

Abstract
Voltage-gated Ca2+ (CaV) channels are dynamically modulated by G protein-coupled receptors (GPCR). The M1 muscarinic receptor stimulation is known to enhance CaV2.3 channel gating through the activation of protein kinase C (PKC). Here, we found that M1 receptors also inhibit CaV2.3 currents when the channels are fully activated by PKC. In whole-cell configuration, the application of phorbol 12-myristate 13-acetate (PMA), a PKC activator, potentiated CaV2.3 currents by ∼two-fold. After the PMA-induced potentiation, stimulation of M1 receptors decreased the CaV2.3 currents by 52 ± 8%. We examined whether the depletion of phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) is responsible for the muscarinic suppression of CaV2.3 currents by using two methods: the Danio rerio voltage-sensing phosphatase (Dr-VSP) system and the rapamycin-induced translocatable pseudojanin (PJ) system. First, dephosphorylation of PI(4,5)P2 to phosphatidylinositol 4-phosphate (PI(4)P) by Dr-VSP significantly suppressed CaV2.3 currents, by 53 ± 3%. Next, dephosphorylation of both PI(4)P and PI(4,5)P2 to PI by PJ translocation further decreased the current by up to 66 ± 3%. The results suggest that CaV2.3 currents are modulated by the M1 receptor in a dual mode—that is, potentiation through the activation of PKC and suppression by the depletion of membrane PI(4,5)P2. Our results also suggest that there is rapid turnover between PI(4)P and PI(4,5)P2 in the plasma membrane.
Keywords: CaV2.3 channel; Danio rerio voltage-sensitive phosphatase (Dr-VSP); M1 muscarinic receptor; PI(4,5)P2; Pseudojanin
INTRODUCTION
Voltage-gated calcium (CaV) channels are expressed in most excitable cells and facilitate Ca2+ entry in response to membrane depolarization. Among the 10 types of CaV channels, R-type CaV2.3 channels belong to the high voltage-activated (HVA) calcium channel family and are broadly expressed in the brain area, including the hippocampus, amygdala, olfactory bulb, and frontal cortex (Niidome et al., 1992; Soong et al., 1993; Williams et al., 1994). The CaV2.3 channels play important roles in neurotransmitter release, pain transmission, and fear (Lee et al., 2002; Saequsa et al., 2000; Wu et al., 1998). Despite high sequence homology among α1 subunits of the CaV2 family, CaV2.3 channels have different gating properties and pharmacological characteristics from those of CaV2.1 and CaV2.2 channels. CaV2.3 channels are activated at a lower voltage than other CaV2 channels. In addition, the kinetics for activation and inactivation of CaV2.3 currents are faster than those of CaV2.2 channels. From a pharmacological perspective, CaV2.3 channels are insensitive to CaV2.1 and CaV2.2 channel blockers (Soong et al., 1993; Williams et al., 1994). Another significant difference between CaV2.3 channels and other CaV2 channels is the modulation by G-protein-coupled receptors (GPCRs). As a Gq-protein-coupled receptor, M1 muscarinic receptor (M1R) activation results in degradation of plasma membrane PI(4,5)P2. According to previous studies, CaV2.3 channel gating is enhanced by M1R stimulation, probably through the activation of Ca2+-independent protein kinase C (PKC) (Bannister et al., 2004; Melliti et al., 2000; Tai et al., 2006). On the other hand, CaV2.1 and CaV2.2 channels are known to be suppressed by M1R activation, and this suppression turned out to be owing to Gβγ-mediated signaling pathways and/or PI(4,5)P2 depletion (Gamper et al., 2004; Kammermeier et al., 2000; Keum et al., 2014; Melliti et al., 2001; Perez-Burgos et al., 2008; 2010; Shapiro et al., 1999). One previous study reported that CaV2.3 channels were slowly inhibited by Gq/11-coupled neurokinin 1 receptors (Meza et al., 2007). The molecular mechanism of slow inhibition was not identified, but they proposed that the depletion of membrane PI(4,5)P2 may be involved in the inhibitory pathway.
In this paper, we further investigated whether CaV2.3 channels are sensitive to plasma membrane PI(4,5)P2 depletion. PI(4,5)P2 hydrolysis by M1R-mediated phospholipase C (PLC) activation results in the generation of several intracellular secondary molecules, such as inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG), as well as the increase of intracellular Ca2+ concentration and PKC activity. Hence, here we employed voltage-sensitive phosphatase from zebrafish (Dr-VSP) and chemically inducible dimerization (CID) systems, which directly and selectively dephosphorylate PI(4,5)P2 in the plasma membrane without producing any other second messengers. By using these methods, we observed that CaV2.3 channels are modulated by M1R through the modification of the membrane PI(4,5)P2 level. Together, our data demonstrate that CaV2.3 channels are regulated by M1R through dual modulatory pathways: activation through PKC activation and inhibition through PI(4,5)P2 depletion.
MATERIALS AND METHODS
Materials
The following cDNAs were gifted to us: rat α1E (accession number NM_019294) from Terrance P. Snutch, University of British Columbia; rat α1B (accession number NM_001195199), β3 (accession number NM_012828), and α2δ1 (accession number NM_012919) from Diane Lipscombe, Brown University, Providence, RI; rat M1-muscarinic receptor (accession number NM_080773) from Neil N. Nathanson, University of Washington, WA; Dr-VSP with EGFP from Yasushi Okamura, Osaka University, Osaka, Japan; Lyn11-FRB, PJ-Dead, PJ-Sac, INPP5E, PJ, and PH-PLCδ-GFP from Bertil Hille, University of Washington School of Medicine, Seattle, Washington.
Cell culture and transfection
Human embryonic kidney cell-derived tsA201 cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen, USA), and 0.2% penicillin/streptomycin (Invitrogen, USA) in 100-mm culture dishes. In all experiments, for calcium channel expression, the α1B or α1E of CaV, β3, and α2δ1 subunits were transiently transfected into tsA201 cells in a 1:1:1 ratio. In some cases, 1 μg of M1R or 1 μg of Dr-VSP was co-transfected. For the rapamycin-inducible dimerization experiment, 200 ng of Lyn11-FRB and 300 ng of translocatable enzymes (PJ-Dead, PJ-Sac, INPP5E, and PJ) were co-transfected. In addition, for the confocal experiment, 200 ng of PH-PLCδ-GFP was co-transfected. The cells were allowed to grow on a 35-mm culture dish and transfection was performed when the confluency of cells reached 60–70%. Lipofectamine 2000 (10 μl; Invitrogen, USA) was added to 250 μl of DMEM and then left for 5 min. cDNA was applied with another 250 μl DMEM. Both solutions were mixed and incubated for 15 min in a dark space, then the transfectant mixture was added to cells. After 4 h, fresh culture media containing FBS and antibiotics was exchanged. Transfected cells were plated on the poly-L-lysine-coated (0.1 mg/ml, Sigma-Aldrich, USA) chip 48 h later for the electrophysiological experiment or 24 h later for the confocal experiment after transfection.
Solutions
The bath solution used to record Ba2+ currents contained (in mM) 10 BaCl2, 150 NaCl, 1 MgCl2, 10 HEPES, and 8 glucose (adjusted to pH 7.4 with NaOH). The pipette solution contained (in mM) 175 CsCl2, 5 MgCl2, 5 HEPES, 0.1 1,2-bis(2-aminophenocy)ethane N,N,N′,N′-tetraacetic acid (BAPTA), 3 Na2 ATP, and 0.1 Na3GTP (adjusted to pH 7.4 with CsOH). The external solution for confocal imaging contained (in mM) 160 NaCl, 2.5 KCl, 2 CaCl2 H2O, 1 MgCl2, 10 HEPES, and 8 glucose (adjusted to pH 7.4 with NaOH). The bath solutions were stored in a refrigerator at 4°C. The pipette solution was stored in a freezer at −20°C. BAPTA, Na2ATP, Na3GTP, CsOH, and BaCl2 reagents were obtained from Sigma-Aldrich (USA), HEPES was from Calbiochem (USA), and other chemicals were obtained from Merck (Germany).
Chemicals
Oxotremorine-M (Oxo-M, Sigma-Aldrich, USA) was dissolved in sterile water to make a 10 mM stock and was stored at −20°C. Both phorbol 12-myristate 13-acetate (PMA, Enzo life sciences, USA) and rapamycin (LC Laboratories, USA) were dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, USA) to make 100 μM and 5 mM stocks, respectively. All chemicals were stored at −20°C. They were diluted with bath solution before being applied to cells.
Current recording
All currents were obtained at room temperature (22–25°C). Patch pipettes (1–4 MΩ) were pulled from borosilicate glass micropipette capillaries (1.5 mm outer diameter; 1.1 mm inner diameter; and 10 cm length) (Sutter Instrument Company, USA). The whole-cell configuration was used to record Ba2+ currents. In cell attached mode, a gigaohm seal was formed, and the plasma membrane was ruptured by negative pressure. Series resistance was 3.6–6 MΩ and was compensated by 60%. A HEKA EPC-10 amplifier with pulse software (HEKA Elektronik) was used for current recording. Ba2+ currents were recorded with a membrane holding potential of −80 mV, and a 100-ms test pulse (+ 10 mV for CaV2.2 channels and 0 mV for CaV2.3 channels) was applied every 4 s.
For Dr-VSP experiments, the following protocol was used. First, test pulse a (+10 mV for CaV2.2 channels and 0 mV for CaV2.3 channels) was applied for 10 ms. This current became the baseline. Then, +120 mV was generated for 1 s to activate Dr-VSP and to deplete PI(4,5)P2. Following the large depolarizing pulse, −150 mV hyperpolarizing pulse was applied for 400 ms to remove calcium channel inactivation. Finally, test pulse b was applied. Currents a and b, before and after PI(4,5)P2 depletion by Dr-VSP activation, were compared to calculate the ratio of current inhibition.
Confocal imaging
Confocal images were obtained with a Carl Zeiss Inverted LSM 700 confocal microscope (Carl Zeiss AG, GFP by argon-ion laser and mRFP by blue diode laser) at room temperature (22–25°C). In time course, images were obtained by scanning cells with a 40× (water) objective lens at 512 × 512 pixels, and were taken every 10 s for 5 min. For the single image, cells were scanned with a 40× (water) objective lens at 1024 × 1024 pixels. Cytosolic fluorescence intensity was measured by using ZEN2010 software (Carl Zeiss) and was processed with Microsoft Office Excel 2010 (Microsoft) or Igor Pro (WaveMetrics, Inc.).
Data analysis
For data acquisition and analysis, a HEKA EPC-10 amplifier (HEKA Elektronik) was used. Additional data processing was accomplished with Igor Pro (WaveMetrics, Inc.) and Microsoft Office Excel 2010 (Microsoft). Time constants for the responses were obtained by fitting the data to a single-exponential function. All quantitative data were expressed as the mean ± standard error of the mean (SEM). Student’s t-test was used for comparisons between two groups. One-way ANOVA was used for comparisons between more than two groups.
RESULTS
To record calcium channel currents, tsA201 cells were transfected with α1B for CaV2.2 currents or α1E for CaV2.3 currents plus auxiliary subunits β3 and α2δ1. We used Ba2+ as a charge carrier instead of Ca2+ to rule out calcium-dependent inactivation and other unexpected events triggered by Ca2+ ions (Liang et al., 2003). In all experiments, we measured the channel activity of both CaV2.2 and CaV2.3, where the CaV2.2 current was measured as a control for PI(4,5)P2 regulation because it is known to be inhibited by M1R activation (Kim et al., 2015; Suh et al., 2012). To obtain the peak currents, +10 mV and 0 mV were applied for CaV2.2 and CaV2.3 channels, respectively.
Differential modulation of CaV2.2 and CaV2.3 channels by M1 muscarinic receptors
Whereas most HVA calcium channels are known to be inhibited by M1R activation, CaV2.3 channels are further activated by M1R activation (Melliti et al., 2000; Suh et al., 2010). When tsA201 cells co-transfected with M1R and either CaV2.2 or CaV2.3 channels were treated with muscarinic receptor agonist Oxo-M (10 μM) for 60 s, CaV2.2 (N-type) currents were rapidly decreased by 55 ± 2% (n = 13, Figs. 1A and 1C), while CaV2.3 (R-type) currents were increased by 83 ± 7% (n = 9, Figs. 1B and 1C). These differential results were consistent with those of previous studies (Melliti et al., 2000; Perez-Burgos et al., 2008; Perez-Rosello et al., 2004; Suh et al., 2010).
Fig. 1.
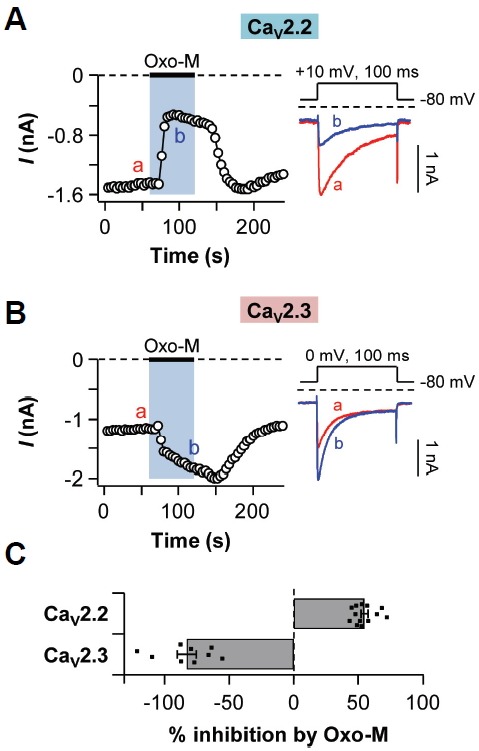
Differential regulation of CaV2.2 and CaV2.3 currents by M1R activation. TsA201 cells co-transfected with M1 muscarinic receptor (M1R) and either CaV2.2 or CaV2.3 channels were treated with 10 μM of Oxo-M for 60 s. (A) Left: Time course of CaV2.2 current regulation. Right: Protocol generating CaV2.2 currents (Upper) and selected current traces designated in left graph (Lower). (B) Left: Time course of CaV2.3 current regulation. Right: Protocol generating CaV2.3 currents (Upper) and selected current traces designated in the left graph (Lower). (C) Summary of % inhibition by Oxo-M treatment in CaV2.2 (n = 13) and CaV2.3 (n = 9) channels. Data are mean ± SEM.
According to previous studies, phosphorylation of CaV α1 subunits by PKC could activate CaV2.3 channels (Fang et al., 2005; Kamatchi et al., 2003; 2004; Rajagopal et al., 2008; Stea et al., 1995). Based on these studies, we decided to verify the effect of PKC on both CaV2.2 and CaV2.3 currents. The bath solution containing 1 μM phorbol 12-myristate 13-acetate (PMA), which is a DAG analog recruiting PKC to the plasma membrane, was applied to the cells for 120 s. While CaV2.2 currents were not significantly changed by PMA application (Fig. 2A), CaV2.3 currents increased almost two-fold (Fig. 2B). Interestingly, we found that after full activation of CaV2.3 channels by PKC activation, M1R activation decreased the CaV2.3 currents by 52 ± 8% (n = 5), similarly to CaV2.2 currents (47 ± 5%, n = 9) (Fig. 2C). The time constants for Oxo-M-induced inhibition of CaV2.2 currents and CaV2.3 currents were 4 ± 0 s (n = 9) and 12 ± 2 s (n = 5), respectively (Fig. 2D). Collectively, our results showed that after full activation of PKC, CaV2.3 channels were also inhibited by M1R activation, which is very similar to CaV2.2 channels.
Fig. 2.
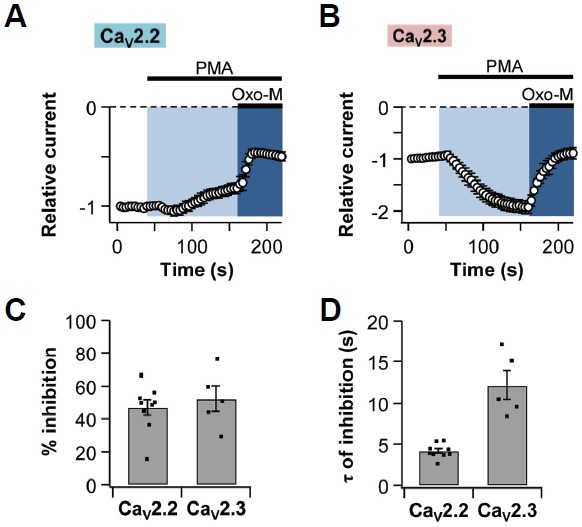
Both CaV2.2 and CaV2.3 currents are suppressed by M1R activation after full-activation of PKC. Phorbol 12-myristate 13-acetate (PMA, 1 μM) was applied for 2 min in tsA201 cells expressing M1R and either CaV2.2 or CaV2.3 channels. Oxo-M was applied for 60 s in the presence of PMA. Normalized current regulation of (A) CaV2.2 channels (n = 9) and (B) CaV2.3 channels (n = 5) by M1R stimulation. (C) Summary of % inhibition by Oxo-M of CaV2.2 (n = 9) and CaV2.3 (n = 5) currents. (D) The time constant for Oxo-M-induced inhibition of CaV2.2 (n = 9) and CaV2.3 (n = 5) currents. Data are mean ± SEM.
CaV2.3 currents are decreased by Dr-VSP activation
Since M1 muscarinic inhibition of voltage-gated calcium channels (VGCCs) is known to be partially due to PI(4,5)P2 depletion through PLCβ enzyme activation (Gamper et al., 2004), we decided to test the effect of PI(4,5)P2 depletion on CaV2.3 channels. Dr-VSP was used to transiently dephosphorylate PI(4,5)P2 in the plasma membrane in response to membrane depolarization and to exclude the effects of the other secondary signaling molecules generated by M1R activation (Okamura et al., 2009; Suh et al., 2010). The protocols used for activating Dr-VSP are represented in Fig. 3A. In tsA201 cells expressing both CaV2.2 channels and Dr-VSP, CaV2.2 currents were decreased by 40 ± 4% (n = 9) after a 1-s depolarizing pulse. In contrast, there was no significant change in the control (-Dr-VSP) (Figs. 3B left and 3C). Similarly, CaV2.3 currents in cells expressing Dr-VSP were decreased by 38 ± 1% (n = 6) in response to PI(4,5)P2 depletion, while the control cells were not (Figs. 3B right and 3D). These results suggest that the depletion of PI(4,5)P2 by Dr-VSP activation inhibits both CaV2.2 and CaV2.3 channels.
Fig. 3.
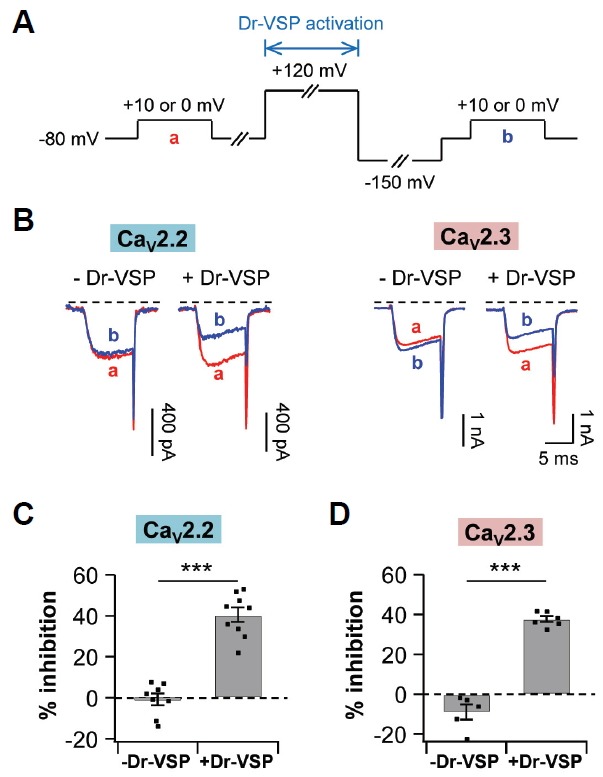
PI(4,5)P2 depletion by Dr-VSP decreases both CaV2.2 and CaV2.3 currents. TsA201 cells were co-transfected with Dr-VSP and either CaV2.2 or CaV2.3 channels. (A) Standard protocol for Dr-VSP activation. Cells received a test pulse (a), then a depolarization to +120 mV for 1 s to activate the Dr-VSP, a hyperpolarization to −150 mV for 0.4 s to remove the voltage-dependent inactivation, and a second test pulse (b). CaV2.2 or CaV2.3 currents were measured before and after the Dr-VSP activation at +10 mV or 0 mV, respectively. (B) Left, CaV2.2 current regulation by a membrane depolarization to +120 mV for 1 s in control (-Dr-VSP, n = 8) and cells expressing Dr-VSP (n = 9). Right, CaV2.3 current regulation in control (n = 6) and cells expressing Dr-VSP (n = 6). (C, D) Summary of % inhibition by Dr-VSP-induced PI(4,5)P2 depletion in CaV2.2 (C) and CaV2.3 (D) channels. Data are mean ± SEM. *** P < 0.001, compared with - Dr-VSP.
CaV2.3 channels are inhibited by rapamycin-inducible pseudojanin systems
To further examine the regulatory effects of membrane PIs on CaV2.3 currents, we employed the recently developed rapamycin-induced translocatable PI phosphatase system (Hammond et al., 2012). In the system, the PI phosphatase is conjugated with FK506-binding protein 12 (FKBP), one of the dimerization subunits. The phosphatase-containing subunit can be recruited to the plasma membrane by application of rapamycin to the cells expressing the plasma membrane-targeting LDR subunit. By using this method, we can selectively and irreversibly deplete the specific PIs in the plasma membrane. Three constructs were used to manipulate the plasma membrane PIs: PJSac, INPP5E, and PJ (Fig. 4A). PJ-Sac is 4-phosphatase from S. cerevisiae sac1. This enzyme dephosphorylates PI(3)P, PI(4)P and PI(3,5)P2, but not PI(4,5)P2 (Guo et al., 1999). INPP5E, inositol polyphosphate-5-phosphatase E, is 5-phosphatase, and its substrates are PI(4,5)P2 and PI(3,4,5)P3 (Bielas et al., 2009). PJ contains both active PJ-Sac and INPP5E domains; thus, this translocatable enzyme can deplete both PI(4,5)P2 and PI(4)P by sequentially dephosphorylating 5-and 4-phosphates. Unlike PJ, PJ-Dead is inactive in both phosphatases. Lyn11, a plasma membrane-targeting motif (Inoue et al., 2005), is fused with FKBP-rapamycin binding protein (FRB). When rapamycin is added, FKBP and FRB form a ternary complex with rapamycin. Hence, the phosphatase conjugated to FKBP is recruited to the plasma membrane and dephosphorylates its substrates (Fig. 4B).
Fig. 4.
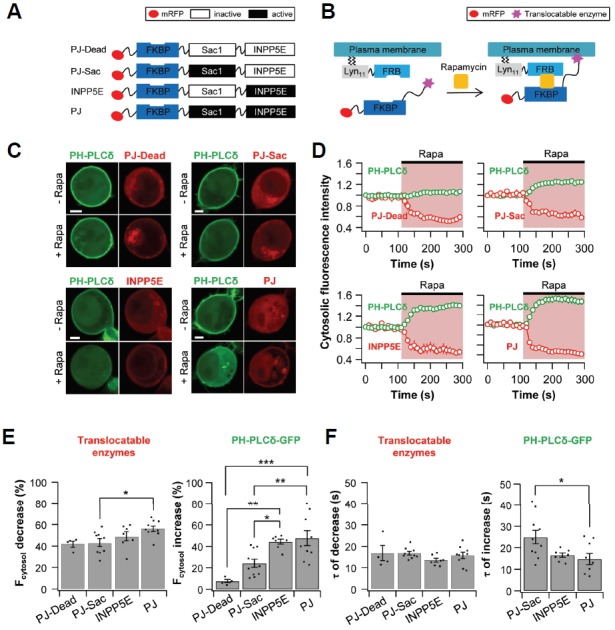
Plasma membrane PI(4,5)P2 levels are reduced by translocation of PJ-Sac, INPP5E, and PJ. TsA201 cells were co-transfected with Lyn11-FRB, PH-PLCδ-GFP, and one of the following four constructs: PJ-Dead, PJ-Sac, INPP5E, or PJ. (A) Construct diagrams of the four translocatable phosphoinositide phosphatases. (B) Diagram of the chemically induced dimerization (CID) system. Rapamycin triggers the translocation of cytoplasmic phosphatases to the plasma membrane. (C) Confocal images of cells expressing PJ-Dead (upper left), PJ-Sac (upper right), INPP5E (lower left), or PJ (lower right) with PH-PLCδ-GFP. Images are from before (Upper) and after (Lower) the application of rapamycin (1 μM) for 180 s (Scale bar, 5 μm). (D) Relative cytosolic intensity of PH-PLCδ-GFP (Green) and translocatable enzyme (Red) for the cells in (C). (E) Summary graph of % decrease in cytosolic translocatable enzymes and of % increase in cytosolic PH-PLCδ-GFP by addition of rapamycin (n = 4 for PJ-Dead; n = 10 for PJ-Sac; n = 7 for INPP5E; and n = 9 for PJ). (F) Summary graph of the time constant for rapamycin-induced decrease in cytosolic translocatable enzymes and for rapamycin-induced increase in cytosolic PH-PLCδ-GFP (n = 4 for PJ-Dead; n = 10 for PJ-Sac; n = 7 for INPP5E; and n = 9 for PJ). * P < 0.05, ** P < 0.01, and *** P < 0.001, with one-way ANOVA followed by Bonferroni post-hoc test.
The movement of translocatable enzymes was monitored using confocal microscopy every 10 s. TsA201 cells were co-transfected with Lyn11-FRB and one of the following four translocatable enzymes tagged with mRFP: PJ-Dead, PJ-Sac, INPP5E, or PJ. The cells were also transfected with the pleckstrin homology (PH) domain of PLCδ labeled with GFP (PH-PLCδ-GFP) as a PI(4,5)P2-specific probe. The PH domain of PLCδ binds to the head group of PI(4,5)P2 so we can detect plasma membrane PI(4,5)P2 in live cells.
Cells expressing both PH-PLCδ-GFP (green) and translocatable enzymes (red) are shown in Fig. 4C. At first, PH-PLCδ-GFP is present in the plasma membrane, while the translocatable enzymes, including PJ-Dead, PJ-Sac, INPP5E, and PJ, exist in the cytosol. After the application of 1 μM rapamycin, all the translocatable enzymes were commonly translocated to the plasma membrane. However, the movement of PH-PLCδ-GFP from the plasma membrane to the cytosol was different depending on the translocatable enzymes. In cells co-transfected with PJ-Dead, the cytosolic fluorescence intensity of PH-PLCδ-GFP was almost the same before and after the rapamycin application (8 ± 2%, n = 4) (Figs. 4D and 4E). However, in cells expressing PJ-Sac, PH-PLCδ-GFP was significantly dissociated from plasma membrane and the cytosolic fluorescence intensity was increased by 24 ± 4% (n = 10). When the INPP5E or PJ systems were applied, the increase in cytosolic fluorescence intensity by INPP5E (44 ± 2%, n = 7) and PJ (48 ± 7%, n = 9) was further increased (Figs. 4D and 4E). We also measured time constants (τ) for the translocation of enzymes as well as PH-PLCδ-GFP. As in a previous study by Dickson et al. (2004), we also used time-series images taken every 10 s for resolving the τ value. There was no difference in τ value between translocatable enzymes. However, when the PJ-Sac transfected, the time constant of the rapamycin-induced increase in cytosolic PH-PLCδ-GFP intensity was 25 ± 3 s (n = 10), relatively slower than those of INPP5E 17 ± 1 s (n = 8) and PJ 15 ± 3 s (n = 9) (Fig. 4F). In summary, our results show that PJ-Sac might be involved in PI(4,5)P2 depletion, but the rate of PI(4,5)P2 dephosphorylation by PJ-Sac was slower than those of INPP5E and PJ. Based on these data, it suggests that PJSac dephosphorylates PI(4)P, and dephosphorylation of PI(4)P induces PI(4,5)P2 depletion.
We then measured the CaV2.2 and CaV2.3 current changes when the translocatable enzymes moved to the plasma membrane and dephosphorylated their PI substrates. The tsA201 cells were transfected with CaV2 channel, Lyn11-FRB, and one of the following phosphatases: PJ-Dead, PJ-Sac, INPP5E, and PJ. The external solution containing 1 μM of rapamycin was perfused for 60 s. The recruitment of PJ-Dead had no significant effects on the currents (Figs. 5A and 5B). CaV2.2 currents in cells expressing PJ-Sac were decreased by 39 ± 5% (n = 9) and the currents expressing INPP5E were decreased by 37 ± 3% (n = 5). When the cells were co-transfected with PJ, the currents were inhibited by 56 ± 4% (n = 11). Because of the irreversibility of rapamycin-induced FKBP-FRB dimerization (Suh et al., 2006), the current amplitudes were not recovered and remained stable even after washout of rapamycin. The inhibition of CaV2.2 currents by the recruitment of PJ-Sac took longer time (29 ± 2 s, n = 9) than that of INPP5E (10 ± 1 s, n = 5) or PJ (7 ± 4 s, n=11) (Fig. 5C).
Fig. 5.
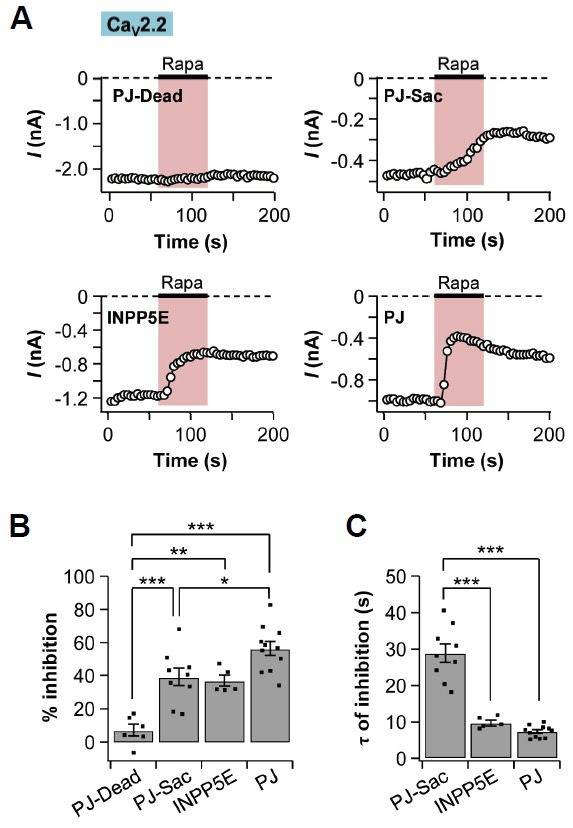
CaV2.2 currents were suppressed by depletion of PI(4,5)P2. TsA201 cells were co-transfected with CaV2.2 channels, Lyn11-FRB (plasma membrane anchoring protein), and one of the following four constructs: PJ-Dead, PJ-Sac, INPP5E, or PJ. Rapamycin was applied for 60 s. (A) Time courses of CaV2.2 currents in cells expressing PJ-Dead, PJ-Sac, INPP5E, or PJ. (B) Summary graph of % inhibition by rapamycin addition in CaV2.2 currents (n = 6 for PJ-Dead; n = 9 for PJ-Sac; n = 5 for INPP5E; and n = 11 for PJ). (C) Summary graph of the time constant for rapamycin-induced inhibition in CaV2.2 currents (n = 9 for PJ-Sac; n = 5 for INPP5E; and n = 11 for PJ). Data are mean ± SEM. * P < 0.05, ** P < 0.01, and *** P < 0.001, with one-way ANOVA followed by Bonferroni post-hoc test.
We also examined the effects of the translocation of pseudojanin constructs on CaV2.3 channel regulation. The tendency for a decrease in CaV2.3 current was similar to that of CaV2.2 channels. The translocation of PJ-Dead had no significant effect on the CaV2.3 currents (3 ± 5%, n = 3). The membrane recruitment of PJ-Sac decreased the CaV2.3 currents by 37 ± 4% (n = 5), while INPP5E decreased the currents by 53 ± 3% (n = 6). Lastly, PJ induced the strongest decrease in CaV2.3 current (66 ± 3%, n = 7) (Figs. 6A and 6B). Like CaV2.2 cur-CaV2.3 current amplitudes were not recovered and remained stable even after rapamycin washout. The time constant for decreasing the CaV2.3 currents by translocation of PJ-Sac was much slower (39 ± 3 s, n = 5) than that of INPP5E (11 ± 1 s, n = 6) or PJ (9 ± 1 s, n = 7) (Fig. 6C). These results also suggested that CaV2.3 currents were suppressed mostly by depletion of PI(4,5)P2 in the plasma membrane.
Fig. 6.
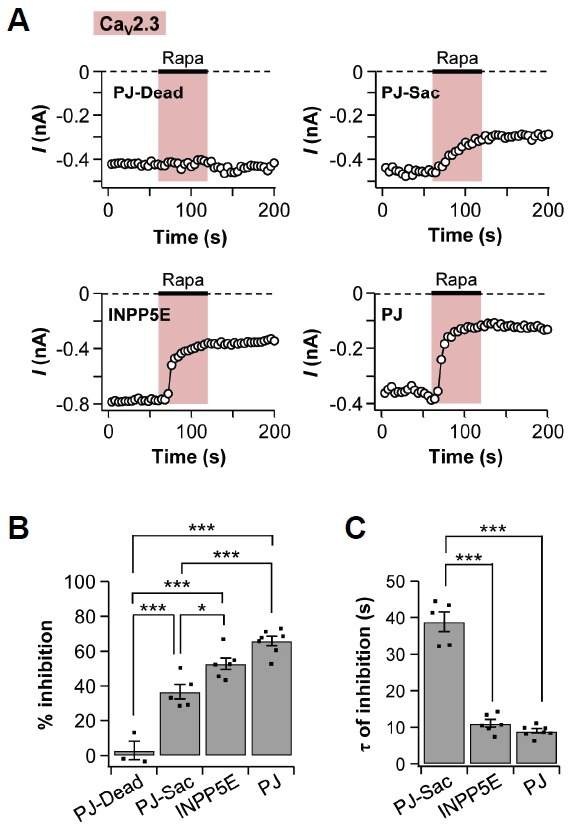
CaV2.3 currents were suppressed by depletion of PI(4,5)P2. CaV2.3 channels were expressed in tsA201 cells with Lyn11-FRB and one of the following four constructs: PJ-Dead, PJ-Sac, INPP5E, or PJ. Rapamycin was added for 60 s. (A) Time courses of CaV2.3 currents in cells expressing PJ-Dead, PJ-Sac, INPP5E, or PJ. (B) Summary of % inhibition by rapamycin in CaV2.3 currents (n = 3 for PJ-Dead; n = 5 for PJ-Sac; n = 6 for INPP5E; and n = 7 for PJ). (C) Summary graph of the time constant for rapamycin-induced inhibition in CaV2.3 currents (n = 5 for PJ-Sac; n = 6 for INPP5E; and n = 7 for PJ). Data are mean ± SEM. * P < 0.05, and *** P < 0.001, with one-way ANOVA followed by Bonferroni post-hoc test.
DISCUSSION
Even though PI(4,5)P2 is known as a crucial regulator of many types of ion channels, including high-voltage activated CaV channels (Hilgemann et al., 2001; Huang, 2007; Rohacs, 2009; Suh and Hille, 2005; 2008), it is not clear whether PI(4,5)P2 in the plasma membrane can regulate CaV2.3 channels. In this study, we showed that CaV2.3 channel can be suppressed by PI(4,5)P2 depletion. This inhibition was proved by direct and selective dephosphorylation of PI(4,5)P2 in the plasma membrane by using Dr-VSP (Fig. 3) and rapamycin-induced translocatable (CID) systems (Figs. 5 and 6).
The α1E gene used in our experiments is rbE-II extracted from rat brain (Soong et al., 1993). The amino-terminus of rbE-II is shorter than other isoforms isolated from mouse, human, and rabbit at about 50 amino acids long. Owing to the short amino terminus, rbE-II is insensitive to voltage-dependent, membrane-delimited inhibition by Gβγ subunits (Page et al., 1998). In our study, we showed that this α1E isoform can be regulated by the voltage-independent, PI(4,5)P2-dependent pathway. Owing to the sequence homology between mammalian α1E (over 93%) (Williams et al., 1994), we speculate that the modulation pattern by M1R activation might be similar.
As shown in Fig. 1B, when M1R is activated, the CaV2.3 current is increased in two phases; an initial steep increase followed by a slow increase. The data suggest that there are two factors involved in CaV2.3 channel regulation. Previous studies showed that M1R induces an increase in the CaV2.3 current through PKC-mediated phosphorylation. However, in our study, we observed that CaV2.3 is also regulated by PI(4,5)P2 depletion. The reason that the inhibitory effect of PI(4,5)P2 depletion on CaV2.3 currents was hidden might be owing to the stronger effect of PKC on CaV2.3 currents. In other words, the PI(4,5)P2 effect seems to be masked by PKC-induced potentiating effects. Why is the PKC effect on CaV2.3 channels stronger than on other types of CaV2 channels? That may be owing to several potential phosphorylation sites in the α1E subunit. As mentioned in the introduction, CaV2.3 channels are potentiated by PKC activation. Previous studies showed that both CaV2.2 and CaV2.3 channels have possible sites for phosphorylation by PMA (Hamid et al., 1999; Zamponi et al., 1997), which are embedded in the I–II loop of the α1 subunit (Fan et al., 2005; Kamatchi et al., 2003; 2004). However, CaV2.3 channels have more phosphorylation sites than CaV2.2 channels in the II–III loop. Indeed, the sequences of the II–III loop between CaV2.3 channels and CaV2.1 or CaV2.2 channels show many differences (Soong et al., 1993). Application of acetyl-β-methylcholine (MCh), another PKC activator, induced phosphorylation in the II–III loop and further increased the CaV2.3 currents (Kamatchi et al., 2004; Rajagopal et al., 2008).
According to our results, the inhibition ratio of CaV2.2 and CaV2.3 currents by the translocation of PJ was greater than that of INPP5E (Figs. 5B and 6B), but the time constants of inhibition by INPP5E and PJ were similar (Figs. 5C and 6C). This might be owing to the rapid turnover between PI(4)P and PI(4,5)P2 (Oude Weernink et al., 2004; Wuttke et al., 2010). In the plasma membrane, PI(4,5)P2 is continuously and rapidly regenerated from PI(4)P by phosphatidylinositol 4-phosphate 5-kinase (Balla, 2013; Wuttke et al., 2010). Since both INPP5E and PJ directly dephosphorylate PI(4,5)P2, the time constants of inhibition in CaV2.2 or CaV2.3 current were similar. However, INPP5E does not deplete PI(4)P, which is a precursor of PI(4,5)P2; thus, PI(4,5)P2 can be rapidly resynthesized from PI(4)P during the INPP5E application and replenished in the plasma membrane. This may be the cause of the lower inhibition of currents by INPP5E compared to PJ.
We also found that in cells expressing PJ-Sac with channels, the CaV2.2 or CaV2.3 currents were decreased by translocation of PJ-Sac to the plasma membrane (Figs. 5B and 6B). However, the time constant (τ) of current inhibition by PJ-Sac was greater compared with that of inhibition by INPP5E or PJ (Figs. 5C and 6C). As shown in the confocal experiments, we observed that four enzymes were translocated to the plasma membrane immediately after the rapamycin application. We also observed that the increase in cytosolic PH-PLCδ-GFP intensity by PJ-Sac was lower than that of INPP5E or PJ (Fig. 4E right), but the τ of PJ-Sac was higher than that of INPP5E or PJ (Fig. 4F right). These data indicate that the translocation of PJ-Sac is also able to induce PI(4,5)P2 depletion. Here, we propose that PJ-Sac dephosphorylates PI(4,5)P2 via continuous turnover to PI(4)P to maintain the equilibrium when the PI(4)P is completely depleted. In the plasma membrane, the amount of PI(4)P and PI(4,5)P2 is maintained at an almost 1:1 ratio by inositol polyphosphate 5-phosphatases (Kwiatkowska, 2010). Altogether, it seems that PI(4)P depletion by PJ-Sac breaks the balance between the amount of PI(4)P and PI(4,5)P2, and thus that PI(4,5)P2 is dephosphorylated to keep the balance between them.
Another regulator of HVA channels is CaV β subunits. They regulate the physiological properties and expression levels of HVA channels. They also regulate the channel sensitivity to PI(4,5)P2, where the sensitivity is different depending on the types of CaV β subunits and their subcellular localization. For example, in cells expressing both CaV2.2 channels and Dr-VSP, currents with β3 subunits were markedly decreased, while currents with β2a subunits showed little effect (Keum et al., 2014; Suh et al., 2012). Therefore, it is meaningful to test the effect of CaV β subunits on the regulation of CaV2.3 channels by PI(4,5)P2 to better understand the regulation mechanism of CaV2.3 channels.
In summary, our study reports that CaV2.3 channels can be regulated by plasma membrane PI(4,5)P2. Like other types of HVA CaV channel, our data demonstrate that CaV2.3 channels are inhibited by PI(4,5)P2 depletion. The present results also show that depletion of PI(4)P, a precursor of PI(4,5)P2, indirectly affects the CaV2.3 channel activity by slowly decreasing the PI(4,5)P2 level in the plasma membrane. This study might contribute to extending our knowledge about regulation of CaV2.3 channels by membrane phosphoinositide.
Acknowledgments
We are grateful to Yeon JH for his valuable discussions. We thank the many labs that provided the plasmids. This work was supported by the Ministry of Education, Science, & Technology (No. 2014R1A1A2044699), the DGIST R&D Program of the Ministry of Science, ICT&Future Planning (No. 14-BD-06), and the DGIST MIREBraiN program (14-01-HRLA-01). The authors declare no conflict of interest. All experiments were conducted in compliance with the ARRIVE guidelines.
REFERENCES
- Balla T. Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013;93:1019–1137. doi: 10.1152/physrev.00028.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister R.A., Melliti K., Adams B.A. Differential modulation of CaV2.3 Ca2+ channels by Gαq/11-coupled muscarinic receptors. Mol. Pharmacol. 2004;65:381–388. doi: 10.1124/mol.65.2.381. [DOI] [PubMed] [Google Scholar]
- Bielas S.L., Silhavy J.L., Brancati F., Kisseleva M.V., Al-Gazali L., Sztriha L., Bayoumi R.A., Zaki M.S., Abdel-Aleem A., Rosti R.O., et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson E.J., Jensen J.B., Hille B. Golgi and plasma membrane pools of PI(4)P contribute to plasma membrane PI(4,5)P2 and maintenance of KCNQ2/3 ion channel current. Proc. Natl. Acad. Sci. USA. 2014;111:E2281–90. doi: 10.1073/pnas.1407133111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H., Franke R., Patanavanich S., Lalvani A., Powell N.K., Sando J.J., Kamatchi G.L. Role of α1 2.3 subunit I–II linker sites in the enhancement of CaV2.3 current by phorbol 12-myristate 13-acetate and acetyl-β-methylcholine. J. Biol. Chem. 2005;208:23559–23565. doi: 10.1074/jbc.M501540200. [DOI] [PubMed] [Google Scholar]
- Gamper N., Reznikov V., Yamada Y., Yang J., Shapiro M.S. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J. Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Stolz L.E., Lemrow S.M., York J.D. SAC1-like domains of yeast SAC1, INP52, and INP53 and of human synaptojanin encode polyphosphoinositide phosphatases. J. Biol. Chem. 1999;274:12990–12995. doi: 10.1074/jbc.274.19.12990. [DOI] [PubMed] [Google Scholar]
- Hamid J., Nelson D., Spaetgens R., Dubel S.J., Snutch T.P., Zamponi G.W. Identification of an integration center for cross-talk between protein kinase C and G protein modulation of N-type calcium channels. J. Biol. Chem. 1999;274:6195–6202. doi: 10.1074/jbc.274.10.6195. [DOI] [PubMed] [Google Scholar]
- Hammond G.R., Fischer M.J., Anderson K.E., Holdich J., Koteci A., Balla T., Irvine R.F. PI4P and PI(4,5)P2 are essential but independent lipid determinants of membrane identity. Science. 2012;337:727–730. doi: 10.1126/science.1222483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D.W., Feng S., Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci. STKE. 2001;2001:re19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- Huang C.L. Complex roles of PIP2 in the regulation of ion channels and transporters. Am. J. Physiol. Renal Physiol. 2007;293:F1761–F1765. doi: 10.1152/ajprenal.00400.2007. [DOI] [PubMed] [Google Scholar]
- Inoue T., Heo W.D., Grimley J.S., Wandless T.J., Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nat. Methods. 2005;2:415–418. doi: 10.1038/nmeth763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamatchi G.L., Tiwari S.N., Chan C.K., Chen D., Do S.H., Durieux M.E., Lynch C., 3rd Distinct regulation of expressed calcium channels 2.3 in Xenopus oocytes by direct or indirect activation of protein kinase C. Brain Res. 2003;968:227–237. doi: 10.1016/s0006-8993(03)02245-5. [DOI] [PubMed] [Google Scholar]
- Kamatchi G.L., Franke R., Lynch C., 3rd, Sando J.J. Identification of sites responsible for potentiation of type 2.3 calcium currents by acetyl-β-methylcholine. J. Biol. Chem. 2004;279:4102–4109. doi: 10.1074/jbc.M308606200. [DOI] [PubMed] [Google Scholar]
- Kammermeier P.J., Ruiz-Velasco V., Ikeda S.R. A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Gαq/11 and Gβγ. J. Neurosci. 2000;20:5623–5629. doi: 10.1523/JNEUROSCI.20-15-05623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum D., Baek C., Kim D.I., Kweon H.J., Suh B.C. Voltage-dependent regulation of CaV2.2 channels by Gq-coupled receptor is facilitated by membrane-localized β subunit. J. Gen. Physiol. 2014;144:297–309. doi: 10.1085/jgp.201411245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.I., Park Y., Jang D.J., Suh B.C. Dynamic phospholipid interaction of β2e subunit regulates the gating of voltage-gated Ca2+ channels. J. Gen. Physiol. 2015;145:529–541. doi: 10.1085/jgp.201411349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska K. One lipid, multiple functions: how various pools of PI(4,5)P2 are created in the plasma membrane. Cell. Mol. Life Sci. 2010;67:3927–3946. doi: 10.1007/s00018-010-0432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.C., Choi S., Lee T., Kim H.L., Chin H., Shin H.S. Molecular basis of R-type calcium channels in central amygdala neurons of the mouse. Proc. Natl. Acad. Sci. USA. 2002;99:3276–3281. doi: 10.1073/pnas.052697799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H., DeMaria C.D., Erickson M.G., Mori M.X., Alseikhan B.A., Yue D.T. Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron. 2003;39:951–960. doi: 10.1016/s0896-6273(03)00560-9. [DOI] [PubMed] [Google Scholar]
- Melliti K., Meza U., Adams B. Muscarinic stimulation of α1E Ca channels is selectively blocked by the effector antagonist function of RGS2 and phospholipase C-β1. J. Neurosci. 2000;20:7167–7173. doi: 10.1523/JNEUROSCI.20-19-07167.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melliti K., Meza U., Adams B.A. RGS2 blocks slow muscarinic inhibition of N-type Ca2+ channels reconstituted in a human cell line. J. Physiol. 2001;532:337–347. doi: 10.1111/j.1469-7793.2001.0337f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza U., Thapliyal A., Bannister R.A., Adams B.A. Neurokinin 1 receptors trigger overlapping stimulation and inhibition of CaV2.3 (R-type) calcium channels. Mol. Pharmacol. 2007;71:284–293. doi: 10.1124/mol.106.028530. [DOI] [PubMed] [Google Scholar]
- Niidome T., Kim M.S., Friedrich T., Mori Y. Molecular cloning and characterization of a novel calcium channel from rabbit brain. FEBS Lett. 1992;308:7–13. doi: 10.1016/0014-5793(92)81038-n. [DOI] [PubMed] [Google Scholar]
- Okamura Y, Murata Y., Iwasaki H. Voltage-sensing phosphatase: actions and potentials. J. Physiol. 2009;587(Pt 3):513–520. doi: 10.1113/jphysiol.2008.163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oude Weernink P.A., Schmidt M., Jakobs K.H. Regulation and cellular roles of phosphoinositide 5-kinases. Eur. J. Pharmacol. 2004;500:87–99. doi: 10.1016/j.ejphar.2004.07.014. [DOI] [PubMed] [Google Scholar]
- Page K.M., Cantí C., Stephens G.J., Berrow N.S., Dolphin A.C. Identification of the amino terminus of neuronal Ca2+ channel α1 subunits α 1B and α1E as an essential determinant of G-protein modulation. J. Neurosci. 1998;18:4815–4824. doi: 10.1523/JNEUROSCI.18-13-04815.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Burgos A., Perez-Rosello T., Salgado H., Flores-Barrera E., Prieto G.A., Fugueroa A., Galarraga E., Bargas J. Muscarinic M1 modulation of N- and L-types of calcium channels is mediated by protein kinase C in neostriatal neurons. Neuroscience. 2008;155:1079–1097. doi: 10.1016/j.neuroscience.2008.06.047. [DOI] [PubMed] [Google Scholar]
- Perez-Burgos A., Prieto G.A., Galarraga E., Bargas J. CaV2.1 channels are modulated by muscarinic M1 receptors through phosphoinositied hydrolysis in neostriatal neurons. Neuroscience. 2010;165:293–299. doi: 10.1016/j.neuroscience.2009.10.056. [DOI] [PubMed] [Google Scholar]
- Perez-Rosello T., Figueroa A., Salgado H., Vilchis C., Tecuapetia F., Guzman J.N., Galarraga E., Bargas J. Cholinergic control of firing pattern and neurotransmission in rat neostriatal projection neurons: role of CaV2.1 and CaV2.2 Ca2+ channels. J. Neurophysiol. 2004;93:2507–2519. doi: 10.1152/jn.00853.2004. [DOI] [PubMed] [Google Scholar]
- Rajagopal S., Fang H., Patanavanich S., Sando J.J., Kamatchi G.L. Protein kinase C isozyme-specific potentiation of expressed CaV2.3 currents by acetyl-β-methylcholine and phorbol-12-myristate, 13-acetate. Brain Res. 2008;1210:1–10. doi: 10.1016/j.brainres.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohacs T. Phosphoinositide regulation of non-canonical transient receptor potential channels. Cell Calcium. 2009;45:554–565. doi: 10.1016/j.ceca.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saequsa H., Kurihara T., Zong S., Minowa O., Kazuno A., Han W., Matsuda Y., Yamanaka H., Osanai M., Noda T., et al. Altered pain responses in mice lacking α1E subunit of the voltage-dependent Ca2+ channel. Proc. Natl. Acad. Sci. USA. 2000;97:6132–6137. doi: 10.1073/pnas.100124197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro M.S., Loose M.D., Hamilton S.E., Nathanson N.M., Gomeza J., Wess J., Gille B. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knockout mice. Proc. Natl. Acad. Sci. USA. 1999;96:10899–10904. doi: 10.1073/pnas.96.19.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong T.W., Stea A., Hodson C.D., Dubel S.J., Vincent S.R., Snutch T.P. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- Stea A., Soong T.W., Snutch T.P. Determinants of PKC-dependent modulation of a family of neuronal calcium channels. Neuron. 1995;15:929–940. doi: 10.1016/0896-6273(95)90183-3. [DOI] [PubMed] [Google Scholar]
- Suh B.C., Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr. Opin. Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- Suh B.C., Hille B. PIP2 is a necessary cofactor for ion channel function: How and why? Annu. Rev. Biophys. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B.C., Inoue T., Meyer T., Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B.C., Leal K., Hille B. Modulation of high-voltage activated Ca2+ channels by membrane phosphatidylinositol 4,5-bisphosphate. Neuron. 2010;67:224–238. doi: 10.1016/j.neuron.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh B.C., Kim D.I., Falkenburger B.H., Hille B. Membrane-localized β-subunits alter the PIP2 regulation of high-voltage activated Ca2+ channels. Proc. Natl. Acad. Sci. USA. 2012;109:3161–3166. doi: 10.1073/pnas.1121434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C., Kuzmiski J.B., MacVicar B.A. Muscarinic enhancement of R-type calcium currents in hippocampal CA1 pyramidal neurons. J. Neurosci. 2006;26:6249–6258. doi: 10.1523/JNEUROSCI.1009-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M.E., Marubio L.M., Deal C.R., Hans M., Brust P.F., Philipson L.H., Miller R.J., Johnson E.C., Harpold M.M., Ellis S.B. Structure and functional characterization of neuronal α1E channel subtypes. J. Biol. Chem. 1994;269:22347–22357. [PubMed] [Google Scholar]
- Wu L.G., Borst J.G., Sakmann B. R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc. Natl. Acad. Sci. USA. 1998;95:4720–4725. doi: 10.1073/pnas.95.8.4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke A., Sågetorp J., Tengholm A. Distinct plasma-membrane PtdIns(4)P and PtdIns(4,5)P2 dynamics in secretagogue-stimulated β-cells. J. Cell Sci. 2010;123:1492–1502. doi: 10.1242/jcs.060525. [DOI] [PubMed] [Google Scholar]
- Zamponi G.W., Bourinet E., Nelson D., Nargeot J., Snutch T.P. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]