

Abstract
The receptor activator of nuclear factor κB (RANK) and its ligand RANKL are key regulators of osteoclastogenesis and well-recognized targets in developing treatments for bone disorders associated with excessive bone resorption, such as osteoporosis. Our previous work on the structure of the RANK-RANKL complex revealed that Loop3 of RANK, specifically the non-canonical disulfide bond at the tip, performs a crucial role in specific recognition of RANKL. It also demonstrated that peptide mimics of Loop3 were capable of interfering with the function of RANKL in osteoclastogenesis. Here, we reported the structure-based design of a smaller peptide with enhanced inhibitory efficiency. The kinetic analysis and osteoclast differentiation assay showed that in addition to the sharp turn induced by the disulfide bond, two consecutive arginine residues were also important for binding to RANKL and inhibiting osteoclastogenesis. Docking and molecular dynamics simulations proposed the binding mode of the peptide to the RANKL trimer, showing that the arginine residues provide electrostatic interactions with RANKL and contribute to stabilizing the complex. These findings provided useful information for the rational design of therapeutics for bone diseases associated with RANK/RANKL function.
Keywords: bone resorption, docking, molecular dynamics simulation, peptide inhibitor, receptor activator of nuclear factor-κB (RANK), structure
INTRODUCTION
Receptor activator of nuclear factor κB (RANK) and its corresponding ligand (RANKL) belong to the tumor necrosis factor (TNF)-receptor ligand family and are known as key regulators of the development and bone-resorbing activity of osteoclasts (Wada et al., 2006). The interaction of RANKL expressed in osteoblasts with the osteoclast cell-surface receptor RANK gives rise to maturation of osteoclast precursors. Osteoprotegerin (OPG) also interacts with RANKL and prevents it from binding to RANK, thereby inhibiting osteoclastogenesis (Burgess et al., 1999). In this way, their complex interplay modulates bone remodeling, and dysregulation of this system may result in imbalance between bone resorption and synthesis (Guerrini and Takayanagi, 2014). Increased bone resorption is associated with disorders such as postmenopausal osteoporosis, osteolytic bone metastases, and rheumatoid arthritis (Michigami et al., 2001; Recker et al., 2004; Tanaka, 2013; Wittrant et al., 2004).
Due to the crucial roles of RANK and RANKL in osteobiology, they are recognized as interesting targets for treatment of bone-metabolic diseases, and many studies have been performed to develop effective ways to modulate their function. RANK-Fc, OPG-Fc, and anti-RANKL antibodies have been developed as therapeutics for osteoporosis (Dempster et al., 2012; Jules et al., 2010; Schieferdecker et al., 2014). For example, a RANKL-specific humanized antibody, denosumab, has been approved for the treatment of osteoporosis in postmenopausal women (Cohen et al., 2008; Cummings et al., 2009; Geusens, 2009; Neuprez et al., 2014). The protein kinase 2 inhibitor CX-4945 has also been reported to inhibit RANKL-induced osteoclast (Son et al., 2013). However, due to complicated manufacturing and disadvantages associated with the use of recombinant proteins for therapeutic intervention, we sought to develop a way to control RANKL signaling using small peptides that efficiently block protein-protein interactions. Furthermore, peptide mimics of OPG (OP3-4) and TNF receptor (WP9QY) developed as inhibitors of RANK-RANKL binding were shown to suppress RANKL-induced osteoclastogenesis in vivo (Ta et al., 2010).
Our previous work on the crystal structure of the complex of RANK-RANKL ectodomains led us to design a 13-amino-acid peptide, L3-3, which mimics Loop3 of RANK and interferes with its function in osteoclastogenesis (Ta et al., 2010). It was designed to cyclize through a disulfide linkage involving the C2 and C10 residues (Table 1) in order to maintain the overall loop structure. Based on a concern that its relatively large size would limit clinical applications, we report the development of a smaller peptide shown to possess enhanced inhibitory activity according to in vitro binding and osteoclastogenesis assays. Additionally, its binding mode to RANKL has been proposed from computational docking and molecular dynamics simulations. Our data showed the potency of the new peptide as an inhibitor of RANKL and provided useful information for the development of antiresorptive therapeutics.
Table 1.
Amino acid sequence of the inhibitory peptides that mimic Loop3 of RANK.
| Peptide name | Sequence |
|---|---|
| Loop3 in RANK | 118-GYH WNSDCECCRR N-131 |
| L3-3 | 1-YC WNSDCECCYRR-13 |
| L3-3S | 1-YC SNSDCECCYRR-13 |
| L3-3n | 1-YC WNSDCECCRR-12 |
| L3-3A | 1-SDCECS-6 |
| L3-3B | 1-SDCECSRR-8 |
Important residues that participate in cyclization or phi-phi interactions are shown in bold letters. Substituted or deleted residues are also indicated.
MATERIAL AND METHODS
Protein and peptide preparation
The pVFT3s (Korean patent 10-0690230) harboring the ectodomain region (residues 157–316) of mouse RANKL cDNA was transformed into Escherichia coli BL21 (DE3) cells (Novagen, USA). The protein was expressed overnight at 18°C following induction with 0.2 mM isopropyl-b-D-thiogalactosidase. After Ni-NTA column (GE Healthcare, USA) chromatography, purified protein was digested overnight with TEV protease at 4°C and loaded onto a Superdex-200 gel-filtration column (GE Healthcare) equilibrated with 25 mM Tris-HCl (pH 7.5) and 50 mM NaCl. The peptides were custom-synthesized (BIO-FD&C, Korea) and dissolved in water to prepare the stock.
Osteoclast differentiation assay
The maturation of mouse osteoclast-precursor cells was examined by the tartrate-resistant acid phosphatase (TRAP) assay (Oshiro et al., 2002). Briefly, osteoclast precursors were cultured in a 96-well plate (2 × 104 cells/well) in α-Minimal Essential Medium supplemented with 10% fetal bovine serum in the presence of 20 ng/ml of RANKL and 30 ng/ml human macrophage colony-stimulating factor. RANK or the peptide to be tested was added to a final concentration of ∼10–100 μM. activity of treated cells was assessed using naphthol AS phosphate (Sigma Aldrich, USA) as a substrate, and its reaction product was quantified by absorbance at 405 nm. The histo-chemical staining of TRAP-expressing cells employed the hydrolysis of naphthol AS BI phosphate (Sigma Aldrich) and subsequent formation of insoluble maroon dye deposits. Multinucleated TRAP-stained cells were counted under the microscope. Each experiment was repeated three times for statistical analysis.
Cell viability measurement
The possible cytotoxicity of peptides was estimated by quantitative colorimetric MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] (Sigma Aldrich) assay as previously described (Denizot and Lang, 1986). Cells were cultured in a 96-well plate (3 × 103 cells/well), and each peptide was added to the medium to varying final concentrations (∼10–100 μM). Cell viability was represented by the reduction of MTT to formazan, which was quantitated by absorbance at 570 nm.
Statistical analysis
All data were expressed as the mean ± SD. Statistical analyses were performed using the unpaired Student t test, and p < 0.05 was considered statistically significant.
Biolayer interferometry (BLI)
All measurements were made using a ForteBio BLItz instrument at 25°C (Pall ForteBio Corp., USA). The peptide (5 μM) was immobilized onto the amine-reactive biosensors using N-hydroxysuccinimide and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (Sigma Aldrich). Amine coupling was performed in 10 mM MES (pH 5.8) after pH optimization. Unoccupied amine groups were quenched with 1 M of ethanolamine. The binding affinity of peptide was calculated using Rmax values that were estimated with 0.2, 1, 5, 10, and 15 μM of purified RANKL. In each cycle of measurements, the baseline was adjusted with 10 mM 2-ethanesulfonic acid (MES; pH 5.8) for 30 s, followed by biosensor activation and peptide immobilization for 150 s. After ethanolamine quenching for 150 s, the baseline was set again for 30 s, and association and dissociation steps of 300 s were each performed sequentially. Kinetic parameters were calculated using ForteBio and SigmaPlot software (Systat Software Inc., USA).
Western blot analysis
Bone marrow-derived macrophage-like cells (BMMs) were washed with ice-cold phosphate-buffered saline (PBS) and lysed in extraction buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid, 0.5% Nonidet P-40, and protease inhibitors]. Cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (Millipore, USA). Membranes were blocked with TBS-T [10 mM Tri-HCl (pH 7.6), 150 mM NaCl, and 0.1% Tween 20] containing 5% skim milk and probed with the primary antibodies against phospho-P38, phospho-JNK, P38, AKT, JNK, and actin (Cell Signaling Technology, USA). Blots were subsequently probed with horse-radish peroxidase-conjugated secondary antibodies (Abcam, UK). Signals were detected with enhanced chemiluminescence (Millipore) and analyzed with an LAS3000 luminescent image analyzer (GE Healthcare).
Modeling and docking of the protein-peptide interaction
The coordinates of the structure of a trimetric biological unit of the RANK-RANKL complex (PDB: 3QBQ) were used as templates in modeling and docking analyses (Ta et al., 2010). The binding mode of Loop3 to the binding cavity of the RANKL trimer guided the homology modeling of each peptide complexed with RANKL. The homology-based multichain-modeling tool of MODELLER version 9.13 was used for this purpose (Eswar et al., 2006). The peptide-RANKL complexes were supplied as a docking input to the Rosetta FlexPepDock ab initio docking server (http://flexpepdock.furmanlab.cs.huji.ac.il/index.php) with default parameters (London et al., 2011). No specific constraints were given to the peptide during docking, so that the peptide was in constraint-free and fully flexible conditions. In subsequent steps of molecular dynamics simulations, the highest-scoring docked structure was used as input for each peptide.
Molecular dynamics simulation
Molecular dynamics simulation was performed using the docking solution obtained from the FlexPepDock docking protocols (London et al., 2011). GROMACS version 5.0.2 was used (Tate, 2014) for molecular dynamics simulations using the DGIST supercomputing cluster facility (Daegu Gyeongbuk Institute of Science and Technology, Korea). An OPLS-AA/L force field was used to generate topology files (George et al., 2001). To define the simulation box, the RANKL-peptide complex was extended by 1 nm using the tool editconf. Water and ions were added to the simulation box and the protein surface using the solvate and genion tools, respectively, in GROMACS. The protein surface was neutralized automatically with the neutral option of genion. The peptide-RANKL complex was energy minimized for several cycles (50,000 steps/cycle) and applied for isothermal-isochoric equilibration (NVT-equilibration) for 100 ps to stabilize the temperature of the system to 300 K. The output of the NVT-equilibration was subjected to constant temperature (300K) and pressure equilibration (NPT-equilibration) to stabilize the pressure of the system. The system was stabilized under a constant average pressure of 1 bar. Next, using the model from the NPT-equilibration, molecular dynamics simulation was performed for 30 ns. For all peptides, a temperature of 300 K and a predetermined constant pressure were applied. Pressure coupling was maintained at the reference pressure of 1 bar and isothermal compressibility of 4.5e-5. Finally, the root-mean-square deviation (RMSD) values of both separate peptides and peptide-protein complexes were calculated.
RESULTS AND DISCUSSION
Design of Loop3-mimic peptides
In our previous work, the crystal structure of the RANKL/RANK complex and biochemical assays of RANK mutants revealed that Loop3 of RANK (118-GYH WNSDCECCRR N-131) was essential for binding to RANKL (Ta et al., 2010). From these observations, we designed several peptides that mimic Loop3 and investigated their inhibitory effects on complex formation with RANKL and osteoclastogenesis. Among the tested peptides, L3-3 (1-YCWNSDCECC YRR-13; Table 1) was shown to be the most potent inhibitor and had higher affinity for RANKL as compared to OP3-4, a previously characterized OPG-mimic inhibitor (Ta et al., 2010). In L3-3, H120 of RANK loop3 was exchanged with a cysteine (C2) for the purpose of cyclization through the C2–C10 disulfide bond, and a tyrosine (Y11) was inserted between C10 and R12 (R129 in RANK) to enable formation of a phi-phi interaction with Y1 (Y119 in RANK). L3-3 was expected to retain an overall loop structure similar to Loop3; however, it was desirable to reduce its size and improve the inhibitory efficiency for clinical applications, given that elucidation of the structural features required for inhibitory function will facilitate the rational design of peptides. Therefore, we modified specific residues in L3-3 (Table 1) and tested the activities of the derivative peptides.
First, W3, a tryptophan corresponding to W121 in RANK, was replaced with a serine in L3-3S in order to relieve the steric hindrance of the indole group. In L3-3n, Y11, the C-terminal tyrosine that was introduced in L3-3 for a phi-phi interaction, was deleted to see how phi-phi stacking affects activity. Two shorter peptides, L3-3A and L3-3B, were designed to determine the minimum requirements for interaction with the Loop3-binding pocket in the RANKL trimer. L3-3A and L3-3B both lacked the N-terminal cysteine residue needed for cyclization (corresponding to C2 in L3-3); however, the formation of an internal disulfide bond (corresponding to C7–C9 in L3-3 and C125–C127 in RANK), which is the critical feature of Loop3, would not be affected. Moreover, the cysteine corresponding to C10 of L3-3 (C128 in RANK) was replaced with a serine to preclude formation of undesired linkages with either cysteine residue (C7 or C9 in L3-3). In L3-3A, two arginine residues at the N-terminus (RR) were deleted in order to enable examination of their roles in the inhibitory activity. The RR residues, corresponding to R129 and R130 in RANK, were shown to interact with E225 and D266 of RANKL through ionic and hydrogen bonding, respectively, in the crystal structure of the complex (Ta et al., 2010).
Inhibition of RANKL-induced osteoclastogenesis
MTT- and TRAP-activity assays were performed for the newly designed peptides, as well as L3-3 as a control, in order to estimate their cytotoxicity and inhibitory effects on osteoclastogenesis, respectively. In the MTT assay, none of the peptides displayed noticeable cytotoxicity on mouse osteoclast-precursor cells (Fig. 1A). The results from the TRAP activity assay indicated that L3-3S and L3-3B were more effective relative to the original L3-3 peptide at inhibiting the maturation of osteoclast precursors (Fig. 1B). The elimination of steric hindrance by the tryptophan residue in L3-3S enhanced the inhibitory effect of the peptide. Most noticeably, the activity of L3-3B was improved significantly, causing almost complete cessation of osteoclastogenesis according to the TRAP-staining assay (Fig. 2). However, inhibitory activity was diminished by the deletion of the RR residues in L3-3A, and no substantial change was observed for L3-3n. These results showed the potential value of L3-3B for therapeutic applications, indicated that overall cyclization was not absolutely required, and that the RR residues contributed to RANKL binding.
Fig. 1.
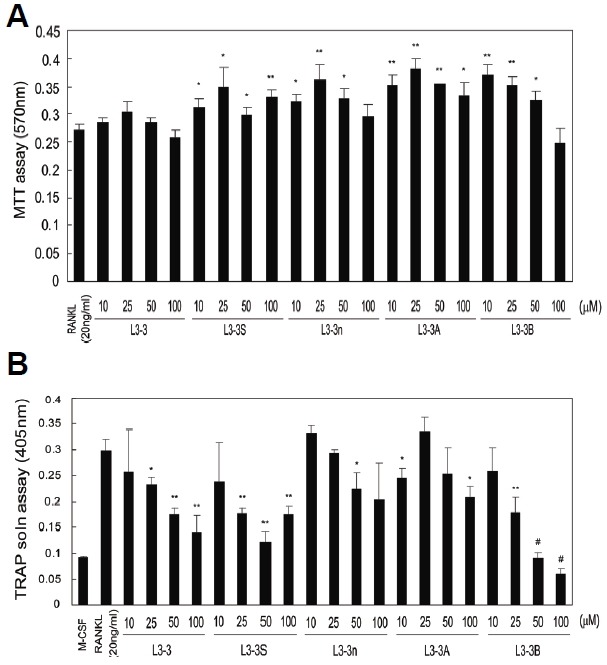
Cytotoxic and inhibitory activities of RANKL-binding peptides. (A) Cytotoxicity of RANKL-binding peptides was estimated by MTT assay. Data represent the mean ± SD of triplicate samples. Results are representative of at least three independent sets of similar experiments. *p < 0.05; **p < 0.005 vs. RANKL (20 ng/ml). (B) The inhibition of RANKL-induced differentiation of osteoclasts was examined by TRAP-activity assay. Mouse osteoclast precursors were treated with 20 ng/ml RANKL and varying concentrations (10–100 μM) of peptides. Data represent the mean ± SD of triplicate samples. *p < 0.05; **p < 0.005; #p < 0.0005 vs. RANKL (20 ng/ml).
Fig. 2.
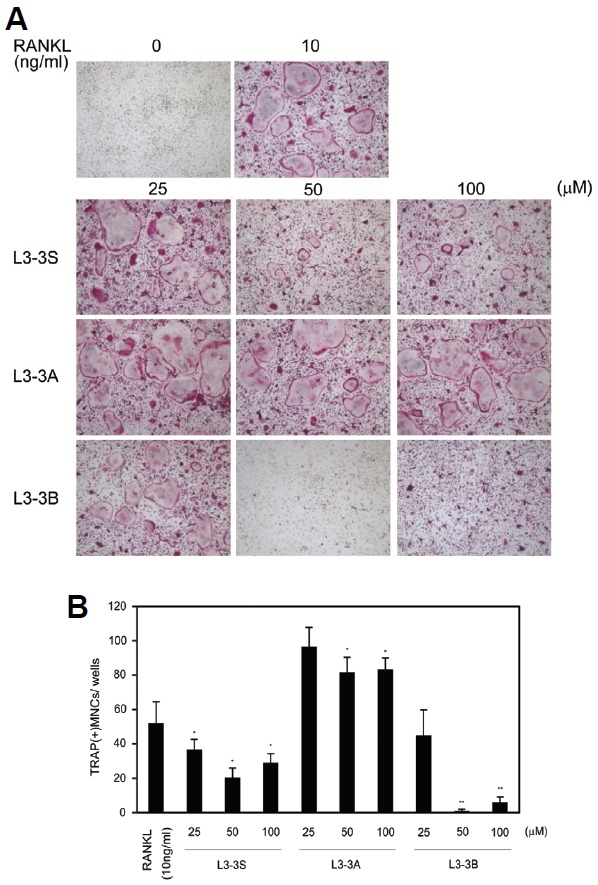
TRAP staining of osteoclast precursors treated with inhibitory peptides. To estimate the inhibition of osteoclastogenesis, multi-nucleated TRAP-stained cells were counted, and L3-3B turned out to be the most efficient blocker. Data represent means ± SD of triplicate samples. Results are representative of at least three independent sets of similar experiments. *p < 0.05, **p < 0.005 vs. RANKL (10 ng/ml).
L3-3B binding affinity for RANKL
We analyzed the binding affinity of L3-3B for RANKL using BLI to corroborate the differentiation assay results. After conjugating a fixed concentration of L3-3B to an amine-reactive biosensor chip, different concentrations of RANKL were applied for association, followed by the dissociation step. The representative graphs at three concentrations are shown in Fig 3A. The Rmax value is defined as the peak point where association ends and dissociation begins at a given concentration of RANKL (Fig. 3A). The steady-state dissociation constant (Kd) of L3-3B for RANKL was calculated using Rmax values at five different concentration points (0.2–15 μM; Fig. 3B). The obtained value (Kd = 6.08 μM) was significantly lower than that of L3-3, which was reported in our previous work (Kd = 17.3 μM; Ta et al., 2010), thereby confirming its improved potency to block formation of the RANK-RANKL complex.
Fig. 3.
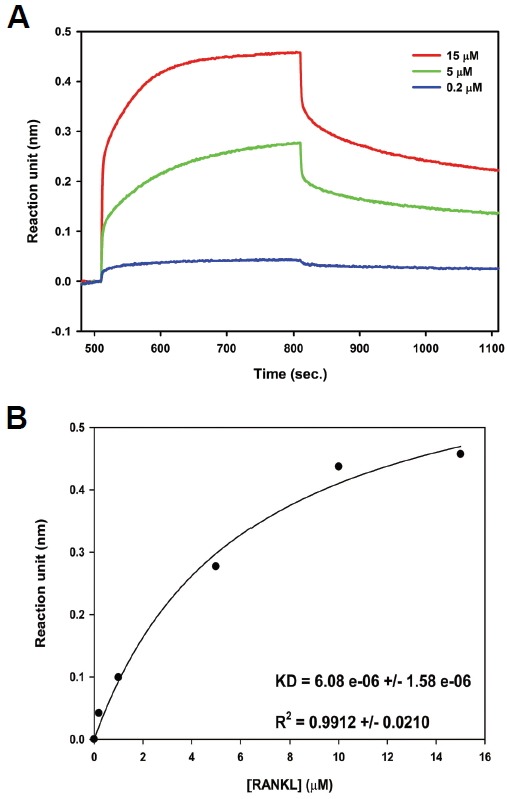
L3-3B binding affinity for RANKL. (A) Diagram of binding between RANKL and L3-3B as determined by biolayer interferometry. (B) Steady-state analysis of the binding affinity between L3-3B and RANKL; f = Bmax * abs(x) / (Kd + abs(x)) was used for the fit-curve analysis.
Specific interference of RANKL signaling by L3-3B
RANKL and macrophage colony-stimulating factor (M-CSF) are two essential cytokines that activate respective intracellular signaling pathways during osteoclast differentiation. In order to investigate the specificity of RANKL inhibition by L3-3B, we performed western blot analyses to examine the interference of RANKL- and M-CSF-signaling pathways in BMM cells. When BMM cells were treated with RANKL that was pre-incubated with L3-3B for 30 min, phosphorylation of p38 and AKT was significantly diminished (Fig. 4A). However, M-CSF-mediated activation of AKT and JNK in BMM cells was not effectively blocked by treatment with L3-3B (Fig. 4B). These results indicated that L3-3B specifically interfered with the RANKL-signaling pathway in BMM cells.
Fig. 4.
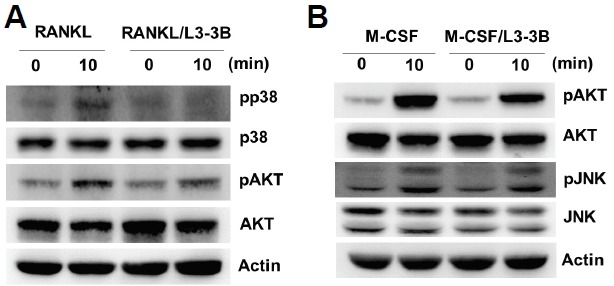
L3-3B inhibition of intracellular RANKL signaling. BMM cells were serum-starved for 5 h and stimulated for 10 min with (A) RANKL (1 μg/ml) or (B) M-CSF (50 ng/ml). L3-3B (1 μg/ml) was pre-incubated with RANKL or M-CSF for 30 min prior to stimulation. Whole-cell lysates were harvested and immunoblotted with indicated antibodies.
Docking of the modeled peptides
The osteoclast differentiation and BLI analyses highlighted the importance of the sharp turn induced by the C-C disulfide linkage (C125 and C127 in RANK) and the RR residues (R129 and R130 in RANK). In order to investigate their interaction modes in the Loop3-binding pocket, peptides were modeled in the RANKL trimer using homology modeling and docking. The binding affinity of each peptide to RANKL was estimated using two scores: a FlexPepDock score, which represented the total Rosetta energy of the model, and an interface score (I_sc), which indicated the difference between the total energy of the complex and the total energy of partners when separated (http://flexpepdock.furmanlab.cs.huji.ac.il/usage.php). We observed that L3-3B possessed the lowest FlexPepDock score of −233.223 Rosetta Energy Units (REUs). Only the score of the shortest peptide, L3-3A (−231.901 REU), was comparable to that of L3-3B (Table 2). Considering that FlexPepDock scores decrease with the length of peptide, it appeared that L3-3B yielded the most stable docking model among the tested peptides. The comparison of I_sc values showed that L3-3B had a much lower score as compared to L3-3A, which can be interpreted as L3-3B having a significantly higher binding affinity to RANKL (Table 2). Generally, the I_sc value is negatively correlated with the length of peptide, assuming that the peptide maintains the same degree of interactions. Other peptides, L3-3, L3-n, and L3-3S, showed similar I_sc values as that of L3-3B, though they were almost twice as long. Therefore, these results indicated that L3-3B possessed the highest binding affinity for the binding pocket of the RANKL timer.
Table 2.
The docking scores for five inhibitory peptides.
| Peptide | FlexPepDock score (REU) | Interface score (I_sc) |
|---|---|---|
| L3-3 | −182.367 | −19.05 |
| L3-3A | −231.901 | −7.803 |
| L3-3B | −233.223 | −15.62 |
| L3-3n | −170.286 | −14.76 |
| L3-3S | −203.767 | −16.02 |
Top docking results (scores) for each peptide are given in Table 2, and the docking of L3-3B in the Loop3-binding pocket of RANKL is depicted in detail in Fig. 5A. In the best docking solution for the L3-3B/RANKL complex, residues S18, H19, K20, Y74, E76, M78, Y80, and S133 from one monomer of RANKL and H64, E108, and F109 from the other interacting monomer of the dimeric interface were found to be oriented within 4 Å of the peptide side chains.
Fig. 5.
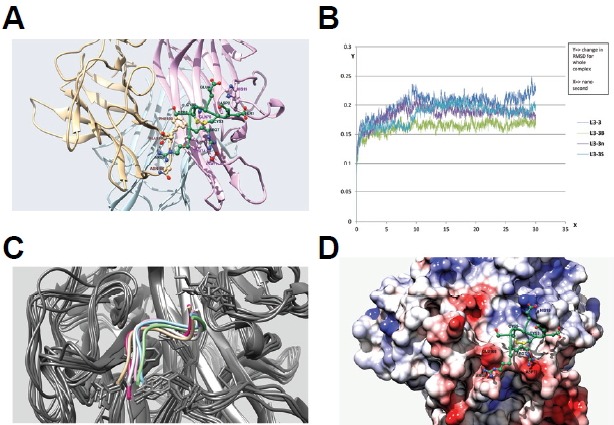
Docking and molecular dynamics simulation of RANKL inhibitory peptides. (A) Docking simulation of the L3-3B peptide. L3-3B is depicted in green, and monomers of the RANKL trimer are colored light brown, light pink, and sky blue. Residues are labeled using the same color scheme with darker shading. (B) Comparison of RMSD graphs from 30-ns molecular dynamics simulation results for four peptides: L3-3, L3-3B, L3-3n, and L3-3S. (C) Overlap of six coordinates of the L3-3B-RANKL complex captured over the course of a 50-ns molecular dynamics simulation. Different backbone orientations of the L3-3B peptide are shown in different colors, with all RANKL backbones colored grey. (D) Binding mode of the L3-3B peptide on the RANKL surface. The RANKL surface is mapped using Coulombic surface-coloring gradient (Chimera 1.10.1), where deep red and deep blue indicate surface charges of −10 and +10, respectively, and white indicates a neutral surface charge. The L3-3B peptide backbone is shown in green, and positive and negative side-chain atoms are depicted in blue and red, respectively. The C3–C5 disulfide bond is colored yellow.
Molecular dynamics simulation
To verify the binding modes of the peptides to RANKL suggested from the docking analyses, further investigation of binding stability was performed using molecular dynamics simulations. The docking model of each peptide bound to RANKL with the highest docking score was used to generate a 30-ns molecular dynamics simulation under an OPLS-AA/L force field. L3-3A showed a high degree of displacement from its initial docking position, the Loop3-binding pocket. Specifically, changes occurring in the first 0.6 ns were the most drastic, with RMSD values close to 5 Å. Throughout the remainder of the simulation, a high degree of fluctuation was observed, indicating the instability of the peptide in the Loop3-binding pocket (Fig. 5B). It is believed that this instability was largely due to the lack of the RR residues, which mediate key interactions involved in RANKL binding (Fig. 5A). However, L3-3B appeared to maintain stable interactions in the binding pocket, which was represented by the minimal changes in RMSD of backbone positions throughout most of the simulation period (30 ns; Fig. 5B). Other peptides showed considerable fluctuation throughout the simulation. These simulation results demonstrated the stable nature of the L3-3B backbone bound to RANKL. These results were further corroborated in a 50-ns simulation, showing the superposition of six L3-3B orientations captured during the simulation and demonstrating the rigidity of its backbone over time (Fig. 5C).
In the molecular dynamics simulation, the interaction between L3-3B and the RANKL trimer was stabilized by interactions between positively charged R7 and R8 residues in L3-3B (R129 and R130 in RANK) and negatively charged residues in RANKL (D73 and E108; Figs. 5A and 5D). Additionally, the sharp loop region, which is maintained by a C3–C5 disulfide bond (C125–C127 in RANK) provided stable packing onto the pocket, consisting of Y74, E76, T100, and F109 in RANKL. Taken together, we concluded that peptide binding to RANKL largely depended upon the packing of the C-E-C loop in RANKL and electrostatic interactions provided by the RR residues.
CONCLUSION
In this study, we designed a new peptide inhibitor of RANKL based on the crystal structure of a RANK loop bound to RANKL. From these efforts, we identified L3-3B as the most promising inhibitor in terms of its length and inhibitory activity. The in vitro binding and osteoclast differentiation assays demonstrated that its binding and inhibitory activities, respectively, were significantly improved as compared to the original peptide, L3-3. The specific inhibition of RANKL by L3-3B was confirmed based on western blot analysis showing that L3-3B was capable of interfering with RANKL-induced phosphorylation of p38 and AKT in BMM cells.
Additionally, the proposed L3-3B binding mode to RANKL according to molecular docking and molecular dynamics simulations was consistent with the experimental results. Our studies confirmed that the two cysteine residues that constitute a sharp turn via a disulfide bond that fits into the Loop3-binding pocket, as well as RR residues that form salt bridges with the negatively charged residues of RANKL, were crucial for stable binding. These interactions appear to be essential requirements for RANKL binding and inhibition of RANKL-mediated osteoclastogenesis. We expect that these findings and strategies will be useful for the rational development of antiresorptive therapeutics. Subsequent investigation of the peptide described here may potentiate development and design of potent peptide therapeutics specific to RANKL modulation.
Acknowledgments
We acknowledge the DGIST Supercomputing & Big Data Center for the allocation of supercomputing time. This work was supported by the Postdoctoral Research Program of Sungkyunkwan University to HK (2014).
REFERENCES
- Burgess T.L., Qian Y., Kaufman S., Ring B.D., Van G., Capparelli C., Kelley M., Hsu H., Boyle W.J., Dunstan C.R., et al. The ligand for osteoprotegerin (OPGL) directly activates mature osteoclasts. J. Cell Biol. 1999;145:527–538. doi: 10.1083/jcb.145.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S.B., Dore R.K., Lane N.E., Ory P.A., Peterfy C.G., Sharp J.T., van der Heijde D., Zhou L., Tsuji W., Newmark R., et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis Rheumatism. 2008;58:1299–1309. doi: 10.1002/art.23417. [DOI] [PubMed] [Google Scholar]
- Cummings S.R., San Martin J., McClung M.R., Siris E.S., Eastell R., Reid I.R., Delmas P., Zoog H.B., Austin M., Wang A., et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Y J. Med. 2009;361:756–765. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- Dempster D.W., Lambing C.L., Kostenuik P.J., Grauer A. Role of RANK ligand and denosumab, a targeted RANK ligand inhibitor, in bone health and osteoporosis: a review of pre-clinical and clinical data. Clin. Ther. 2012;34:521–536. doi: 10.1016/j.clinthera.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Denizot F., Lang R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 1986;89:271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Eswar N., Webb B., Marti-Renom M.A., Madhusudhan M.S., Eramian D., Shen M.Y., Pieper U., Sali A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006;Chapter 5 doi: 10.1002/0471250953.bi0506s15. Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski George A., F. R.A., Tirado-Rives Julian, Jorgensen William L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B. 2001;105:6474–6487. [Google Scholar]
- Geusens P. Emerging treatments for postmenopausal osteoporosis - focus on denosumab. Clin. Interv. Aging. 2009;4:241–250. doi: 10.2147/cia.s3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini M.M., Takayanagi H. The immune system, bone and RANKL. Arch. Biochem. Biophys. 2014;561:118–123. doi: 10.1016/j.abb.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Jules J., Ashley J.W., Feng X. Selective targeting of RANK signaling pathways as new therapeutic strategies for osteoporosis. Expert Opin. Ther. Targets. 2010;14:923–934. doi: 10.1517/14728222.2010.511179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N., Raveh B., Cohen E., Fathi G., Schueler-Furman O. Rosetta FlexPepDock web server--high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011;39:W249–253. doi: 10.1093/nar/gkr431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michigami T., Ihara-Watanabe M., Yamazaki M., Ozono K. Receptor activator of nuclear factor kappaB ligand (RANKL) is a key molecule of osteoclast formation for bone metastasis in a newly developed model of human neuroblastoma. Cancer Res. 2001;61:1637–1644. [PubMed] [Google Scholar]
- Neuprez A., Coste S., Rompen E., Crielaard J.M., Reginster J.Y. Osteonecrosis of the jaw in a male osteoporotic patient treated with denosumab. Osteoporos. Int. 2014;25:393–395. doi: 10.1007/s00198-013-2437-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiro T., Shiotani A., Shibasaki Y., Sasaki T. Osteoclast induction in periodontal tissue during experimental movement of incisors in osteoprotegerin-deficient mice. Anat. Rec. 2002;266:218–225. doi: 10.1002/ar.10061. [DOI] [PubMed] [Google Scholar]
- Recker R., Lappe J., Davies K.M., Heaney R. Bone remodeling increases substantially in the years after menopause and remains increased in older osteoporosis patients. J. Bone Miner. Res. 2004;19:1628–1633. doi: 10.1359/JBMR.040710. [DOI] [PubMed] [Google Scholar]
- Schieferdecker A., Voigt M., Riecken K., Braig F., Schinke T., Loges S., Bokemeyer C., Fehse B., Binder M. Denosumab mimics the natural decoy receptor osteoprotegerin by interacting with its major binding site on RANKL. Oncotarget. 2014;5:6647–6653. doi: 10.18632/oncotarget.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son Y.H., Moon S.H., Kim J. The protein kinase 2 inhibitor CX-4945 regulates osteoclast and osteoblast differentiation in vitro. Mol. Cells. 2013;36:417–423. doi: 10.1007/s10059-013-0184-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta H.M., Nguyen G.T., Jin H.M., Choi J., Park H., Kim N., Hwang H.Y., Kim K.K. Structure-based development of a receptor activator of nuclear factor-kappaB ligand (RANKL) inhibitor peptide and molecular basis for osteopetrosis. Proc. Natl. Acad. Sci. USA. 2010;107:20281–20286. doi: 10.1073/pnas.1011686107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S. Regulation of bone destruction in rheumatoid arthritis through RANKL-RANK pathways. World J. Orthop. 2013;4:1–6. doi: 10.5312/wjo.v4.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate A., Kamil A., Dubey A., Größlinger A., Chamberlain B., Goglin B., Edwards C., Newburn C.J., Padua D. Programming abstractions for data locality. PADAL Workshop 2014; Swiss National Supercomputing Center (CSCS), Lugano, Switzerland. 2014. [Google Scholar]
- Wada T., Nakashima T., Hiroshi N., Penninger J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006;12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Wittrant Y., Theoleyre S., Chipoy C., Padrines M., Blanchard F., Heymann D., Redini F. RANKL/RANK/OPG: new therapeutic targets in bone tumours and associated osteolysis. Biochim. Biophys. Acta. 2004;1704:49–57. doi: 10.1016/j.bbcan.2004.05.002. [DOI] [PubMed] [Google Scholar]