

Abstract
To determine the genetic diversity and geographic distribution of Cao Bang virus (CBNV) and to ascertain the existence of CBNV-related hantaviruses, natural history collections of archival tissues from Chinese mole shrews (Anourosorex squamipes) and Taiwanese mole shrews (Anourosorex yamashinai), captured in Guizho Province, People’s Republic of China, and in Nantou County, Taiwan, in 2006 and 1989, respectively, were analyzed for hantavirus RNA by RT-PCR. Pair-wise alignment and comparison of the S-, M- and L-segment sequences indicated CBNV in two of five Chinese mole shrews and a previously unrecognized hantavirus, named Xinyi virus (XYIV), in seven of 15 Taiwanese mole shrews. XYIV was closely related to CBNV in Vietnam and China, as well as to Lianghe virus (LHEV), recently reported as a distinct hantavirus species in Chinese mole shrews from Yunnan Province in China. Phylogenetic analyses, using maximum-likelihood and Bayesian methods, showed that XYIV shared a common ancestry with CBNV and LHEV, in keeping with the evolutionary relationship between Anourosorex mole shrews. Until such time that tissue culture isolates of CBNV, LHEV and XYIV can be fully analyzed, XYIV and LHEV should be regarded as genetic variants, or genotypes, of CBNV.
Keywords: Hantavirus, Shrew, Evolution
1. Introduction
Formerly believed as being hosted exclusively by rodents (order Rodentia, families Muridae and Cricetidae), hantaviruses (family Bunyaviridae, genus Hantavirus) are now known to be harbored by multiple species of shrews and moles (order Eulipotyphla, families Soricidae and Talpidae) throughout Europe, Asia and North America (Yanagihara et al. 2014). Hantaviruses have also been found in crocidurine and myosoricine shrews unique to the African subcontinent (Gu et al., 2013; Kang et al., 2011, 2014; Klempa et al., 2007). And phylogenetically divergent lineages of hantaviruses have been identified recently in insectivorous bats (order Chiroptera) in Asia (Arai et al., 2013; Guo et al., 2013; Xu et al., 2015) and Africa (Sumibcay et al., 2012; Weiss et al., 2012). Thus, the previously unimaginable host diversity of hantaviruses now provides a rich palette from which to draw hypothesis-driven studies on their evolutionary origins and phylogeography (Bennett et al., 2014).
Ample opportunities also abound for validating earlier reports of hantaviruses in non-rodent hosts. In this regard, decades-old reports of suspected hantavirus infection in the Eurasian common shrew (Sorex araneus), Eurasian pygmy shrew (Sorex minutus), Eurasian water shrew (Neomys fodiens), northern short-tailed shrew (Blarina brevicauda) and European mole (Talpa europaea) (Gavrilovskaya et al., 1983; Gligic et al., 1992; Lee et al., 1985; Tkachenko et al., 1983) have been confirmed using powerful gene-amplification techniques, with the discovery of Seewis virus (Song et al., 2007a), Asikkala virus (Radosa et al., 2013), Boginia virus (Gu et al., 2014), Camp Ripley virus (Arai et al., 2007) and Nova virus (Kang et al., 2009), respectively. Also, a hantavirus, isolated from the Chinese mole shrew (Anourosorex squamipes) captured in Sichuan Province in 1986 (Chen et al., 1986) and reported to be closely related to Hantaan virus (HTNV), the prototype virus of hemorrhagic fever with renal syndrome, prompted our studies in Vietnam, where we detected a genetically distinct hantavirus, designated Cao Bang virus (CBNV), in the Chinese mole shrew (Song et al., 2007b). Based on sequence and phylogenetic analyses, CBNV was strikingly different from HTNV, suggesting that the conclusion reached previously was premature. Unfortunately, the unavailability of the original hantavirus isolate from the Chinese mole shrew made impossible any in-depth comparisons.
The discovery of CBNV raises questions about its genetic diversity and geographic distribution, as well as conjectures regarding the existence of CBNV-related hantaviruses, particularly in the Taiwanese mole shrew (Anourosorex yamashinai), formerly classified as a subspecies of the Chinese mole shrew but now considered a distinct species (Hutterer, 2005). In this multi-institutional international collaboration, we demonstrate that genetic variants of CBNV are extant in Chinese mole shrews in the People’s Republic of China, as well as in Taiwanese mole shrews in Taiwan.
2. Materials and methods
Archival frozen tissues from five Chinese mole shrews, captured in Kuankuoshui Nature Reserve in Guizho Province, People’s Republic of China, during April 2006 (Lim et al., 2008), and from 15 Taiwanese mole shrews, captured along Sha-Li-Xian Trail, in Xin-Yi Township, in Nantou County, Taiwan, during September 1989 (Yu, 1993, 1994) (Figure 1), were analyzed for hantavirus RNA by RT-PCR, using newly designed and previously employed oligonucleotide primers (Arai et al., 2007; Gu et al., 2013; Song et al., 2007b). Tissues from 10 Asian gray shrews (Crocidura attenuata) and six Taiwanese gray shrews (Crocidura tanakae), captured in China, were also tested. Total RNA was extracted from tissues using the PureLink Micro-to-Midi total RNA purification kit (Invitrogen, San Diego, CA), and cDNA was synthesized using the SuperScript III First-Strand Synthesis Systems (Invitrogen) with a highly conserved primer and/or random hexamers by two-step RT-PCR cycles. As described previously, first- and second-round PCR were performed in 20-μL reaction mixtures, containing 250 μM dNTP, 2.5 mM MgCl2, 1 U of Takara LA Taq polymerase (Takara, Shiga, Japan) and 0.25 μM of each primer. Initial denaturation at 94°C for 2 min was followed by two cycles each of denaturation at 94°C for 30 sec, two-degree step-down annealing from 46°C to 38°C for 40 sec, and elongation at 72°C for 1 min, then 30 cycles of denaturation at 94°C for 30 sec, annealing at 42°C for 40 sec, and elongation at 72°C for 1 min, in a GeneAmp PCR 9700 thermal cycler (Perkin-Elmer, Waltham, MA). PCR products were separated, using MobiSpin S-400 spin columns (MoBiTec, Goettingen, Germany), and amplicons were sequenced directly using an ABI Prism 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA).
Figure 1.
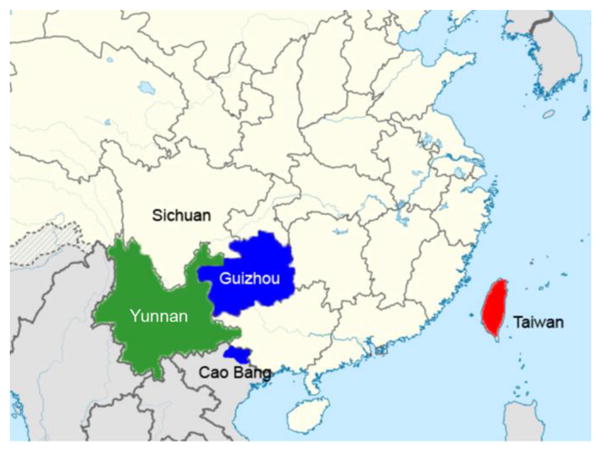
Map of China and neighboring countries, showing sites where Anourosorex mole shrews were trapped. Chinese mole shrew samples were collected in Guizhou (blue) and Yunnan provinces (green) in the People’s Republic of China and in Cao Bang province in Vietnam (blue). Taiwanese mole shrew samples were collected in Taiwan (red).
Phylogenetic trees were generated by maximum likelihood and Bayesian methods, implemented in PAUP* (Phylogenetic Analysis Using Parsimony, 4.0b10) (Swofford, 2003), RAxML Blackbox webserver (Stamatakis et al., 2008) and MrBayes 3.1 (Ronquist and Huelsenbeck, 2003), under the best-fit GTR+I+Γ model of evolution selected by hierarchical likelihood-ratio test in MrModeltest v2.3 (Posada and Crandall, 1998) and jModelTest version 0.1 (Posada, 2008). Two replicate Bayesian Metropolis–Hastings Markov Chain Monte Carlo runs, each consisting of six chains of 10 million generations sampled every 100 generations with a burn-in of 25,000 (25%), resulted in 150,000 trees overall. The S, M and L segments were treated separately in phylogenetic analyses. Topologies were evaluated by bootstrap analysis of 100 iterations (using the RAxML BlackBox), and posterior node probabilities were based on 2 million generations and estimated sample sizes over 100 (implemented in MrBayes).
To verify the species of the hantavirus-infected shrew hosts, the cytochrome b or cytochrome oxidase subunit I gene of mitochondrial DNA (mtDNA) was amplified from tissue DNA by PCR, using conventional primers and methods (Arai et al., 2008; Borisenko et al., 2008).
3. Results and discussion
In employing RT-PCR to analyze archival frozen tissues from natural history collections, comprising Chinese mole shrews, Taiwanese mole shrews, Asian gray shrews and Taiwanese gray shrews, hantavirus RNA was found in two of five Chinese mole shrews from China and in seven of 15 Taiwanese mole shrews from Taiwan (Table 1).
Table 1.
Sequence analysis of Cao Bang virus (CBNV), Lianghe virus (LHEV) and Xinyi virus (XYIV) in Anourosorex shrews.
| Virus | Strain | Host species | Country | S | M | L |
|---|---|---|---|---|---|---|
| CBNV | CBN-3 | Anourosorex squamipes | Vietnam | 1,833 bp EF543524 |
3,637 bp EF543526 |
6,533 bp EF543525 |
| ROM117784 | China | 1,818 bp KJ162406 |
3,628 bp KJ162397 |
3,276 bp KJ162404 |
||
| ROM117730 | 365 bp KJ162407 |
734 bp KJ162405 |
||||
| LHEV | As-1 | Anourosorex squamipes | China | 1,804 bp JX465404 |
||
| As-216 | 1,805 bp JX465405 |
330 bp JX465371 |
||||
| As-217 | 1,806 bp JX465406 |
3,628 bp JX465390 |
1,465 bp JX465372 |
|||
| As-220 | 1,804 bp JX465407 |
3,628 bp JX465391 |
||||
| As-221 | 1,804 bp JX465408 |
3,628 bp JX465392 |
336 bp JX465374 |
|||
| As-222 | 1,814 bp JX465409 |
3,629 bp JX465393 |
330 bp JX465375 |
|||
| As-238 | 1,814 bp JX465410 |
3,632 bp JX465394 |
324 bp JX465376 |
|||
| As-255 | 1,804 bp JX465411 |
3,628 bp JX465395 |
330 bp JX465377 |
|||
| As-311 | 1,804 bp JX465412 |
330 bp JX465378 |
||||
| XYIV | MVZ180988 | Anourosorex yamashinai | Taiwan | 1,805 bp KF705677 |
2,390 bp KF705678 |
6,535 bp KF705679 |
| MVZ180979 | 1,311 bp KT901295 |
594 bp KT901296 |
309 bp KJ162403 |
|||
| MVZ180981 | 1,574 bp KT901297 |
309 bp KJ162402 |
||||
| MVZ180982 | 1,802 bp KT901298 |
1,574 bp KT901299 |
309 bp KJ162401 |
|||
| MVZ180983 | 594 bp KT901300 |
309 bp KJ162398 |
||||
| MVZ180986 | 581 bp KT901301 |
1,574 bp KT901302 |
309 bp KJ162399 |
|||
| MVZ181004 | 1,311 bp KJ191754 |
1,574 bp KT901303 |
309 bp KJ162400 |
The full-length 1,818-nucleotide S-genomic segment of CBNV strain ROM117784, from a Chinese mole shrew captured in Guizhou province, contained a single open reading frame, encoding a predicted nucleocapsid (N) protein of 428 amino acids (nucleotide positions 39 to 1,322), and 5′- and 3′-noncoding regions of 38- and 493-nucleotides, respectively. Although the S-segment nucleotide sequence of CBNV strain ROM117784 differed from prototype CBNV strain CBN-3 from Vietnam by 15.3%, the amino acid sequence difference of the N protein was only 2.6% (Table 2). Also, in analyzing the full-length S segment of Lianghe virus (LHEV), recently reported as a genetically distinct hantavirus in Chinese mole shrews from Yunnan Province (Guo et al., 2013), the amino acid sequence of the N protein differed from CBNV by only 4.4–5.1% (Table 2), well below the 7% difference, set by the International Committee on Taxonomy of Viruses (ICTV) (Plyusnin et al., 2012), to qualify as a hantavirus species. By comparison, the N protein amino acid sequence of XYIV, encoded by the full-length S-genomic segment of XYIV strains MVZ 180982 and MVZ180988 from the Taiwanese mole shrews, differed by 7.2–7.5% from that of the prototype CBNV strain CBN-3 (Table 2). However, the predicted N protein secondary structures of CBNV, LHEV and XYIV, as determined using software available on the @NPS structure server (Combet et al., 2000), were virtually indistinguishable (data not shown).
Table 2.
Nucleotide and amino acid sequence similarity (%) between CBNV strain CBN-3 and representative rodent-, shrew- and talpid-borne hantaviruses.
| Virus strain | S segment
|
M segment
|
L segment
|
|||
|---|---|---|---|---|---|---|
| 1,284nt | 428aa | 3,417nt | 1,139aa | 6,453nt | 2,151aa | |
| CBNV ROM117784 | 84.7 | 97.4 | 82.3 | 93.3 | 83.5 | 96.5 |
| CBNV ROM117730 | 81.3 | 94.2 | – | – | 82.1 | 97.1 |
| LHEV As-1 | 82.5 | 95.1 | – | – | – | – |
| LHEV As-216 | 82.3 | 95.1 | – | – | 77.2 | 94.5 |
| LHEV As-217 | 82.5 | 94.9 | 80.0 | 92.7 | 77.6 | 94.7 |
| LHEV As-220 | 82.6 | 95.3 | 80.2 | 92.4 | – | – |
| LHEV As-221 | 82.5 | 95.3 | 80.1 | 92.5 | 77.0 | 93.8 |
| LHEV As-222 | 82.4 | 95.3 | 79.7 | 91.4 | 79.3 | 92.7 |
| LHEV As-238 | 82.4 | 95.6 | 80.2 | 92.7 | 79.9 | 93.5 |
| LHEV As-255 | 82.4 | 95.3 | 79.0 | 89.6 | 77.2 | 94.5 |
| LHEV As-311 | 82.4 | 95.1 | – | – | 76.0 | 91.8 |
| XYIV MVZ180988 | 77.6 | 92.8 | 77.2 | 86.8 | 80.0 | 93.9 |
| XYIV MVZ180979 | 76.5 | 90.6 | 77.6 | 87.4 | 77.6 | 92.2 |
| XYIV MVZ 180981 | – | – | 79.1 | 88.2 | 77.6 | 92.2 |
| XYIV MVZ 180982 | 80.0 | 92.5 | 79.2 | 88.2 | 77.6 | 92.2 |
| XYIV MVZ 180983 | – | – | 77.8 | 87.4 | 77.6 | 92.2 |
| XYIV MVZ 180986 | 76.5 | 87.0 | 79.0 | 88.2 | 77.6 | 92.2 |
| XYIV MVZ 181004 | 76.3 | 90.6 | 79.1 | 88.2 | 77.6 | 92.2 |
| ARRV MSB73418 | 70.7 | 69.3 | – | – | 78.0 | 90.4 |
| JMSV MSB144475 | 72.0 | 77.1 | 74.0 | 79.6 | 77.5 | 89.3 |
| SWSV mp70 | 65.8 | 69.9 | 77.1 | 81.9 | 73.7 | 82.1 |
| QHSV YN05-284 | 67.3 | 69.9 | 72.0 | 77.5 | 78.6 | 87.9 |
| KKMV MSB148794 | 66.7 | 67.3 | 71.2 | 76.3 | 75.1 | 84.8 |
| MJNV Cl05-11 | 50.8 | 47.7 | 42.9 | 41.3 | 64.5 | 62.3 |
| TPMV VRC66412 | 49.8 | 48.4 | 41.3 | 42.4 | 64.0 | 61.9 |
| BOWV VN1512 | 69.2 | 74.5 | 66.7 | 66.2 | 72.2 | 78.2 |
| TGNV Tan826 | 65.8 | 64.6 | – | – | 72.5 | 77.4 |
| AZGV KBM15 | 68.3 | 63.3 | 70.0 | 76.9 | 72.2 | 77.8 |
| JJUV 10-11 | 69.6 | 70.3 | 66.3 | 63.9 | 71.2 | 77.5 |
| OXBV Ng1453 | 69.3 | 74.5 | 68.2 | 66.4 | 72.8 | 80.6 |
| ASAV N10 | 69.8 | 71.5 | 68.8 | 71.0 | 74.8 | 83.6 |
| RKPV MSB57412 | 64.3 | 61.4 | 59.1 | 51.8 | 68.0 | 68.3 |
| NVAV MSB95703 | 58.5 | 50.2 | 57.4 | 43.2 | 65.2 | 62.0 |
| HTNV 76-118 | 65.1 | 64.3 | 63.7 | 61.0 | 71.6 | 77.9 |
| SEOV 80-39 | 64.0 | 62.6 | 63.9 | 60.8 | 70.7 | 77.5 |
| SOOV SOO-1 | 65.4 | 65.0 | 63.8 | 60.6 | 71.5 | 78.1 |
| DOBV Greece | 65.0 | 62.4 | 63.6 | 61.6 | 72.0 | 78.3 |
| PUUV Sotkamo | 64.3 | 61.9 | 59.1 | 51.6 | 68.5 | 70.2 |
| TULV 5302v | 63.0 | 61.4 | 60.9 | 52.7 | 68.4 | 68.9 |
| PHV PH-1 | 62.6 | 58.2 | 58.2 | 52.2 | 67.1 | 68.5 |
| ANDV Chile9717869 | 63.8 | 61.4 | 61.5 | 54.4 | 67.8 | 69.1 |
| SNV NMH10 | 53.9 | 59.3 | 61.5 | 54.1 | 68.1 | 69.5 |
Abbreviations: ANDV, Andes virus; ARRV, Ash River virus; ASAV, Asama virus; AZGV, Azagny virus; BOWV, Bowé virus; CBNV, Cao Bang virus; DOBV, Dobrava virus; HTNV, Hantaan virus; JJUV, Jeju virus; JMSV, Jemez Spring virus; KKMV, Kenkeme virus; LHEV, Lianghe virus; MJNV, Imjin virus; NVAV, Nova virus; OXBV, Oxbow virus; PHV, Prospect Hill virus; PUUV, Puumala virus; QHSV, Qian Hu Shan virus; RKPV, Rockport virus; SEOV, Seoul virus; SNV, Sin Nombre virus; SOOV, Soochong virus; SWSV, Seewis virus; TGNV, Tanganya virus; TPMV, Thottapalayam virus; TULV, Tula virus; XYIV, Xinyi virus. nt, nucleotides; aa, amino acids.
– sequences unavailable
Analysis of the amino acid sequences of the M segment-encoded glycoproteins of CBNV and LHEV showed differences of 7.3–10.4%. Unfortunately, the full-length amino acid sequence of XYIV was unavailable for comparison. However, in applying the more stringent criteria of 10% and 12% amino acid sequence difference for the N protein and envelope glycoproteins, neither XYIV nor LHEV would qualify as a distinct hantavirus species (Maes et al., 2009). But having at least a 7% amino acid sequence difference in the complete N protein and glycoproteins is only one of four ICTV criteria. The other three taxonomic criteria include a unique ecological niche of the reservoir host; at least a 4-fold difference in two-way cross-neutralization antibody tests; and no naturally occurring reassortants (Plyusnin et al., 2012). In the absence of cell-culture isolates of CBNV, LHEV and XYIV, it is impossible to address the third critical criterion. Thus, for the time being, the conservative stance would be to consider XYIV and LHEV as genetic variants of CBNV, rather than as distinct hantavirus species.
Alignment and comparison of the full-length 6533- and 6535-nucleotide L-genomic segment of CBNV and XYIV, respectively, showed equal divergence from other shrew-borne hantaviruses, differing by as much as 38.1% at the amino acid level. However, the L-segment amino acid sequences of CBNV and XYIV exhibited a high degree of sequence conservation, differing by only 6.1%, presumably due to the functional constraints on the RNA-dependent RNA polymerase.
Phylogenetic analyses, based on S-, M- and L-segment sequences, using maximum-likelihood and Bayesian methods, with the GTR+I+Γ model of evolution, showed that XYIV shared a common ancestry with CBNV (Figure 2), in keeping with the evolutionary relationship between these Anourosorex mole shrews. Despite small differences between trees based on each genomic segment, the topologies were generally congruent and highly supported by bootstrap values (>70%) and posterior node probabilities (>0.70). The phylogenetic positions of XYIV, LHEV and CBNV in the S- and M-segment trees were identical, whereas in the L-segment tree, CBNV and XYIV segregated apart from LHEV. Possibly the limited L-segment sequences of LHEV might account for this different topology.
Figure 2.

Phylogenetic trees were generated by maximum-likelihood and Bayesian methods, using the GTR+I+Γ model of evolution, based on the alignment of the S-, M- and L-segment sequences of hantavirus strains. Since tree topologies were very similar using RAxML and MrBayes, the trees generated by MrBayes were displayed. The phylogenetic positions of Cao Bang virus (CBNV CBN-3, S: EF543524, M: EF543526, L: EF543525) and CBNV ROM117784 (S: KJ162406, M: KJ162397, L: KJ162404) and CBNV ROM117730 (S: KJ162407, L: KJ162405) (blue) are shown in relationship to Xinyi virus (XYIV) (red) and Lianghe virus (LHEV) (green). GenBank numbers for XYIV and LHEV are provided in Table 1. Also shown are the phylogenetic positions of Nova virus (NVAV MSB95703, S: FJ539168, M: HQ840957, L: FJ593498), Thottapalayam virus (TPMV VRC66412, S: AY526097, M: EU001329, L: EU001330), Imjin virus (MJNV Cl05-11, S: EF641804, M: EF641798, L: EF641806), Seewis virus (SWSV mp70, S: EF636024, M: EF636025, L: EF636026), Kenkeme virus (KKMV MSB148794, S: GQ306148, M: GQ306149, L: GQ306150), Ash River virus (ARRV MSB 73418, S: EF650086, L: EF619961), Jemez Springs virus (JMSV MSB144475, S: FJ593499, M: FJ593500, L: FJ593501), Qian Hu Shan virus (QHSV YN05-284, S: GU566023, M: GU566022, L: GU566021), Tanganya virus (TGNV Tan826, S: EF050455, L: EF050454), Azagny virus (AZGV KBM15, S: JF276226, M: JF276227, L: JF276228), Jeju virus (JJUV 10–11, S: HQ834695, M: HQ834696, L: HQ834697), Bowé virus (BOWV VN1512, S: KC631782, M: KC631783, L: KC631784), Asama virus (ASAV N10, S: EU929072, M: EU929075, L: EU929078), Oxbow virus (OXBV Ng1453, S: FJ5339166, M: FJ539167, L: FJ593497) and Rockport virus (RKPV MSB57412, S: HM015223, M: HM015219, L: HM015221). Also shown are representative Murinae rodent-borne hantaviruses, including Hantaan virus (HTNV 76–118, S: NC_005218, M: Y00386, L: NC_005222), Soochong virus (SOOV SOO-1, S: AY675349, M: AY675353, L: DQ056292), Dobrava virus (DOBV Greece, S: NC_005233, M: NC_005234, L: NC_005235), Seoul virus (SEOV 80–39, S: NC_005236, M: NC_005237, L: NC_005238); Arvicolinae rodent-borne hantaviruses, including Tula virus (TULV 5302v, S: NC_005227, M: NC_005228, L: NC_005226), Puumala virus (PUUV Sotkamo, S: NC_005224, M: NC_005223, L: NC_005225) and Prospect Hill virus (PHV PH-1, S: Z49098, M: X55129, L: EF646763); and Neotominae rodent-borne hantaviruses, Sin Nombre virus (SNV NMH10, S: NC_005216, M: NC_005215, L: NC_005217) and Andes virus (ANDV Chile9717869, S: NC_003466, M: NC_003467, L: NC_003468). The numbers at each node are posterior node probabilities (left) based on 150,000 trees implemented in MrBayes and rapid bootstrap values (right) based on 100 replicates executed on the RAxML BlackBox webserver, respectively. The scale bar indicates nucleotide substitutions per site.
As determined by analysis of the cytochrome b or cytochrome oxidase subunit I mtDNA genes, the taxonomic identities of the CBNV- and XYIV-infected shrews were confirmed as Anourosorex squamipes and Anourosorex yamashinai, respectively. The evolutionary relationship between Anourosorex mole shrews (Figure 3) was consistent with the phylogenetic positions of CBNV and XYIV.
Figure 3.
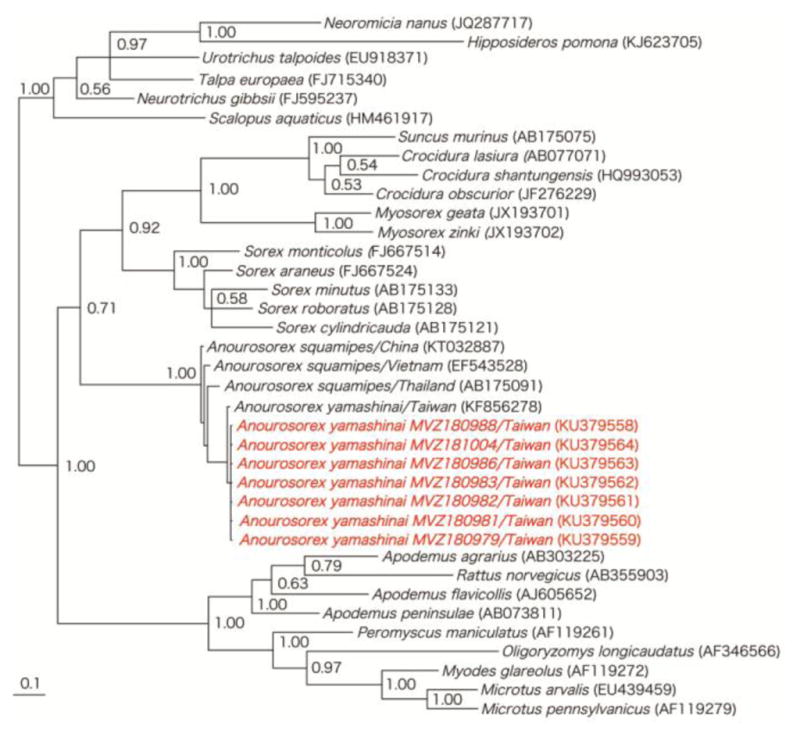
Unrooted phylogenetic tree, using Bayesian method, based on 551- to 1140-nucleotides of the cytochrome b mtDNA of rodents, shrews, moles and bats known to harbor hantaviruses. GenBank accession numbers for cytochrome b mtDNA sequences of Anourosorex yamashinai MVZ180979, MVZ180981, MVZ180982, MVZ180983, MVZ180986, MVZ180988 and MVZ181004 and of other taxa are shown on the tree. Numbers at nodes indicate posterior probability values based on 150,000 trees: two replicate Markov chain Monte Carlo runs, consisting of six chains of 10 million generations each sampled every 100 generations with a burn-in of 25,000 (25%).
The Chinese mole shrew is a forest-dwelling soricine shrew species typically residing at elevations between 1,500 and 3,000 meters, in western and central China, northern Myanmar, northern Thailand, Assam, Bhutan, northern Vietnam and possibly Laos. Endemic to Taiwan, the Taiwanese mole shrew is a semi-fossorial species which is widely distributed in mountainous regions from about 300 m elevation, it is most abundant in hardwood deciduous forests at 1,500–2,500 m elevation, but can be found in agricultural fields, riparian woodlands, and dwarf bamboo groves. In Yushan National Park, the Taiwanese mole shrew is widely distributed across most elevations and is typically found in moist microhabitats, especially along streams in both broad-leaf and conifer forests (Yu, 1993, 1994).
Based on morphological and karyotypic features, the Taiwanese mole shrew is now considered a distinct species (Hutterer, 2005). Previously, sequence analysis of the mitochondrial cytochrome b gene of Taiwanese mole shrews from three mountain ranges in Taiwan disclosed 36 haplotypes, which constituted three geographic-specific phylogroups, presumably the result of interglacial refugia (Northern, Houhuan and Southern) in Taiwan during the middle Pleistocene (divergence time, 0.63–0.71 Mya) (Yuan et al., 2006). Phylogeographic studies of XYIV in Taiwanese mole shrews representing each of these phylogroups would provide insights into the evolutionary history of this newly identified soricid-borne hantavirus. Finally, the isolation of XYIV in cell culture will better clarify its biology, as well as its pathogenic potential for humans (Yanagihara et al., 2015).
Highlights.
Cao Bang virus is harbored by the Chinese mole shrew (Anourosorex squamipes) in Vietnam and China.
Xinyi virus in the Taiwanese mole shrews (Anourosorex yamashinai) appears to represent a genetic variant of Cao Bang virus.
Anourosorex mole shrew species and the hantaviruses they harbor share common ancestries.
Acknowledgments
We thank Eileen Lacey and Chris Conroy, of the Museum of Vertebrate Zoology, for providing archival tissues from Taiwanese mole shrews. The technical assistance of Laarni Sumibcay is also gratefully acknowledged. This work was supported in part by U.S. Public Health Service grants R01AI075057, P20GM103516 and P30GM114737 from the National Institutes of Health, and grant S13205 from the Japan Society for the Promotion of Science. The services provided by the Genomics Core Facility, funded partially by the Centers of Biomedical Research Excellence program (P30GM103341), are acknowledged. The funding agencies had no role in the study design, data collection and analysis, manuscript preparation, and/or decision to publish.
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arai S, Song JW, Sumibcay L, Bennett SN, Nerurkar VR, Parmenter C, Cook JA, Yates TL, Yanagihara R. Hantavirus in northern short-tailed shrew, United States. Emerg Infect Dis. 2007;13:1420–1423. doi: 10.3201/eid1309.070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai S, Ohdachi SD, Asakawa M, Kang HJ, Mocz G, Arikawa J, Okabe N, Yanagihara R. Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides) Proc Natl Acad Sci USA. 2008;105:16296–16301. doi: 10.1073/pnas.0808942105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai S, Nguyen ST, Boldgiv B, Fukui D, Araki K, Dang CN, Ohdachi SD, Nguyen NX, Pham TD, Boldbaatar B, Satoh H, Yoshikawa Y, Morikawa S, Tanaka-Taya K, Yanagihara R, Oishi K. Novel bat-borne hantavirus, Vietnam. Emerg Infect Dis. 2013;19:1159–1161. doi: 10.3201/eid1907.121549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SN, Gu SH, Kang HJ, Arai S, Yanagihara R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014;22:473–482. doi: 10.1016/j.tim.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisenko AV, Lim BK, Ivanova NV, Hanner RH, Hebert PDN. DNA barcoding in surveys of small mammal communities: a field study in Suriname. Mol Ecol Resour. 2008;8:471–479. doi: 10.1111/j.1471-8286.2007.01998.x. [DOI] [PubMed] [Google Scholar]
- Chen SZ, Chen LL, Tao GF, Fu JL, Zhang CA, Wu YT, Luo LJ, Wang YZ. Strains of epidemic hemorrhagic fever virus isolated from the lungs of C. russula and A. squamipes. Chin J Prev Med. 1986;20:261–263. [PubMed] [Google Scholar]
- Combet C, Blanchet C, Geourjon C, Deléage G. NPS@: Network Protein Sequence Analysis. Trends Biochem Sci. 2000;25:147–150. doi: 10.1016/s0968-0004(99)01540-6. [DOI] [PubMed] [Google Scholar]
- Gavrilovskaya IN, Apekina NS, Myasnikov YA, Bernshtein AD, Ryltseva EV, Gorbachkova EA, Chumakov MP. Features of circulation of hemorrhagic fever with renal syndrome (HFRS) virus among small mammals in the European U.S.S.R. Arch Virol. 1983;75:313–316. doi: 10.1007/BF01314898. [DOI] [PubMed] [Google Scholar]
- Gligic A, Stojanovic R, Obradovic M, Hlaca D, Dimkovic N, Diglisic G, Lukac V, Ler Z, Bogdanovic R, Antonijevic B, Ropac D, Avsic T, LeDuc JW, Ksiazek T, Yanagihara R, Gajdusek DC. Hemorrhagic fever with renal syndrome in Yugoslavia: epidemiologic and epizootiologic features of a nationwide outbreak in 1989. Eur J Epidemiol. 1992;8:816–825. doi: 10.1007/BF00145326. [DOI] [PubMed] [Google Scholar]
- Gu SH, Nicolas V, Lalis A, Sathirapongsasuti N, Yanagihara R. Complete genome sequence analysis and molecular phylogeny of a newfound hantavirus harbored by the Doucet’s musk shrew (Crocidura douceti) in Guinea. Infect Genet Evol. 2013;20:118–123. doi: 10.1016/j.meegid.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu SH, Markowski J, Kang HJ, Hejduk J, Sikorska B, Liberski PP, Yanagihara R. Boginia virus, a newfound hantavirus harbored by the Eurasian water shrew (Neomys fodiens) in Poland. Virol J. 2014;10:160. doi: 10.1186/1743-422X-10-160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo WP, Lin XD, Wang W, Tian JH, Cong ML, Zhang HL, Wang MR, Zhou RH, Wang JB, Li MH, Xu J, Holmes EC, Zhang YZ. Phylogeny and origins of hantaviruses harbored by bats, insectivores and rodents. PLoS Pathog. 2013;9:e1003159. doi: 10.1371/journal.ppat.1003159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutterer R. Order Soricomorpha. In: Wilson DE, Reeder DM, editors. Mammal Species of the World. Johns Hopkins University Press; Baltimore, MD: 2005. pp. 220–311. [Google Scholar]
- Kang HJ, Bennett SN, Sumibcay L, Arai S, Hope AG, Mocz G, Song JW, Cook JA, Yanagihara R. Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea) PLoS ONE. 2009;4:e6149. doi: 10.1371/journal.pone.0006149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kadjo B, Dubey S, Jacquet F, Yanagihara R. Molecular evolution of Azagny virus, a newfound hantavirus harbored by the West African pygmy shrew (Crocidura obscurior) in Côte d’Ivoire. Virol J. 2011;8:373. doi: 10.1186/1743-422X-8-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Stanley WT, Esselstyn JA, Gu SH, Yanagihara R. Expanded host diversity and geographic distribution of hantaviruses in sub-Saharan Africa. J Virol. 2014;88:7663–7667. doi: 10.1128/JVI.00285-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempa B, Fichet-Calvet E, Lecompte E, Auste B, Aniskin V, Meisel H, Barriere P, Koivogui L, ter Meulen J, Krüger DH. Novel hantavirus sequences in shrew, Guinea. Emerg Infect Dis. 2007;13:520–522. doi: 10.3201/eid1303.061198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempa B, Avsic-Zupanc T, Clement J, Dzagurova TK, Henttonen H, Heyman P, Jakab F, Krüger DH, Maes P, Papa A, Tkachenko EA, Ulrich RG, Vapalahti O, Vaheri A. Complex evolution and epidemiology of Dobrava-Belgrade hantavirus: definition of genotypes and their characteristics. Arch Virol. 2013;158:521–529. doi: 10.1007/s00705-012-1514-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JG, Gu SH, Baek LJ, Shin OS, Park KS, Kim HC, Klein TA, Yanagihara R, Song JW. Muju virus, harbored by Myodes regulus in Korea, might represent a genetic variant of Puumala virus, the prototype arvicolid rodent-borne hantavirus. Viruses. 2014;6:1701–1714. doi: 10.3390/v6041701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PW, Amyx HL, Yanagihara R, Gajdusek DC, Goldgaber D, Gibbs CJ., Jr Partial characterization of Prospect Hill virus isolated from meadow voles in the United States. J Infect Dis. 1985;152:826–829. doi: 10.1093/infdis/152.4.826. [DOI] [PubMed] [Google Scholar]
- Lim BK, Eger JL, Peterson AT, Robbins MB, Clayton DH, Bush SE, Brown RM. Biodiversity in China: lost in the masses? Harvard Asia Quarterly. 2008;11:12–23. [Google Scholar]
- Maes P, Klempa B, Clement J, Matthijnssens J, Gajdusek DC, Krüger DH, Van Ranst M. A proposal for new criteria for the classification of hantaviruses, based on S and M segment protein sequences. Infect Genet Evol. 2009;9:813–820. doi: 10.1016/j.meegid.2009.04.012. [DOI] [PubMed] [Google Scholar]
- Plyusnin A, Beatty BJ, Elliott RM, Goldbach R, Kormelink R, Lundkvist A, Schmaljohn CS, Tesh RB. Bunyaviridae. In: King AMQ, Lefkowitz EJ, Adams MJ, Carstens EB, editors. Virus Taxonomy: Classification and Nomenclature of Viruses. Ninth Report of the International Committee on Taxonomy of Viruses. Elsevier Academic Press; San Diego, CA: 2012. pp. 725–741. [Google Scholar]
- Posada D. jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008;25:1253–1256. doi: 10.1093/molbev/msn083. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. [DOI] [PubMed] [Google Scholar]
- Radosa L, Schlegel M, Gebauer P, Ansorge H, Heroldová M, Jánová E, Stanko M, Mošanský L, Fričová J, Pejčoch M, Suchomel J, Purchart L, Groschup MH, Krüger DH, Ulrich RG, Klempa B. Detection of shrew-borne hantavirus in Eurasian pygmy shrew (Sorex minutus) in Central Europe. Infect Genet Evol. 2013;19:403–410. doi: 10.1016/j.meegid.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Song JW, Gu SH, Bennett SN, Arai S, Puorger M, Hilbe M, Yanagihara R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus) Virol J. 2007a;4:114. doi: 10.1186/1743-422X-4-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JW, Kang HJ, Song KJ, Truong TT, Bennett SN, Arai S, Truong NU, Yanagihara R. Newfound hantavirus in Chinese mole hantavirus in Chinese mole shrew, Vietnam. Emerg Infect Dis. 2007b;13:1784–1787. doi: 10.3201/eid1311.070492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. A rapid bootstrap algorithm for the RAxML Web-Servers. Syst Biol. 2008;75:758–771. doi: 10.1080/10635150802429642. [DOI] [PubMed] [Google Scholar]
- Sumibcay L, Kadjo B, Gu SH, Kang HJ, Lim BK, Cook JA, Song JW, Yanagihara R. Divergent lineage of a novel hantavirus in the banana pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol J. 2012;9:34. doi: 10.1186/1743-422X-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford DL. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods) Sinauer Associates; Sunderland, Massachusetts: 2003. Version 4. [Google Scholar]
- Tkachenko EA, Ivanov AP, Donets MA, Miasnikov YA, Ryltseva EV, Gaponova LK, Bashkirtsev VN, Okulova NM, Drozdov SG, Slonova RA, Somov GP, Astakhova TI. Potential reservoir and vectors of haemorrhagic fever with renal syndrome (HFRS) in the U.S.S.R. Ann Soc Belg Med Trop. 1983;63:267–269. [PubMed] [Google Scholar]
- Weiss S, Witkowski PT, Auste B, Nowak K, Weber N, Fahr J, Mombouli JV, Wolfe ND, Drexler JF, Drosten C, Klempa B, Leendertz FH, Krüger DH. Hantavirus in bat, Sierra Leone. Emerg Infect Dis. 2012;18:159–161. doi: 10.3201/eid1801.111026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Wu J, He B, Qin S, Xia L, Qin M, Li N, Tu C. Novel hantavirus identified in black-bearded tomb bats, China. Infect Genet Evol. 2015;31:158–160. doi: 10.1016/j.meegid.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara R, Gu SH, Arai S, Kang HJ, Song JW. Hantaviruses: rediscovery and new beginnings. Virus Res. 2014;187:6–14. doi: 10.1016/j.virusres.2013.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara R, Gu SH, Song J-W. Expanded host diversity and global distribution of hantaviruses: implications for identifying and investigating previously unrecognized hantaviral diseases. In: Shapshak P, Sinnott JT, Chiappelli F, Kuhn J, editors. Global Virology I–Identifying and Investigating Viral Diseases. Springer Publishing Company; 2015. pp. 161–198. [Google Scholar]
- Yashina LN, Abramov SA, Dupal TA, Danchinova GA, Malyshev BS, Hay J, Gu SH, Yanagihara R. Hokkaido genotype of Puumala virus in the grey red-backed vole (Myodes rufocanus) and northern red-backed vole (Myodes rutilus) in Siberia. Infect Genet Evol. 2015;33:304–313. doi: 10.1016/j.meegid.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu HT. Natural history of small mammals of subtropical montane areas in central Taiwan. J Zool. 1993;231:403–422. [Google Scholar]
- Yu HT. Distribution and abundance of small mammals along a subtropical elevational gradient in central Taiwan. J Zool. 1994;234:577–600. [Google Scholar]
- Yuan SL, Lin LK, Oshida T. Phylogeography of the mole-shrew (Anourosorex yamashinai) in Taiwan: implications of interglacial refugia in a high-elevation small mammal. Mol Ecol. 2006;15:2119–2130. doi: 10.1111/j.1365-294X.2006.02875.x. [DOI] [PubMed] [Google Scholar]