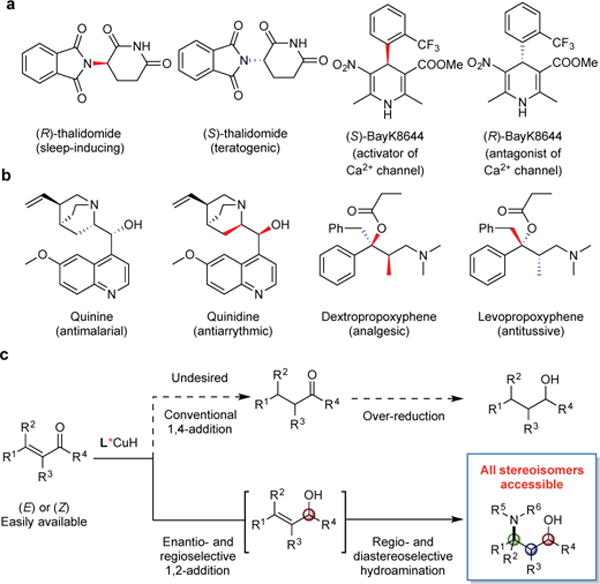
a Different enantiomers give distinct activities in biological systems. b. Stereoisomeric amino alcohols (esters) and their biological activities. c. Top, potential side reactions; Bottom (this work), forming all stereoisomers of amino alcohols via selective hydrosilylation/hydroamination reactions.
In the past few decades, asymmetric synthesis has witnessed tremendous progress, providing numerous chiral bioactive compounds with high levels of selectivity3. Although asymmetric catalysis has allowed enantiomers of a chiral molecule to be obtained with equal ease, relatively few methods are capable of providing a unified route leading to all possible stereoisomers of products containing multiple contiguous stereocenters. Thus, full control of absolute and relative stereochemical configuration remains an unmet synthetic challenge3. Aside from classical techniques in asymmetric catalysis, such as the use of additives4,5, modification of catalyst structure6–10, and variation of substrate protecting group11,12, multi-catalytic approaches13–16 have been advanced to access the full complement of stereoisomers for certain classes of compounds. MacMillan has previously described the novel concept of cycle-specific amino-catalysis in which two chiral catalysts sequentially perform an iminium/enamine catalysis cascade to selectively functionalize enals13,14. Carreira recently demonstrated an elegant dual catalyst system to independently control the two stereocenters for the α-allylation of branched aldehydes15,16. Despite these important developments, a rapid and predictable approach to access complete stereoisomeric sets of important products bearing multiple stereocenters (e.g. three contiguous stereocenters) from readily available precursors based on a single catalyst system would be highly desirable but remains underdeveloped.
Optically pure amino alcohols are important structural elements frequently found in pharmaceutical agents and bioactive natural products (Fig. 1b)17. Moreover, these compounds also serve as important building blocks for catalysts and auxiliaries in asymmetric synthesis. We speculated that our recently disclosed catalytic systems18–23 for enantioselective hydroamination24,25 based on copper hydride (CuH) intermediates26 and electrophilic aminating reagents27 could be applied to the synthesis of this class of compounds. We hypothesized that enals and enones, which are readily available as geometrically pure isomers, could serve as ideal precursors to amino alcohols. In particular, we anticipated that a CuH-based catalyst could reduce enals and enones chemoselectively to the corresponding allylic alcohols, which would undergo regio- and stereoselective hydroamination to afford amino alcohol products bearing multiple contiguous stereocenters in one synthetic operation (Fig. 1c). As a consequence of the synfacial nature of the hydroamination process, we reasoned that this two-step sequence would be stereospecific with respect to olefin geometry. Thus, effective catalyst-control would allow the generation of all diastereomeric possibilities through the appropriate choice of substrate geometry (E or Z) and ligand enantiomer (R or S). Although the asymmetric hydrosilylation of ketones using a copper catalyst is a well-known process28, we were aware that 1,2-reduction of α, β-unsaturated carbonyl compounds by CuH is less favorable than 1,4-reduction due to the inherent preference of Cu to coordinate to the olefin via soft−soft interactions26. Pioneering work by Stryker29 and Lipshutz30 suggested that control of the regioselectivity in the CuH reduction of Michael acceptors was sensitive to subtle variation in steric and electronic properties of the ligand and the substituents of substrates. On the other hand, the control of regio- and stereoselectivity of hydroamination step is also non-trivial25, due to the steric and electronic bias of the allylic silyl ether intermediates and potential matched/mismatched effects between substrates and chiral catalysts. Here, we report a copper-based catalyst system for the execution of this strategy that provides access to all possible amino alcohol stereoisomers in high chemo-, regio-, diastereo- and enantioselectivity starting from readily available enals and enones.
In initial efforts to implement this proposed hydrosilylation/hydroamination sequence, we first treated (E)-2-methyl-cinnamaldehyde (1a) with an excess of dimethoxymethylsilane in the presence of 5 mol % of copper acetate and (S)-DTBM-SEGPHOS (L1) at room temperature for 15 minutes. Neither the conventional 1,4-reduction product nor the over-reduced saturated alcohol was observed and the desired 1,2-adduct was obtained in nearly quantitative yield. Further treatment of the reaction mixture with aminating reagent 2a at 55 °C effectively provided the amino alcohol 3a in 95% yield and with complete diastereo- and enantioselectivity (>20/1 d.r., >99% e.e.). A thorough evaluation of the electrophilic amine source indicated that 2a was the optimal aminating reagent. We attributed the high efficiency of 2a to the electron-rich para-diethylaminobenzoyl group. This group resulted in slower reductive decomposition of 2a and presumably faster regeneration of the CuH catalyst through σ-bond metathesis between the corresponding copper benzoate species and hydrosilane species22,23. L1 was the best ligand among the phosphines screened both in terms of reactivity and selectivity (See Supplementary Information for details).
With these optimized reaction conditions, we investigated the substrate scope of this one-pot transformation. We found that an array of β-aryl substituted (E)-enals could be efficiently transformed to the corresponding chiral amino alcohols in a highly regio-, diastereo- and enantioselective manner (Fig. 2, 3a–3n). A diverse range of hydroxylamine esters and enals with a variety of functional groups were suitable coupling partners for this sequential transformation, including phenols (3g, 3h), an aryl chloride (3k), an ester (3h), an acetal (3d), a trifluoromethoxyl (3e), cis-olefins (3f, 3l) and a trimethylsilanyl group (from the reaction of a vinyl silane) (3l). In addition to acyclic substrates, both a cyclic enal and cyclic aminating reagents were compatible, providing the products with high levels of stereocontrol (3d, 3i, 3k). Furthermore, substrates bearing a broad range of pharmaceutically important heteroaromatic components–including an indole (3g), a thiophene (3f), a pyridine (3h), a pyrrole (3j), a pyrimidylpiperidine (3i), and a chromene (3k)–could be readily converted to the desired products in excellent enantioselectivity. Additionally, an enal substrate bearing a ketone functional group smoothly underwent double hydrosilylation and hydroamination sequence under these conditions, furnishing amino diol (3m) bearing three stereocenters as a single stereoisomer. Lastly, a reaction conducted on a 5 mmol scale with decreased catalyst loading (1 mol%) efficiently provided the desired product (3a) in undiminished yield and stereoselectivity, demonstrating the robustness and practicality of this process.
Figure 2. Asymmetric hydrosilylation/hydroamination of enals.
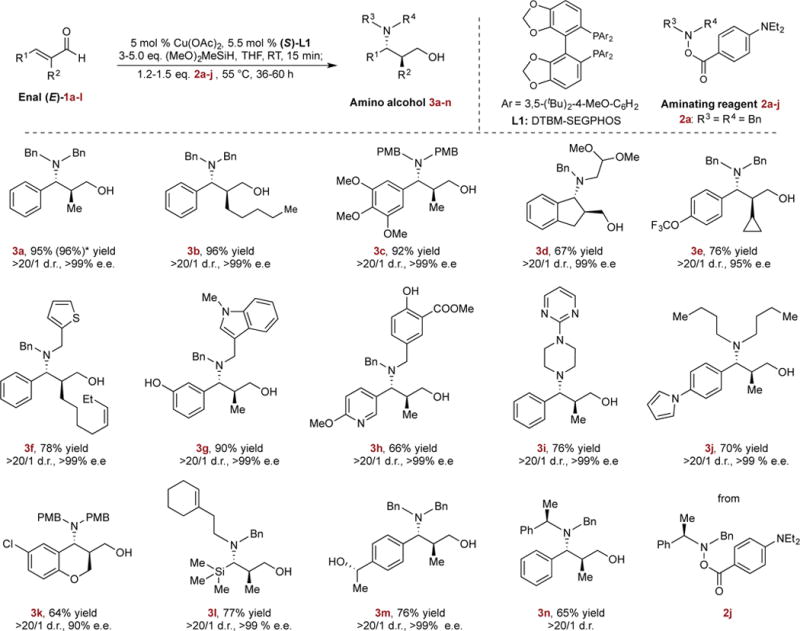
Top row, reaction studied; Bottom rows, substrate scope. Isolated yields are reported (average of two runs on 0.5 mmol scale). Diastereomeric ratios (d.r.) were determined by GC and NMR analysis. Enantiomeric excesses (e.e.) were determined by HPLC analysis using chiral stationary phases. *Reaction was conducted in 5 mmol scale. See Supplementary Information for details. Bn, benzyl; PMB, p-methoxylbenzyl; RT, room temperature.
As previously described, we were particularly interested in applying this approach to selectively access all of the possible stereoisomers, with high diasterero- and enantiocontrol for a given substrate. We felt that the use of the (Z)-enal substrate would lead to the formation of the complementary pair of diastereomers as that realized from the (E)-enal substrate. Accordingly, (Z)-2-amyl-cinnamaldehyde (1b) was subjected to the current reaction conditions, and produced the desired diastereomeric syn-amino alcohol in high chemical yield with complete diastereo- and enantioselectivity. Thus, by using the proper combinations of the enantiomer of ligand for the CuH catalyst and the olefin geometry of the enal substrate, all four stereoisomers of the corresponding amino alcohol could be readily prepared with full control of absolute and relative stereochemistry (Fig. 3a).
Figure 3. All stereoisomers of amino alcohols from enals.
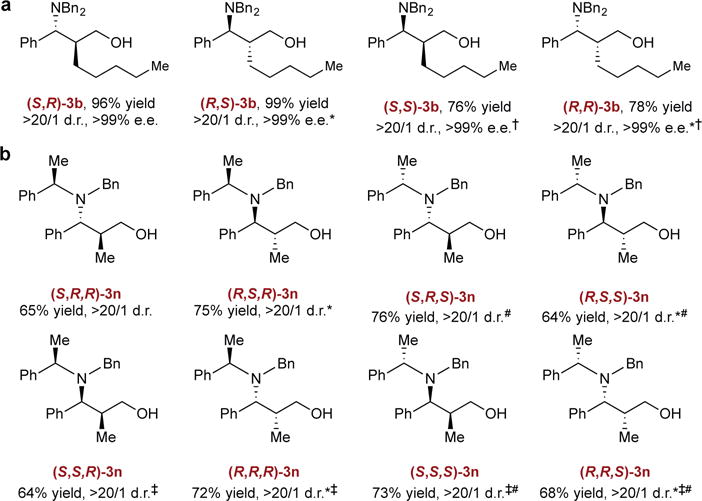
a Reaction using (S)-L1 and (E)-2-amyl-cinnamaldehyde (1b) under the standard conditions; *using (R)-L1 instead of (S)-L1; †using (Z)-1b instead of (E)-1b. b Reaction using (S)-L1, (E)-2-methyl-cinnamaldehyde (1a) and 2j under the standard conditions; *using (R)-L1 instead of (S)-L1; ‡using (Z)-1a instead of (E)-1a; #using ent-2j instead of 2j.
We also found that amino alcohols bearing three stereogenic centers could be generated in excellent catalyst-controlled diastereoselectivity when chiral hydroxylamine esters were used. The existing α-stereocenter on chiral aminating reagents did not interfere with the stereoselectivity when applied to the current catalyst system. Thus, all eight stereoisomers of the amino alcohol 3n were easily constructed in good yield and excellent diastereoselectivity in one step by selecting the appropriate enantiomer of the chiral aminating reagent and geometric isomer of the enal substrate as starting materials and employing either enantiomer of the chiral catalyst (Fig. 3b).
We then sought to examine the possibility of using enones as substrates for the rapid synthesis of chiral amino alcohols with three contiguous stereocenters. Based on Lipshutz’s exceptional work on the asymmetric CuH-catalyzed 1,2-reduction of enones30, we found that readily accessible enone 4a could be effectively converted to the corresponding chiral allylic alcohol in quantitative yield and 92 % e.e. at −60 °C in the presence of 5 mol % of Cu(OAc)2-L1 complex. Upon addition of the aminating reagent to the reaction mixture while heated at 55 °C, 5a was furnished in 76% chemical yield with complete diastereo- and enantioselectivity (>20/1 d.r., >99% e.e.). Under these reaction conditions, a variety of enones were transformed successfully to the respective amino alcohol products (Fig. 4, 5a–5f) with excellent absolute and relative stereoselectivity (>20/1 d.r., >99% e.e.). In addition, a cyclic enone was converted into indanyl amino alcohol 5g in high diastereoselectivity (10/1 d.r.) and outstanding enantioselectivity (>99% e.e.) for both diastereomers. Lastly, we found that α-substitution on the enone was not crucial for selective 1,2-reduction. For instance, less substituted product 5h could be obtained in high enantioselectivity and synthetically useful diastereomeric ratio from benzylidenacetone.
Figure 4. Asymmetric hydrosilylation/hydroamination of enones.
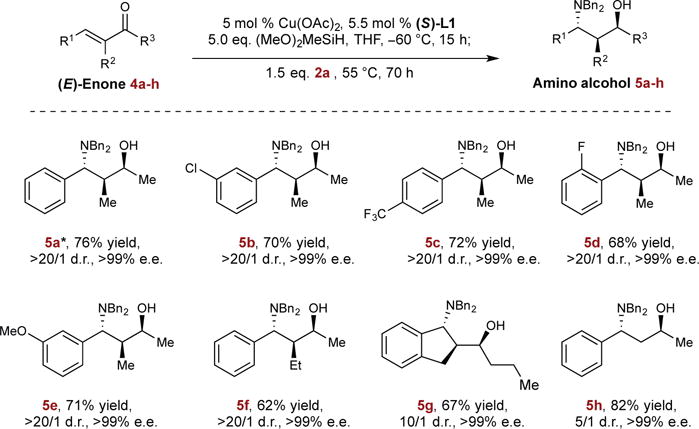
Top row, reaction studied; Bottom rows, substrate scope. Isolated yields are reported (average of two runs on 1.0 mmol scale). Diastereomeric ratios (d.r.) were determined by GC and NMR analysis. Enantiomeric excesses (e.e.) were determined by HPLC analysis. * The absolute and relative stereochemistry of 5a was determined to be (S,S,R) by single crystal X-ray diffraction. See Supplementary Information for details.
Subsequently, we wondered whether these conditions could be adapted to the stereodivergent construction of all eight stereoisomers of 5a. Above (Fig. 4), we prepared (S,S,R)-5a from (E)-4a in a one-pot sequence by employing (S)-L1 as the ligand. To prepare (S,R,S)-5a, we developed a modified protocol involving a ligand switch in which (E)-4a was first hydrosilylated using (S)-L1 as the ligand, and the resulting chiral allylic alcohol was isolated and subjected to hydroamination conditions using copper catalyst based on (R)-L1. Use of enantiomeric ligands for the one-pot and ligand switch protocols yielded (R,R,S)- and (R,S,R)-5a, respectively, allowing four of the possible stereoisomers of 5a to be prepared from (E)-4a. We sought to prepare the four remaining stereoisomers of 5a from (Z)-4a by employing a similar strategy. The same ligand-switch protocol applied to (Z)-4a furnished (R,S,S)-5a and (S,R,R)-5a. Initial attempts to prepare (S,S,S)-5a and (R,R,R)-5a were unsuccessful, presumably due to the unfavorable steric interactions between the intermediate chiral (Z)-allylic alcohol and L1-based catalyst. Accordingly, a less bulky ligand, DM-SEGPHOS (L2), was selected for the hydroamination step to obtain these products. Ultimately, all eight stereoisomers of 5a were prepared expediently in one to two steps in useful isolated yields (33 to 76%), with complete enantioselectivity (>99% e.e.) and good to excellent diastereoselectivity (7:1 - >20:1 produced in the reaction mixture prior to purification) (Fig. 5a). Upon isolation and chromatographic, all isomers were obtained in >20/1 d.r., as confirmed by the HPLC traces displayed in Fig. 5b. These results indicate that the enantioselective hydroamination steps proceeded through excellent catalyst-control in all eight cases.
Figure 5. All eight stereoisomers of amino alcohols from enones.
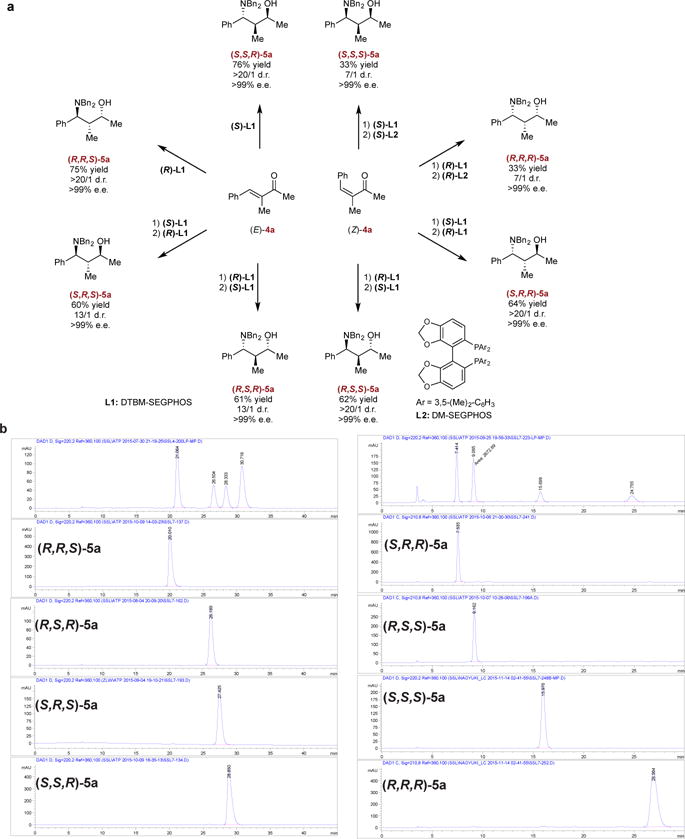
a Access to all stereoisomers via reaction using (E)- or (Z)-4a with catalyst permutation in each step. Isolated yields are reported. Diastereomeric ratios (d.r.) were determined by NMR analysis using a crude reaction mixture. b HPLC traces of all stereoisomeric amino alcohol samples. See Supplementary Information for details.
In conclusion, we have developed a unified and stereodivergent strategy for the rapid and predictable construction of amino alcohols amenable to the synthesis of all possible stereoisomers of a given product. This practical protocol assembles two or three contiguous stereocenters utilizing enal and enone substrates in a highly selective copper catalyzed hydrosilylation/hydroamination sequence. The application of a stereospecific process to readily obtained pure geometric isomers of alkenes and highly effective catalyst control were critical aspects of our approach and may prove to be generally applicable to the development of other enantio- and diastereodivergent hydrofunctionalization reactions.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health (grant GM-58160 for S.L.B.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Dr. Yi-Ming Wang and Dr. Michael T. Pirnot for assistance with the preparation of the manuscript. The departmental X-ray diffraction instrumentation was purchased with the help of funding from NSF (CHE-0946721). We are grateful to P. Müller for X-ray crystallographic analysis of 5a.
Footnotes
Supplementary Information is available in the online version of the paper.
Author contributions S.-L.S. and S.L.B. conceived the idea and designed the research. S.-L.S. and Z.W. performed the experiments. S.-L.S. and S.L.B. wrote the manuscript. All authors commented on the final draft of the manuscript and contributed to the analysis and interpretation of the data.
The authors declare no competing financial interests.
References
- 1.Jozwiak K, Lough WJ, Wainer IW. Drug Stereochemistry: Analytical Methods and Pharmacology. Third. Informa; New York: 2012. [Google Scholar]
- 2.Wermuth CG. The Practice of Medicinal Chemistry. Third. Elsevier; New York: 2008. [Google Scholar]
- 3.Jacobsen EN, Pfaltz A, Yamamoto H, editors. Comprehensive Asymmetric Catalysis: Vol I–III, Suppl I–II. Springer; New York: 1999. [Google Scholar]
- 4.Tian X, et al. Diastereodivergent asymmetric sulfa-Michael additions of α-branched enones using a single chiral organic catalyst. J Am Chem Soc. 2011;133:17934–17941. doi: 10.1021/ja207847p. [DOI] [PubMed] [Google Scholar]
- 5.McInturff EL, Yamaguchi E, Krische MJ. Chiral-anion-dependent inversion of diastereo- and enantioselectivity in carbonyl crotylation via ruthenium-catalyzed butadiene hydrohydroxyalkylation. J Am Chem Soc. 2012;134:20628–20631. doi: 10.1021/ja311208a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang B, Wu F, Wang Y, Liu X, Deng L. Control of diastereoselectivity in tandem asymmetric reactions generating nonadjacent stereocenters with bifunctional catalysis by cinchona alkaloids. J Am Chem Soc. 2007;129:768–769. doi: 10.1021/ja0670409. [DOI] [PubMed] [Google Scholar]
- 7.Yan X-X, et al. Highly diastereoselective switchable enantioselective Mannich reaction of glycine derivatives with imines. J Am Chem Soc. 2008;130:14362–14362. doi: 10.1021/ja804527r. [DOI] [PubMed] [Google Scholar]
- 8.Luparia M, et al. Catalytic asymmetric diastereodivergent deracemization. Angew Chem Int Ed. 2011;50:12631–112635. doi: 10.1002/anie.201106321. [DOI] [PubMed] [Google Scholar]
- 9.Lu G, Yoshino T, Morimoto H, Matsunaga S, Shibasaki M. Stereodivergent direct catalytic asymmetric mannich-type reactions of α-isothiocyanato ester with ketimines. Angew Chem Int Ed. 2011;50:4382–4385. doi: 10.1002/anie.201101034. [DOI] [PubMed] [Google Scholar]
- 10.Mechler M, Peters R. Diastereodivergent asymmetric 1,4-addition of oxindoles to nitroolefins by using polyfunctional nickel-hydrogen-bond-azolium catalysts. Angew Chem Int Ed. 2015;54:10303–10307. doi: 10.1002/anie.201502930. [DOI] [PubMed] [Google Scholar]
- 11.Lee EC, Hodous BL, Bergin E, Shih C, Fu GC. Catalytic asymmetric Staudinger reactions to form β-lactams: an unanticipated dependence of diastereoselectivity on the choice of the nitrogen substituent. J Am Chem Soc. 2005;127:11586–11587. doi: 10.1021/ja052058p. [DOI] [PubMed] [Google Scholar]
- 12.Huber JD, Leighton JL. Highly enantioselective imine cinnamylation with a remarkable diastereochemical switch. J Am Chem Soc. 2007;129:14552–14553. doi: 10.1021/ja076035h. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, Walji AM, Larsen CH, MacMillan DWC. Enantioselective organo-cascade catalysis. J Am Chem Soc. 2005;127:15051–15053. doi: 10.1021/ja055545d. [DOI] [PubMed] [Google Scholar]
- 14.Simmons B, Walji AM, MacMillan DWC. Cycle-specific organocascade catalysis: application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew Chem Int Ed. 2009;48:4349–4353. doi: 10.1002/anie.200900220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Enantio- and diastereodivergent dual catalysis: α-allylation of branched aldehydes. Science. 2013;340:1065–1068. doi: 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]
- 16.Schindler CS, Jacobsen EN. A new twist on cooperative catalysis. Science. 2013;340:1052–1053. doi: 10.1126/science.1238769. [DOI] [PubMed] [Google Scholar]
- 17.Nugent TC. Chiral Amine Synthesis: Methods, Developments and Applications. Wiley VCH; Weinheim: 2010. [Google Scholar]
- 18.Zhu S, Niljianskul N, Buchwald SL. Enantio- and regioselective CuH-catalyzed hydroamination of alkenes. J Am Chem Soc. 2013;135:15746–15749. doi: 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi S-L, Buchwald SL. Copper-catalyzed selective hydroamination reactions of alkynes. Nature Chem. 2015;7:38–44. doi: 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu S, Buchwald SL. Enantioselective CuH-catalyzed anti-Markovnikov hydroamination of 1, 1-disubstituted alkenes. J Am Chem Soc. 2014;136:15913–15916. doi: 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Shi S-L, Niu D, Liu P, Buchwald SL. Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science. 2015;349:62–66. doi: 10.1126/science.aab3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu D, Buchwald SL. Design of modified amine transfer reagents allows the synthesis of α-chiral secondary amines via CuH-catalyzed hydroamination. J Am Chem Soc. 2015;137:9716–9721. doi: 10.1021/jacs.5b05446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bandar JS, Pirnot MT, Buchwald SL. Mechanistic studies lead to dramatically improved reaction conditions for the Cu-catalyzed asymmetric hydroamination of olefins. J Am Chem Soc. 2015;137 doi: 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miki Y, Hirano K, Satoh T, Miura M. Copper-catalyzed intermolecular regioselective hydroamination of styrenes with polymethylhydrosiloxane and hydroxylamines. Angew Chem Int Ed. 2013;52:10830–10834. doi: 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]
- 25.Hannedouche J, Schulz E. Asymmetric hydroamination: a survey of the most recent developments. Chem Eur J. 2013;19:4972–4985. doi: 10.1002/chem.201203956. [DOI] [PubMed] [Google Scholar]
- 26.Deutsch C, Krause N, Lipshutz BH. CuH-catalyzed reactions. Chem Rev. 2008;108:2916–2927. doi: 10.1021/cr0684321. [DOI] [PubMed] [Google Scholar]
- 27.Barker TJ, Jarvo ER. Developments in transition-metal-catalyzed reactions using electrophilic nitrogen sources. Synthesis. 2011;24:3954–3964. [Google Scholar]
- 28.Lipshutz BH, Noson K, Chrisman W, Lower A. Asymmetric hydrosilylation of aryl ketones catalyzed by copper hydride complexed by nonracemic biphenyl bis-phosphine ligands. J Am Chem Soc. 2003;125:8779–8782. doi: 10.1021/ja021391f. [DOI] [PubMed] [Google Scholar]
- 29.Chen JX, et al. Highly chemoselective catalytic hydrogenation of unsaturated ketones and aldehydes to unsaturated alcohols using phosphine-stabilized copper(I) hydride complexes. Tetrahedron. 2000;56:2153–2166. [Google Scholar]
- 30.Moser R, Boskovic ZV, Crowe CS, Lipshutz BH. CuH-catalyzed enantioselective 1,2-reductions of α, β-unsaturated ketones. J Am Chem Soc. 2010;132:7852–7853. doi: 10.1021/ja102689e. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.