

Abstract
Purpose
To make comparative analyses of the common three purification protocols for retinal ganglion cells (RGCs), providing a solid practical basis for selecting the method for purifying RGCs for use in subsequent experiments.
Methods
Rat RGCs were isolated and purified using three methods, including two-step immunopanning (TIP) separation, two-step immunopanning-magnetic (TIPM) separation, and flow cytometric (FC) separation. Immunocytochemical staining, quantitative real-time PCR, flow cytometry, electrophysiology, and Cell Counting Kit-8 (CCK-8) analyses were performed to compare the purity, yield, and viability of the RGCs.
Results
The RGC yields from the TIP, TIPM, and FC methods were 24.60±15.98 × 104, 5.28±4.42 × 104, and 5.4±2.7 × 103 per retina, respectively. We easily controlled the relative purity of the RGCs with the FC method and even reached 100% of the maximum expected purity. However, the RGC purity was only 80.97±5.45% and 95.41±3.23% using the TIP and TIPM methods, respectively. The contaminant cells were mainly large, star-shaped, glial fibrillary acidic protein (GFAP)-positive astrocytes and small, round, syntaxin 1-positive amacrine cells with multiple short neurites. The RGCs purified with FC could not be cultured successively in our study; however, the TIP-RGCs survived more than 20 days with good viability, while the TIPM-RGCs survived less than 9 days.
Conclusions
The three protocols for purifying the RGCs each had its own pros and cons. The RGCs isolated by the TIP method exhibited the highest viability and yield but had low purity. The purity of the RGCs isolated with the FC method could reach approximately 100% but had a low yield and cell viability. The TIPM method was reliable and produced RGCs with considerable purity, yield, and viability. This study provides a solid practical basis for selecting the method for purifying RGCs for use in subsequent experiments.
Introduction
Retinal ganglion cells (RGCs) are the sole output neurons that aid in extending axons throughout the optic nerve to receive, process, and relay light-evoked signals to the brain via the optic nerve [1]. RGCs are one of the most important retinal cells. Their anatomic or functional impairment is associated with or a consequence of many ophthalmic disorders, such as diabetic retinopathy or glaucomatous optic neuropathy [2-4], central retinal artery or vein occlusion, etc. [5], and may eventually result in optic neuropathy and vision loss [6]. Unfortunately, why and how the disease-associated RGCs degenerate are largely unknown [5]. Therefore, it is of vital importance to obtain an in-depth understanding of the mechanisms of RGC death to identify new therapeutic strategies for protecting RGCs.
An in vitro analysis of RGCs will be a crucial and almost indispensable tool for the study of retinal visual physiology and pathophysiology associated with various retinopathies and neuropathies, which cannot easily be realized in animal models. For instance, RGCs can be studied in isolation and observed over time, ruling out the effects of other types of cells in the retina. The RGC receptors and signaling pathways can be precisely and quantitatively perturbed using specific chemical factors or pharmacological agents or by introducing genes of interest, and the consequences for cell biology could be evaluated using molecular biology, electrophysiological, or imaging techniques. Using these techniques in situ within an animal model would be technically challenging. Based on their high research value and urgent need, several types of culture models, including mixed retinal cells [7], purified RGCs [8], transformed RGC cell lines [9,10], retinal explant cells [11,12], embryonic stem (ES) cells, and induced pluripotent stem (iPS) cell cultures [13-15] have been established. However, most studies have limitations. For example, the immortalized RGC-5 cell line has been widely used to study the neurobiology of RGCs. However, Krishnamoorthy et al. demonstrated that the purported rat ganglion cell line RGC-5 is in fact of mouse origin and contaminated with 661W cells; therefore, any findings using RGC-5 cells as an in vitro model for RGCs must be carefully interpreted [16], thus largely limiting their usefulness [7]. RGC explant cultures are a mixed culture of different retinal cell types, and studies have shown that RGCs constitute only 5% of the total retinal cells in the mixed culture, thus limiting the application of RGCs in the study of RGC function [17]. IPS cells can directly differentiate into RGCs but require highly sophisticated techniques, and the cells often exhibit a low differentiation rate. Therefore, there is a mounting need to establish an effective system for isolating primary RGCs.
RGCs comprise the innermost layer of the retina and represent less than 1% of the total population of various types of retinal neurons and non-neuronal cells [8], making the purification of the cells more difficult. There have been many methods for the isolation, purification, and culture of RGCs from retinas [18-24]. As each method has its pros and cons, identifying the optimal method for purifying RGCs for specific applications can be difficult. Until recently, no systematic study has analyzed the strengths and weaknesses of every approach. In this work, we used the three most common methods for purifying RGCs from newborn rat retinas and compared the efficiency of each method, providing a practical basis for selecting the method for purifying RGCs for use in subsequent experiments.
Methods
Experiments were approved by the Shanghai Jiao Tong University Institutional Animal Care and Use Committee (IACUC) and adhered to the ARVO Statement for Use of Animals. Here we describe the three most common methods for purifying the RGCs from newborn rat retinas and compared the efficiency of each method with different experimental methods. An overview of the method is provided in the flowchart (Figure 1).
Figure 1.

The protocol steps for isolating and analyzing RGCs are outlined and illustrated in this schematic procedure workflow.
Preparation of the retinal cell suspensions
The retinal tissues were separated from the enucleated eyeballs of newborn Sprague-Dawley rats on postnatal days 1 to 4 and incubated in precooled calcium-free and magnesium-free Earle’s Balanced Salt Solution (EBSS; Gibco, Grand Island, NY) and Hank’s Balanced Salt Solution (Life Technologies, Grand Island, NY) containing 5 mg/ml of papain, 0.24 mg/ml of L-cysteine, and 10 U/ml of DNase І for 30 min. Then, an ovomucoid solution containing 0.1% bovine serum albumin (BSA, Sigma-Aldrich, St. Louis, MO), 0.1% ovomucoid (Sigma-Aldrich), and 1% DNase І (4 mg/ml, Sigma-Aldrich) in minimum essential media (MEM, Gibco) was subsequently used to fully quench any residual papain activity. After centrifugation at 200 ×g for 10 min, the cells were resuspended in MEM containing 0.5 mg/ml of BSA, and then the cell suspension was filtered through the mesh filter (pore size 40 μm, BD Falcon, Franklin Lakes, NJ) to yield a single cell suspension. The procedures were conducted at room temperature in a laminar flow hood.
RGC purification
Preparation of panning dishes and cell culture dishes/plates
The antibody-coated 10-cm Petri dishes (one dish per every eight rats) were prepared for negative or positive selection by adding 15 μl of a rabbit anti-rat macrophage/Thy-1 antibody and 7 ml of 50 mM Tris-HCl (pH 9.5) per dish. The plates were swirled until the surfaces were evenly coated with the antibody-Tris solution. The panning plates were incubated overnight at 4 °C. Immediately before use, the plates were rinsed three times with Dulbecco’s PBS (1X; 0.9 mM CaCl2, 0.49 mM MgCl2-6H2O, 137.9 mM NaCl, 2.67 mM KCl, 8.06 mM Na2HPO4-7H2O, 1.47 mM KH2PO4, pH 7.4; D-PBS, Gibco). Then 1×Poly-D-lysine stock (PDL, Sigma-Aldrich) was added to the cell culture plates (50 μl for the 96-well plates, 100 μl for the 24-well plates, and 500 μl for the six-well plates), and the plates were incubated overnight at room temperature. The plates were rinsed three to four times with sterile H2O and aspirated to dryness. Mouse laminin (1 mg/ml) was diluted to a final concentration of 50 μg/ml by adding 10 μl of the laminin stock to 5 ml of Neurobasal medium (Gibco, Grand Island, NY). The diluted laminin solution was mixed well, added to the dried cell culture plates, and incubated in a 37 °C incubator for 2 h. The plates were rinsed with D-PBS three times before use. To isolate RGCs with immunopanning (IP), we adapted a protocol from Cold Spring Harbor Protocols [25].
Removing the retinal macrophages
In the retina, at least two types of cells, RGCs and macrophages, express Thy-1; most are macrophages [8]. Thus, it was necessary to deplete the macrophages and other Thy l-positive contaminants that are not RGCs and then select the Thy l-positive RGCs. Therefore, the retinal cell suspensions were first incubated in flasks coated with an anti-rat macrophage monoclonal antibody (OX-41; 1:50) for 45–60 min to exclude the macrophages; the plate was agitated every 15 min to ensure good cell adhesion. The non-adherent cells were collected. This procedure allowed us to rapidly isolate a population of pure RGCs with excellent viability.
IP for RGCs
The RGCs were isolated as previously described [8,25]. The retinal cell suspension excluding macrophages (or not) was incubated in a 100-mm Petri dish coated with a mouse anti-rat Thy-1 antibody (1:50 dilution; Abcam, Cambridge, MA) for 1 h; the plate was shaken every 15 min during this time period. The supernatant was discarded, and the plate was washed six times with D-PBS. The adherent cells were collected as RGCs. (Note: we abbreviated the direct immunopanning in a plate coated with an anti-rat Thy-1 antibody as IP and the immunopanning in two plates coated with anti-rat macrophage and Thy-1 antibodies as TIP.)
Immunopanning-magnetic (IPM) for RGCs
The retinal cell suspension excluding macrophages was centrifuged at 300 g for 10 min at 4 °C. After the supernatant had been completely aspirated, the cell pellet was resuspended in a concentration of 107 cells per 90 μl buffer. Subsequently, 10 μl of CD90.1 MicroBeads (Miltenyi Biotec, GmbH, Bergisch Gladbach, Germany) per 107 total cells was added, mixed well, and incubated at 4 °C for 15 min. Then, the cells were carefully washed twice with magnetic cell sorter (MACS) separation buffer. After being resuspended, the cell suspension was applied onto the MS column (Miltenyi Biotec GmbH) in the magnetic fields of a MiniMACS (Miltenyi Biotec GmbH). When the column reservoir was empty, the column was washed with 1.5 ml of MACS separation buffer and then removed from the magnetic field. To increase the purity of the RGCs, the eluted fraction was enriched over a second MS column. The magnetic separation procedure was repeated as described with a new column. Upon completion, the column was removed from the separator, and the retained cells were eluted as a magnetically labeled RGC fraction. (Note: we abbreviated the direct magnetic separation as IPM; if the retina macrophages had been removed before the magnetic separation, we referred to it as TIPM.)
Flow cytometry (FC) method for RGCs
The non-adherent cell suspension in flasks coated with an anti-rat macrophage monoclonal antibody was centrifuged at 300 ×g for 10 min at 4 °C. After the supernatant had been completely aspirated, up to 106 cells were resuspended in 45 μl of buffer and incubated with 5 μl of phycoerythrin (PE)-conjugated anti-rat CD90.1 or PE-conjugated lgG1 isotype control antibodies (eBioscience, San Diego, CA) for 15 min at 4 °C. The cells were then carefully washed twice. After being resuspended, the cell suspension was sorted with a flow cytometer (FACS Caliber; Becton Dickinson, San Jose, CA). The cells were counted automatically, and cells that were not incubated with the PE-conjugated anti-rat CD90.1 antibody served as the blank (Figure 2A). To improve the RGCs’ yield, the sort gates were set as R10 and R21 (Figure 2); the RGCs in R21 were high purity (HP-RGCs), while the cells in R10 were low-purity RGCs (LP-RGCs).
Figure 2.

Design of FC cell sorting gates. A: Blank control. Staining with control antibodies conjugated with phycoerythrin (PE). B: Retinal ganglion cell (RGC) sorting strategy. RGCs were identified by staining with PE-conjugated mAb directed against Thy-1 (R10 and R21). R10 is regarded as a low-purity group and R21 as a high-purity group. R16 consists mainly of non-RGCs. Y axes = side scatter (SSC), X axes = forward scatter (FSC), fluorescence intensity on the FL2 channel.
Cell culture
The purified RGCs were seeded at the desired density on the PDL- and laminin-coated coverslips in 24-well or six-well plates in prewarmed RGC growth medium and maintained in a 37 °C cell culture incubator with a humidified atmosphere containing 5% CO2 and 95% air. The RGC growth medium was improved from the formulation described in the Cold Spring Harbor Protocols [25], based on our repeated experiments, and contained Neurobasal medium, BSA (0.1 mg/ml; Sigma), transferrin (0.1 mg/ml; Sigma), progesterone (60 ng/ml; Sigma), putrescine (16 µg/ml; Sigma), selenium (40 ng/ml; Sigma), 3,5,3-triiodothyronine T3 (40 ng/ml; Sigma), thyroxine T4 (40 ng/ml; Sigma), B27 (20 µl/ml; Invitrogen), sodium pyruvate (1 mM; Gibco), glutamine (2 mM; Gibco), N-acetyl-L-cysteine (NAC, 5 µg/ml; Sigma), insulin (5 µg/ml; Sigma), forskolin (5 µM; Sigma), brain-derived neurotrophic factor (BDNF, 50 ng/ml; PeproTech, Rocky Hill, NJ), ciliary neurotrophic factor (CNTF, 10 ng/ml; PeproTech), basic fibroblast growth factor (bFGF, 10 ng/ml; PeproTech), and penicillin-streptomycin (100 U/ml; Gibco). Half of the medium was replenished every 3 days. However, although a high level of purity was obtained with FACS, the level of cell survival was low. The causes of this low survival are unclear.
Cell Counting Kit-8 assay for RGC viability
The RGCs were seeded in 96-well plates at a density of 3×104 cells/100 μl and were cultured for 1, 3, 5, 7, 9, 11, 14, 17, and 20 days. Then, 10 μl of Cell Counting Kit-8 solution (Dojindo Laboratories, Kumamoto, Japan) was added to each well of the plate. After a 4-h incubation at 37 °C, the plates were analyzed with a Tecan Genios (Synergy H1, BIOtAK) at 450 nm. All values are reported as the mean ± standard error of the mean (SEM) of at least three wells and at least three separate experiments.
RGC purity and identification
Immunocytochemical staining
Two days after seeding, the RGCs isolated using the TIP and TIPM methods were fixed with 4% paraformaldehyde for 30 min, treated with 0.1% Triton X-100 (Sigma) for 20 min, and then blocked with 3% BSA (Sigma-Aldrich) for 1 h. The cells were incubated with anti-rat γ-synuclein (1:400 dilution; Abcam), anti-rat Thy-1 (1:800 dilution; Abcam), anti-rat microtubule associated protein-2 (MAP2, 1:800 dilution; Abcam), anti-rat neuron-specific class III beta tubulin (TUJ-1,1:400 dilution; Abcam), anti-rat Brn3b (1:400 dilution; Abcam), anti-rat Islet1 (1:400 dilution; Abcam), astrocyte-specific anti-rat glial fibrillary acidic protein (GFAP; 1:100 dilution; Abcam), and amacrine-specific anti-rat syntaxin 1 antibodies (1:50 dilution; Abcam) overnight at 4 °C. They were then exposed to the appropriate fluorescent secondary antibodies (1:400 dilution, Life Technologies) for 1 h at room temperature. The nuclei were counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Life Technologies). The cells were imaged with a confocal microscope (Leica SP8).
Quantitative real-time PCR analysis
The RNA extraction and cDNA synthesis were performed as previously described [26]. The gene-specific primers for β-actin, Thy-1, TUJ1, Brn3b, syntaxin 1, and GFAP were verified before use (Table 1 for primer sequences). Quantitative real-time PCR (Applied Biosystems, 7500 Fast) was performed in duplicate with 10 ng of cDNA and 10 pmol of each primer. The thermal conditions were 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 60 s at 60 °C. The specificity of the detected signals was confirmed with a dissociation curve, which consisted of a single peak. Using the SYBR Green data, the relative RNA expression was normalized to β-actin. All samples were run in triplicate in each experiment. The data were analyzed using the 2-△△CT method.
Table 1. List of the primer sequences for PCR studies.
| Target | Sequence (5'-3') |
|---|---|
| β-actin |
F: TGACGTGGACATCCGCAAAG |
| |
R: CTGGAAGGTGGACAGCGAGG |
| Thy1.1 |
F: GGCAGTGAAGAGGCAGGATA |
| |
R: AGGCACAGACACAGTCCAAC |
| TUJ1 |
F: TGCTGGCCATTCAGAGTAAGA |
| |
R: TGCTGGCCATTCAGAGTAAGA |
| Brn3b |
F: GGCTGGAGGAAGCAGAGAAA |
| |
R: TTGGCTGGATGGCGAAGTAG |
| syntaxin 1 |
F: AGGCACGCAGGAAGAAGAT |
| |
R: CAGGGAGACCCATCCAGAA |
| GFAP |
F: ACCGCATCACCATTCCTGTA |
| R: GCATCTCCACCGTCTTTACC |
Flow cytometry analysis
The RGCs’ purity was evaluated using a flow cytometer (FACSCaliber; Becton Dickinson). As most of the Thy-1 antigens on the surface of the RGCs were bound to the corresponding antibody during the separation and purification process, we adopted another RGC-specific marker, Brn3a, as a surrogate marker for the RGCs. The steps are summarized as follows: The RGCs that had been separated and purified using the three methods were separately fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. Then, the cells were centrifuged and resuspended in PBS containing 0.4% Triton X-100 (Sigma), and 3% BSA was added to block non-specific binding of the antibodies. Each sample was subsequently centrifuged and incubated with a rabbit anti-rat Brn3a antibody (1:50, Abcam) for 60 min at 4 °C. After washing with PBS, the cells were exposed to fluorescein isothiocyanate (FITC)–conjugated goat anti-rabbit immunoglobulin G (IgG; 1:400, Life Technologies) in 1.0 ml of PBS for 1 h at RT. After washing twice with PBS, the cells were excited with a 488-nm laser and collected in the FITC (515–545 nm) channels. Cell Quest Acquisition and Analysis software (Becton Dickinson) was used to quantify the intensities of the fluorescent signals and to construct dot density plots.
Electrophysiology of purified RGCs
Electrophysiological recordings were performed on the primary cultured RGCs that had been purified with the TIP and TIPM methods on the second to fourth day. The bath solution contained the following components: 125 mM NaCl, 3 mM KCl, 1 mM MgCl2.6H2O, 2 mM CaCl2.2H2O, 26 mM NaHCO3, 1.25 mM NaH2PO4, and 15 mM glucose, pH 7.4. The pipette solution for the current-clamp recordings consisted of the following components: 120 mM K-gluconate, 10 mM HEPES, 1 mM EGTA, 1 mM MgCl2.6H2O, 0.1 mM CaCl2.2H2O, 4 mM ATP-Mg, 0.3 mM GTP-Na, and 10 mM phosphocreatine. The pH was adjusted to 7.2 with KOH, and the osmolarity was adjusted to 280–290 mOsm/l. The pipette resistance was typically 3–7 MΩ after it was filled with the internal solution. The spontaneous action potentials were recorded in the whole-cell current-clamp configuration using a Multiclamp 700B amplifier (Molecular Devices, Sunnyvale, CA) with a Digidata 1440A data acquisition board. The data were digitized at 10 kHz with a 1 kHz low-pass filter. The data were analyzed using Clampfit 10.02 (Axon Instruments, Foster City, CA). The membrane potentials were maintained at approximately −55 mV.
Statistical analysis
The TIP, TIPM, and FC methods were separately repeated 18, 15, and five times, respectively, and approximately 50–60 Sprague-Dawley rats were used for each experiment. The RGC yield, purity, and quantitative RT–PCR data were expressed as the mean ± standard error of the mean (SEM) and compared with the Kruskal–Wallis one-way ANOVA (ANOVA) using the PASW Statistics 18 for Windows, version 18.0.0 (SPSS, Chicago, IL). A p value of less than 0.05 was considered statistically significant.
Results
RGC yield
Approximately 4.24±0.62 million cells were collected per retina. First, we tried to leave the macrophages in the suspension and directly incubated the retinal cells with the Thy l antibodies. The results (Figure 3A and Table 2) show that 136.29±50.20 × 104 and 64.40±29.98 × 104 RGCs were achieved per retina using the IP and IPM methods, respectively, which accounted for 33.04±9.23% and 14.26±5.90% of the total retinal cells. These values were far higher than the actual numbers [8]. However, when the macrophages were removed first, the RGC yields were 24.60±15.98 × 104 (5.91±3.38%) and 5.28±4.42 × 104 (1.31±0.94%) using the TIP and TIPM methods, respectively. This yield is within (TIP) or below (TIPM) the statistical error of the 110,000 ganglion cells per adult rat retina that was previously determined [27-29]. Flow cytometry is a versatile technique for cell sorting. It is quantitative, highly reproducible, and relatively easy to perform. As the density of Thy-1 on the surface varies greatly in specific subsets of RGCs, two regions, R10 and R21, would appear when flow cytometry is used to measure the Thy-1 protein, as shown in Figure 2B. We regarded the cells in the R21 region as high-purity RGCs (HP-RGCs) and the cells in the R10 region as low-purity RGCs (LP-RGCs). In our experiment, we obtained only approximately 2.7 × 103 RGCs per retina in the R21 region, while 5.4 × 103 RGCs were obtained in R21+R10, comprising only 0.05–0.35% of the total retinal cells.
Figure 3.
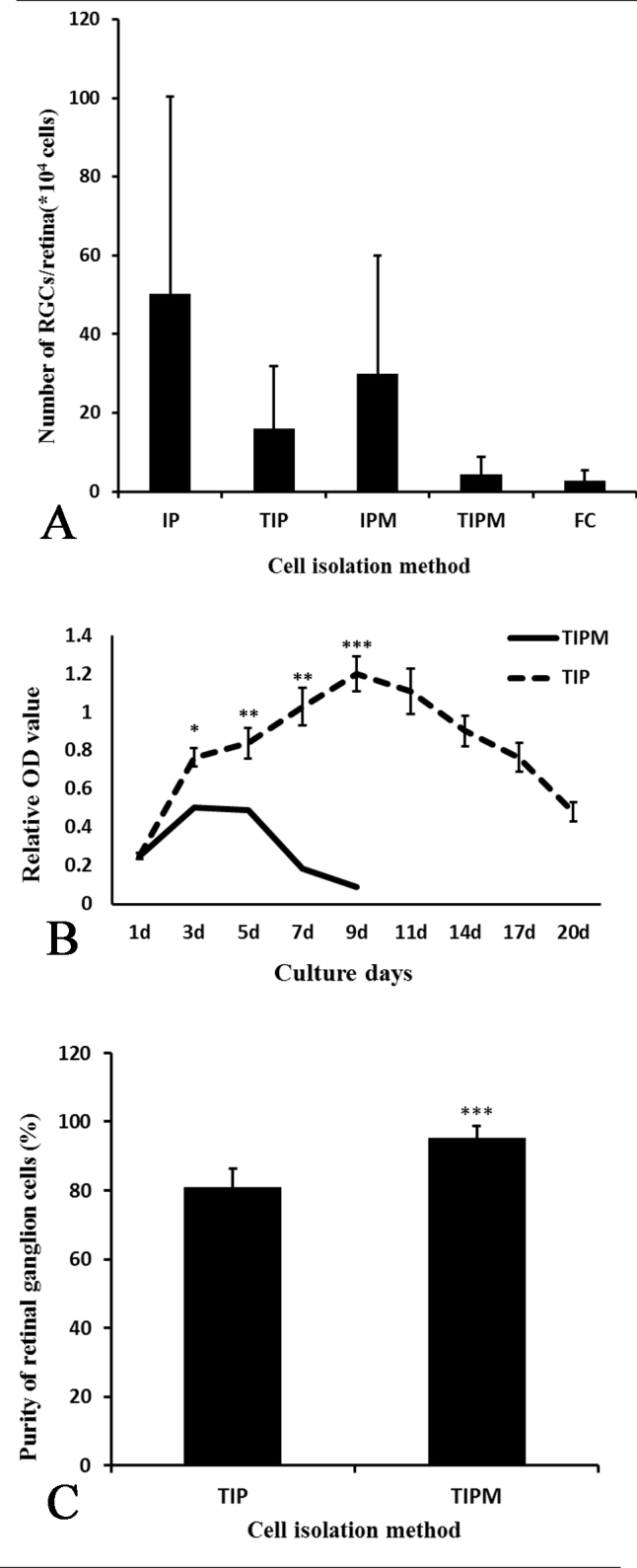
The yield, purity, and viability of RGCs from different methods. A: The yield of retinal ganglion cells (RGCs) from IP is the highest, while that from flow cytometry (FC) is the lowest. B: The viability of two-step immunopanning–retinal ganglion cells (TIP-RGCs) was increased at the first 9 days with culture time and was much higher than that of the two-step immunopanning-magnetic–retinal ganglion cells (TIPM-RGCs). Although the activity of the TIP-RGCs began to decline slowly, they still survived for 20 days, which was far longer than the TIPM-RGCs. C: The purity of the RGCs isolated from TIP is around 81% and much lower than that from the TIPM method (about 95%).
Table 2. Comparative analysis of three purification protocols for RGCs.
| Various | TIP | TIPM | FC |
|---|---|---|---|
| Yield(*104) |
24.60±15.98 |
5.28±4.42 |
0.54±0.27 |
| RGCs/retina cell (%) |
5.91±3.38 |
1.31±0.94 |
0.05–0.35 |
| Purity (%) |
80.97±5.45 |
95.41±3.23 |
Controlled, up to 100 |
| viability |
+++ |
++ |
- |
| Longevity (days) |
>20 |
≤9 |
- |
| operation time (hour) |
3–4 |
2–3 |
3–4 |
| stability |
+ |
++ |
+++ |
| cost | + | ++ | +++ |
+:low, ++: medium, +++:maximum -:we did not get.
RGC viability and longevity
The data from the Cell Counting Kit-8 (CCK-8) assay showed (Figure 3B) that there was no significant difference in RGC5 viability between the TIP and TIPM methods after the cells were cultured for 24 h. Then, the viability of the cells that were purified using both methods increased during the first 3 days and with prolonged culture times. The TIP-RGC viability was always greater than that of the TIPM-RGCs (p<0.05). However, the viability of the TIPM-RGCs began to decrease, and most of the cells had died by the 9th day (the absorption value was 0.09). Interestingly, the viability of the TIP-RGCs reached the highest value at the same time point (the absorption value was 1.2) and began to slowly decrease until the 20th day (the absorption value was 0.48). Therefore, we can see that the TIP-RGCs could survive for more than 20 days with higher viability, while the TIPM-RGCs survived for less than 9 days.
RGC identification
The RGCs were identified by their morphology, immunofluorescence staining and whole-cell patch-clamp recordings. Neurons typically have dendrites and axons that appear at opposing poles of the cell. We screened the survival and morphology of RGC isolated by the two immunopanning methods (TIP, TIPM) every day as shown Figure 4. Under standard culture conditions, the RGCs tended to form cell clusters with small to medium round cell bodies, and some extended fine neurites after a 24-h culture, but there were no significant differences between the two groups. However, from the third day, the RGCs purified using the two methods showed obvious differences. The majority of the TIP-RGCs exhibited uniform, round cell bodies and their neurites gradually extended to connect each other. At day 14, most cells developed a complex dendritic network with numerous neurites and branches. By replenishing half of the medium with fresh medium every 3 to 4 days, the TIP-RGCs were maintained for up to one month in culture, which was the longest time point in our examination (Figure 4A). However, the TIPM-RGCs neurites were obviously shorter than those from the TIP-RGCs beginning at day 3, and its cell body began shrinking. Although its neurites were still extended, they ultimately survived for no more than 9 days (Figure 4B).
Figure 4.
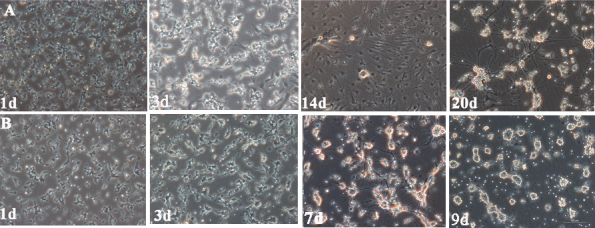
Morphological changes in RGCs. A: Two-step immunopanning–retinal ganglion cells (TIP-RGCs). B: Two-step immunopanning-magnetic–retinal ganglion cells (TIPM-RGCs). Scale bar = 10 μm.
In retinas, many types of cells, such as rod and cone photoreceptors, horizontal, bipolar and amacrine cells, have neurites and round cell bodies, similar to the RGCs [30]. Therefore, we could not identify the RGCs by morphology alone. Several cell-specific markers have been suggested to be expressed in RGCs, such as Thy1 [31], Brn3a/b [32, 33], βIII tubulin [31], MAP2 [34], Islet-1 [35,36], and γ-synuclein [37]. As none of these are specifically expressed in all RGC subgroups, we used six common RGC-specific immunocytochemical markers to provide a more accurate identification of the RGCs and further analyzed their expression in RGCs. γ-Synuclein and MAP2 (Figure 5A-D) were expressed in the soma and neurites in almost all of the RGCs, and their expression perfectly coincided with each other. TUJ1 and Thy1 were expressed in the cytoplasm of the soma and neurites in some of the RGCs, particularly those with long axons, while Brn3b and Islet-1 were predominantly localized in the nuclei in all of the RGCs, and little expression was observed in the cytoplasm (Figure 5E-L).
Figure 5.

RGC identification. Immunofluorescence of retinal ganglion cells (RGCs) at day 2 of culture shows costaining of γ-synuclein (green) and MAP2 (red; A–D), TUJ1 (green), and Brn3b (red; E–H), Thy-1 (green), and Islet 1 (red; I–L). 4’,6-diamidino-2-phenylindole (DAPI) nuclear staining is shown in C, G, and K, and the merged images are shown in D, H and L. Scale bar = 50 μm.
To further investigate whether the primary cultured RGCs have functional membrane properties, we performed whole-cell patch-clamp recordings of the RGCs on day 2 to 4 after they were cultured. For the majority of the analyzed primary cultured RGCs (TIP-RGC: 86.7%; TIPM-RGC: 63.1%, n=15), action potentials could be spontaneously elicited in current-clamp mode (Figure 6A). These action potentials could be blocked by tetrodotoxin (TTX), a specific inhibitor of Na+ ion channels (not shown). For the other mixed cells, such as glial cells, action potentials could not be spontaneously produced in current-clamp mode (Figure 6B). Action potentials could not be elicited by depolarizing the membrane from -90 mV to + 50 mV at 10 mV intervals (not shown).
Figure 6.

Representative traces of membrane potential in current-clamp mode. Spontaneous action potentials recorded from primary cultured RGCs purified by TIP and TIPM method at the second to the fourth day. No current injection was applied. Membrane potential was current-clamped at around -55 mV (A). In some cells action potentials were not produced spontaneously, indicating these were not RGCs (B).
RGC purity
The purity of the RGCs isolated with the two methods was first determined with immunofluorescence staining (Figure 7). Nearly all of the TIPM-RGCs were positive for Thy-1 and MAP2, and only three cells in the field were negative (Figure 7D). The number of negative TIP-RGCs was much higher, as approximately seven cells in equal visual fields were not labeled (Figure 7H). To further analyze the source of these negative cells, we stained the cells for the expression of GFAP (the marker for glia cells) and syntaxin-1 (the marker for amacrine cells). As shown in Figure 8, large, star-shaped, GFAP-positive astrocytes (Figure 8B) and small, round, syntaxin 1-positive amacrine cells with multiple short neurites (Figure 8A) were rarely observed in the entire field.
Figure 7.

RGC purity. Confocal double immunofluorescence of retinal ganglion cell (RGCs) from the two-step immunopanning (TIP) and two-step immunopanning-magnetic (TIPM) methods shows the costaining of Thy-1 (A and E) and MAP2 (B and F). 4’,6-diamidino-2-phenylindole (DAPI) nuclear staining is shown in C and G, and in the merged images in (D) and (H). Cells negative for Thy-1 and MAP2 are regarded as non-RGCs and are marked with Arabic numbers (D and H). Visual counting of immunostained cells seeded at the same density demonstrates that seven cells in a 200X visual field were not labeled by Thy-1 or MAP2 in the TIP groups while only three cells were negative in the TIPM group. Scale bars = 100 μm.
Figure 8.

Rare labeling of astrocytes and amacrine cells. Syntaxin 1-labeled amacrine cells (A, green) and glial fibrillary acidic protein (GFAP)–labeled astrocytes (B, red) cells were rarely detected in the fields of cells. Nuclei were visualized using 4’,6-diamidino-2-phenylindole (DAPI; blue). Scale bars = 100 μm.
The quantitative RT–PCR data for Thy-1, syntaxin 1 and GFAP were calculated as a relative RNA ratio to the housekeeping gene, β-actin (Figure 9A). The GFAP levels in the TIP- and TIPM-RGCs were obviously higher than in the FC-RGCs (p<0.01), whereas the syntaxin 1 levels in the TIP-RGCs were significantly higher than in the other two groups (p<0.05), while there were no significant differences in the TIPM- and FC-RGCs. This is consistent with the immunofluorescence staining results. As we had divided the FC-RGCs into HP-RGCs and LP-RGCs, an additional, more detailed quantitation of the quantitative RT–PCR data was performed to examine whether our assumption was valid (Figure 9B). The expression of the Thy-1, TUJ 1, Brn3b, syntaxin 1, and GFAP mRNAs was analyzed in the HP and LP groups, and to our surprise, there were no significant differences in the mRNA expression levels, except for GFAP.
Figure 9.

Quantitative expression of cell markers in isolated RGCs. A: Quantitative real-time PCR for glial fibrillary acidic protein (GFAP), syntaxin 1, and Thy-1 of retinal ganglion cells (RGCs) isolated with the two-step immunopanning (TIP), two-step immunopanning-magnetic (TIPM), and flow cytometry (FC) methods. The Thy-1 levels in FC were the highest (p<0.05), and the syntaxin 1 levels were the lowest (p<0.05), while GFAP were zero. This indicates that the purity of the RGCs from FC is the highest, followed by TIPM, and the RGC purity of TIP is the lowest of the three methods. B: Relative mRNA levels of Thy-1, TUJ-1, Brn3b, syntaxin 1(syn), and GFAP from RGCs isolated with the FC method (high purity and low purity). There were no significant differences in the mRNA expression levels, except for GFAP. Data expressed as relative expression to the housekeeping gene β-actin. All relative RNA ratios expressed as the mean ± standard error of the mean (SEM; n = 9).
We next used flow cytometry to further compare purity, and RGCs isolated using the TIP and TIPM methods were immunostained with an anti-Brn3a antibody and analyzed with FACS. The results (Figure 10) showed that 80.97±5.45% of the cells from the TIP method and 95.41±3.23% of the cells from the TIPM method were Brn3a-positive, indicating that the TIPM method obtained higher-purity RGCs (Figure 3C).
Figure 10.

Flow cytometry analysis of the purity of RGCs sorted with the TIP and TIPM methods. The retinal ganglion cells (RGCs) were identified by staining with fluorescein isothiocyanate (FITC)–conjugated mAb against Thy-1 (Ex: 490 nm; Em: 520 nm).
Discussion
Cultured RGCs will be an important and almost indispensable tool for the study of retinal visual physiology and pathophysiology. Until recently, numerous approaches, including TIP methods [25], magnetic separation methods [19,23], retrograde labeling with fluorescent dyes combined with a pull-off technique [24,38], and flow cytometric methods [20] have been established to isolate and purify RGCs in attempts to study retinal pathophysiology. Labeling RGCs with retrogradely transported fluorescent marker that had been injected into a fiber tract or target nucleus isolated RGCs from postnatal retinas with nearly 100% purity; however, the yield was sacrificed for purity, and the experiment is time-consuming and requires expensive instrumentation [38]. The TIP, TIPM, and FC methods are currently the most commonly used methods, but they vary greatly in experimental principles, procedures, and RGC purity, yield, and viability. The basis and key to a successful study is selecting the most effective and appropriate method for purifying RGCs. However, there has been no specific comparison with which to benchmark these methods and determine which are the most appropriate.
The TIP method has been widely used to purify RGCs. It has been reported that the purity of RGCs isolated with TIP ranges from 50% to 99.5% [8,21,30], but we achieved approximately 81% in our study, even though we had repeated the whole experiments dozens of times. To find the main reasons for this discrepancy, we coated the panning dishes only with 50 mM Tris-HCl as negative control but found that many cells were still attached to the plate, suggesting that most of the contaminating cells had absorbed on the plate, even though we used a low absorption plate (Nunc, Thermo, Denmark). Therefore, we insist that purity higher than 90% is difficult to achieve, and it is difficult to maintain a constant yield. Furthermore, the binding affinity between the RGCs and the anti- Thy-1 antibody is not strong, and plate swinging, cell clusters, and undigested retinal tissue may, in part, cause the variable yield. However, although the purity of the RGCs from the TIP method was not as high as that from the TIPM method, their viability and longevity were much higher, which are essential and particularly important in some studies. This method is particularly ideal for experiments in which the presence of a small number of contaminating cells could be ignored without any obvious impact on the experimental results but that require a large quantity of highly viable RGCs for a long period.
The TIPM method introduced by Samin Hong [19] is faster and less complicated than the TIP method, as summarized in Table 2. The TIPM method not only has the advantage of a more stable yield but also has a much higher purity compared with the TIP method, even though the TIP-RGCs have a short cell survival time and lower viability. However, if the experimental cycle is short and/or highly pure RGCs with specific activity are required, TIPM-RGCs are the best source. Nevertheless, TIPM requires highly skilled hands, and the cost was slightly higher than that of the TIP method.
Flow cytometry has been used for cell sorting for many years. Samin Hong et al. compared the purity of the RGCs from the TIP and TIPM methods in 2012 [20], but they did not analyze FC, which is an ideal and efficient approach for cell sorting. Using FC, several million cells can be screened within a short time, and subpopulations and single cells can be isolated from within mixed-cell populations, even when the cells are present at frequencies as low as 10−6 within the population [39]. In our experiment, 5.4±2.7 × 103 RGCs were obtained per retina, and this reduced yield is due to the increased vulnerability of the cells to the intricate process and the long-term mechanical separation, which may also reduce the activities of RGCs. Although this number is not sufficient for protein analyses or additional biochemical approaches that require a great quantity of cells, their purity can almost reach 100%, and we can easily control the relative purity of RGCs. Therefore, FC is a suitable choice for studies that require extremely pure RGCs. If a large number of RGCs with 100% purity are required, the method could be scaled up by increasing the number of retinas and extending the sorting time. However, the reduced viability of cells collected by this method will limit their use to experiments of short duration only.
Collectively, this is the first report to compare the three most common methods for purifying RGCs from newborn rat retinas by analyzing the purity, yield, and viability. Our study found that the RGCs isolated with the TIP method possess the highest viability and yield but low purity; the TIPM method was the most reliable, as the RGCs produced using this method exhibited considerable purity, yield, and viability. The purity of the RGCs from the FC method can reach approximately 100%, but their yield was low with the same number of retinas. Although factors such as increasing the amount of starting tissue and reducing the isolation and sorting times may improve yield and viability by the FC method, an area that may have greater impact on cell viability of RGCs isolated by all methods is modification of the culture conditions of these cells to permit greater survival.
In summary, our study demonstrates that TIP, TIPM, and FC can isolate RGCs from newborn rats, and each approach has its own pros and cons. The research provided a solid practical basis or at least a reference for selecting the method for purifying RGCs for subsequent experiments.
Acknowledgments
This project was supported by grants from the Research Fund for the National Natural Science Foundation of China (NCFC81070738, NCFC81300775, NSFC81470624). Jihong Wu (jihongwu@fudan.edu.cn) Qiang Wu (Qiang.wu@shsmu.edu.cn) are co-corresponding authors for this study
References
- 1.Maturana MI, Grayden DB, Burkitt AN, Meffin H, Kameneva T. Multicompartment retinal ganglion cells response to high frequency bi-phasic pulse train stimulation: Simulation results. Conference proceedings: Annual International Conference of the IEEE Engineering in Medicine and Biology Society IEEE Engineering in Medicine and Biology Society Annual Conference 2013; 2013:69–72. [DOI] [PubMed] [Google Scholar]
- 2.Wu JH, Zhang SH, Nickerson JM, Gao FJ, Sun ZM, Chen XY, Zhang SJ, Zhang R, Gao F, Chen JY, Luo Y, Wang Y, Sun XH. Cumulative mtDNA damage and mutations contribute to the progressive loss of RGCs in a rat model of glaucoma. Neurobiol Dis. 2015;74:167–79. doi: 10.1016/j.nbd.2014.11.014. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25478814&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu JY, Li TT, Du SS, Chen YD, Wang S, Xiong F, Wu Q. The MAPK signaling pathway mediates the GPR91-dependent release of VEGF from RGC-5 cells. Int J Mol Med. 2015;36:130–8. doi: 10.3892/ijmm.2015.2195. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25936351&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding Y, Yuan ST, Liu XY, Mao PA, Zhao C, Huang Q, Zhang RH, Fang Y, Song QL, Yuan DQ, Xie P, Liu Y, Liu QH. Protective Effects of Astragaloside IV on db/db Mice with Diabetic Retinopathy. PLoS One. 2014;9 doi: 10.1371/journal.pone.0112207. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25411784&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farkas RH, Grosskreutz CL. Apoptosis, neuroprotection, and retinal ganglion cell death: An overview. Int Ophthalmol Clin. 2001;41:111–30. doi: 10.1097/00004397-200101000-00011. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11198138&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 6.Aguirregomozcorta M, Mancini L, Jenkins TM, Hickman SJ, Ciccarelli O, Plant GT, Thompson AJ, Toosy AT. A longitudinal functional MRI study of non-arteritic anterior ischaemic optic neuropathy patients. J Neurol Neurosurg Psychiatry. 2011;82:905–13. doi: 10.1136/jnnp.2009.194563. [DOI] [PubMed] [Google Scholar]
- 7.Fuchs C, Forster V, Balse E, Sahel JA, Picaud S, Tessier LH. Retinal-cell-conditioned medium prevents TNF-alpha-induced apoptosis of purified ganglion cells. Invest Ophthalmol Vis Sci. 2005;46:2983–91. doi: 10.1167/iovs.04-1177. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16043875&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 8.Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803. doi: 10.1016/0896-6273(88)90127-4. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=2908449&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 9.Krishnamoorthy RR, Agarwal P, Prasanna G, Vopat K, Lambert W, Sheedlo HJ, Pang IH, Shade D, Wordinger RJ, Yorio T, Clark AF, Agarwal N. Characterization of a transformed rat retinal ganglion cell line (Retraction of vol 86, pg 1, 2001). Brain Res. 2014;1544:62. doi: 10.1016/s0169-328x(00)00224-2. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24316244&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 10.Krishnamoorthy RR, Agarwal P, Prasanna G, Vopat K, Lambert W, Sheedlo HJ, Pang IH, Shade D, Wordinger RH, Yorio T, Clark AF, Agarwal N. Characterization of a transformed rat retinal ganglion cell line (Retracted article. See vol. 1544, pg. 62, 2014). Brain Res Mol Brain Res. 2001;86:1–12. doi: 10.1016/s0169-328x(00)00224-2. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11165366&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 11.Bahr M, Vanselow J, Thanos S. In vitro regeneration of adult rat ganglion cell axons from retinal explants. Exp Brain Res. 1988;73:393–401. doi: 10.1007/BF00248232. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=3265109&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 12.Thanos S, Bahr M, Barde YA, Vanselow J. Survival and Axonal Elongation of Adult Rat Retinal Ganglion Cells. Eur J Neurosci. 1989;1:19–26. doi: 10.1111/j.1460-9568.1989.tb00770.x. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=12106170&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 13.Meyer JS, Shearer RL, Capowski EE, Wright LS, Wallace KA, McMillan EL, Zhang SC, Gamm DM. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc Natl Acad Sci USA. 2009;106:16698–703. doi: 10.1073/pnas.0905245106. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19706890&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lamba DA, McUsic A, Hirata RK, Wang PR, Russell D, Reh TA. Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells. PLoS One. 2010;5:e8763. doi: 10.1371/journal.pone.0008763. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20098701&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda H, Osakada F, Watanabe K, Mizuseki K, Haraguchi T, Miyoshi H, Kamiya D, Honda Y, Sasai N, Yoshimura N, Takahashi M, Sasai Y. Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells. Proc Natl Acad Sci USA. 2005;102:11331–6. doi: 10.1073/pnas.0500010102. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=16076961&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishnamoorthy RR, Clark AF, Daudt D, Vishwanatha JK, Yorio T. A Forensic Path to RGC-5 Cell Line Identification: Lessons Learned. Invest Ophthalmol Vis Sci. 2013;54:5712–9. doi: 10.1167/iovs.13-12085. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23975727&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 17.Xu ZR, Jiang FG, Zeng YC, Alkhodari HTM, Chen F. Culture of Rat Retinal Ganglion Cells. J Huazhong. U Sci-Med. 2011;31:400–3. doi: 10.1007/s11596-011-0389-0. [DOI] [PubMed] [Google Scholar]
- 18.Winzeler A, Wang JT. Purification and culture of retinal ganglion cells. Cold Spring Harb Protoc. 2013;2013:614–7. doi: 10.1101/pdb.top070961. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23818663&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 19.Hong S, Iizuka Y, Kim CY, Seong GJ. Isolation of primary mouse retinal ganglion cells using immunopanning-magnetic separation. Mol Vis. 2012;18:2922–30. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23233794&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 20.Chang ZY, Lu DW, Yeh MK, Chiang CH. A novel high-content flow cytometric method for assessing the viability and damage of rat retinal ganglion cells. PLoS One. 2012;7:e33983. doi: 10.1371/journal.pone.0033983. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22457807&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi N, Cummins D, Caprioli J. Rat retinal ganglion cells in culture. Exp Eye Res. 1991;53:565–72. doi: 10.1016/0014-4835(91)90214-y. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=1743255&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 22.Hu DN, Ritch R. Tissue culture of adult human retinal ganglion cells. J Glaucoma. 1997;6:37–43. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9075079&dopt=Abstract [PubMed] [Google Scholar]
- 23.Shoge K, Mishima HK, Mukai S, Shinya M, Ishihara K, Kanno M, Sasa M. Rat retinal ganglion cells culture enriched with the magnetic cell sorter. Neurosci Lett. 1999;259:111–4. doi: 10.1016/s0304-3940(98)00918-5. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10025570&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 24.Knop CO, Vowerk CK, Vorwerk P, Mawrin C, Wex H. Purification and separation of retinal ganglion cells by retrograde fluorescence labeling and fluorescent activated cell sorting (FACS). ARVO Annual Meeting; 2004 April 25-29; Fort Lauderdale (FL). [Google Scholar]
- 25.Winzeler A, Wang JT. Purification and culture of retinal ganglion cells from rodents. Cold Spring Harb Protoc. 2013;2013:643–52. doi: 10.1101/pdb.prot074906. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23818667&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez AR, Muller LPD, Brecha NC. The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J Comp Neurol. 2014;522:1411–43. doi: 10.1002/cne.23521. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24318667&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding H, Jin GH, Zou LQ, Zhang XQ, Li HM, Tao XL, Zhang XH, Qin JB, Tian ML. Stromal derived factor-1 in hippocampus radial glial cells in vitro regulates the migration of neural progenitor cells. Cell Biol Int. 2015;39:750–8. doi: 10.1002/cbin.10442. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25604551&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 28.Potts RA, Dreher B, Bennett MR. The loss of ganglion cells in the developing retina of the rat. Brain Res. 1982;255:481–6. doi: 10.1016/0165-3806(82)90013-x. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7066701&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 29.Perry VH, Henderson Z, Linden R. Postnatal changes in retinal ganglion cell and optic axon populations in the pigmented rat. J Comp Neurol. 1983;219:356–68. doi: 10.1002/cne.902190309. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=6619343&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 30.Romano C, Hicks D. Adult retinal neuronal cell culture. Prog Retin Eye Res. 2007;26:379–97. doi: 10.1016/j.preteyeres.2007.03.001. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17482863&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 31.Zhang XM, Liu DTL, Chiang SWY, Choy KW, Pang CP, Lam DSC, Yam GHF. Immunopanning purification and long-term culture of human retinal ganglion cells. Mol Vis. 2010;16:2867–72. [PMC free article] [PubMed] [Google Scholar]
- 32.Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, Nieto-Lopez L, Canovas-Martinez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M. Brn3a as a Marker of Retinal Ganglion Cells: Qualitative and Quantitative Time Course Studies in Naive and Optic Nerve-Injured Retinas. Invest Ophthalmol Vis Sci. 2009;50:3860–8. doi: 10.1167/iovs.08-3267. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19264888&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 33.Xiang M, Zhou L, Macke JP, Yoshioka T, Hendry SH, Eddy RL, Shows TB, Nathans J. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995;15:4762–85. doi: 10.1523/JNEUROSCI.15-07-04762.1995. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7623109&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Camilli P, Miller PE, Navone F, Theurkauf WE, Vallee RB. Distribution of microtubule-associated protein 2 in the nervous system of the rat studied by immunofluorescence. Neuroscience. 1984;11:817–46. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=6377119&dopt=Abstract [PubMed] [Google Scholar]
- 35.Baver SB, Pickard GE, Sollars PJ, Pickard GE. Two types of melanopsin retinal ganglion cell differentially innervate the hypothalamic suprachiasmatic nucleus and the olivary pretectal nucleus. Eur J Neurosci. 2008;27:1763–70. doi: 10.1111/j.1460-9568.2008.06149.x. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18371076&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 36.Mu X, Fu X, Beremand PD, Thomas TL, Klein WH. Gene regulation logic in retinal ganglion cell development: Isl1 defines a critical branch distinct from but overlapping with Pou4f2. Proc Natl Acad Sci USA. 2008;105:6942–7. doi: 10.1073/pnas.0802627105. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18460603&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Surgucheva I, Weisman AD, Goldberg JL, Shnyra A, Surguchov A. gamma-Synuclein as a marker of retinal ganglion cells. Mol Vis. 2008;14:1540–8. [PMC free article] [PubMed] [Google Scholar]
- 38.Simon P, Thanos S. Combined methods of retrograde staining, layer-separation and viscoelastic cell stabilization to isolate retinal ganglion cells in adult rats. J Neurosci Methods. 1998;83:113–24. doi: 10.1016/s0165-0270(98)00060-0. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=9765124&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 39.Kumar N, Borth N. Flow-cytometry and cell sorting: An efficient approach to investigate productivity and cell physiology in mammalian cell factories. Methods. 2012;56:366–74. doi: 10.1016/j.ymeth.2012.03.004. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22426008&dopt=Abstract [DOI] [PubMed] [Google Scholar]