

Summary
STING (stimulator of interferon genes) has been shown to be critical for controlling anti-viral responses, as well as anti-tumor adaptive immunity but little is known regarding its regulation in human tumors. Here, we report that STING-signaling is recurrently suppressed in a wide variety of cancers, including colorectal carcinoma. Loss of STING signaling impeded DNA damage responses accountable for generating key cytokines that facilitate tissue repair and anti-tumor-T cell priming, such as type I interferon (IFN). Correspondingly, defective STING function was also highly predictive of effectual DNA virus-mediated oncolytic activity. Thus, impaired STING responses may enable damaged cells to evade host immunosurveillance processes, although provides a critical prognostic measurement that could help predict the outcome to effective oncoviral therapy.
Graphical Abstract
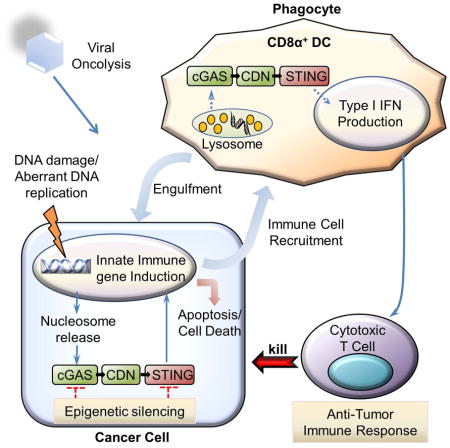
Introduction
Colorectal cancer (CRC) affects about 1.2 million people in the United States with approximately 150,000 new cases are being diagnosed every year. Indeed, CRC is the third most common cause of cancer worldwide, after lung and breast cancer, and the second leading cause of cancer death in adults (DeSantis et al., 2014). Intestine-associated malignant disease frequently develops from colonic epithelial cells that accumulate genetic alterations in key genes involved in the control of cell growth (Fearon, 2011). Multistep genomic damage aggravated alterations can be acquired from environmental factors comprising carcinogens or from genotoxic microbial pathogens including Helicobacter pylori (Arthur et al., 2014; Dzutsev et al., 2015; Kim and Chang, 2014; Louis et al., 2014). Such genetic amendments frequently involve activation of cell growth signaling through mutation of k-ras as well as through mutation or epigenetic silencing of critical tumor suppressor genes (TSGs) such as p53 and adenomatous polyposis coli (APC). Mutated TSGs such as APC can also be inherited, thus increasing the risk of CRC significantly (Fearon, 2011; Hammoud et al., 2013).
Orally administered carcinogens such as the DNA-adduct forming azoxymethane (AOM) induce genomic changes in gastrointestinal epithelial cells, an event which can trigger the activation of DNA damage response (DDR) pathways (Chen and Huang, 2009). While these responses involve repairing DNA breaks and eliminating base mismatches, they can also include activating the production of pro-inflammatory cytokines which alerts the immune surveillance system to the damaged area and facilitates wound repair (Chatzinikolaou et al., 2014). For example, using murine models, it has been demonstrated that the administration of AOM followed by the inflammatory drug dextran sulphate sodium (DSS) can cause epithelial cells to produce IL-1β and IL-18 which becomes processed by the inflammasome, a multiprotein complex comprising nucleotide-binding oligomerization-domain protein like receptors (NLRs) such as NLRP3 and NLRP6 as well as apoptotic speck protein containing a CARD (ASC/PYCARD) and caspase-1, for secretion (Elinav et al., 2013). IL-18, for example, can bind to colonic dendritic cells and signal through MyD88 to prevent the production of growth inhibitory IL-22 binding protein (IL-22BP), which enables unrestricted IL-22 to stimulate tissue repair (Huber et al., 2012; Salcedo et al., 2010). Thus, mice defective in key inflammasome-associated molecules such as ASC or caspase-1 are susceptible to carcinogen induced colitis-associated cancer (CAC) (Elinav et al., 2013). Similarly, loss of key adaptor molecules such as MyD88, required for IL1-R signaling are susceptible to AOM/DSS induced CAC (Salcedo et al., 2013). Plausibly, unrepaired lesions enable the infiltration of microbes with heightened genotoxic aptitude that can chronically aggravate inflammatory processes and the production of DNA damaging radical oxygen species (ROS) (Arthur et al., 2012; Elinav et al., 2013).
While the inflammasome has been shown to be important for processing proinflammatory cytokines such as IL1β and IL-18, it remained to be fully clarified how such wound repair proteins become transcriptionally activated in response to actual genomic damage. However, it has recently been shown that mice lacking the innate immune regulator STING (stimulator of interferon genes) are also sensitive to AOM/DSS-induced CAC (Ahn et al., 2015; Ishikawa and Barber, 2008). STING resides in the endoplasmic reticulum (ER) of hematopoietic cells as well as endothelial and epithelial cells and controls the induction of numerous host defense gene, such as type I IFN as well as pro-inflammatory genes including IL1-β in response to the detection of cyclic dinucleotides (CDNs) such as cyclic-di-AMP (c-di-AMP) generated from intracellular bacteria (Burdette et al., 2011; Ishikawa et al., 2009). STING is also the sensor for CDNs produced from a cellular nucleotidyltransferase referred to as cGAS (cyclic GMP-AMP synthase, also referred to as Mab-21 Domain-Containing Protein and C6orf150) (Sun et al., 2013). Cytosolic DNA species which can constitute the genome of invading pathogens such as HSV-1, or plausibly self-DNA leaked from the nucleus can bind to cGAS to generate non-canonical cGAMP containing one 2′–5′ phosphodiester linkage and a canonical 3–5′ linkage (c[G(2′,5′)pA(3′,5′)p]) (Hornung et al., 2014). The STING pathway may recognize damaged DNA during early response to intestinal damage and may be essential for invigorating tissue repair pathways involving IL1β and IL-18 (Ahn et al., 2015; Ahn et al., 2014a). STING has also been recently reported to play an essential role in dendritic cell recognition of dying tumor cells and the priming of anti-tumor cytotoxic T-cell (CTL) responses (Deng et al., 2014; Woo et al., 2014). Thus, while loss of STING may facilitate tumorigenesis through preventing wound repair and by preventing the production of tumor specific CTLs, the effectiveness of STING signaling in human tumors remains unknown.
Here we report that STING mediated innate immune signaling is largely impaired in human colon cancers as well as many other types of human cancers. In many instances, this was achieved through silencing STING and/or synthase cGAS expression through epigenetic hypermethylation processes. Our findings suggest that STING pathway may have a major function in suppressing colon tumorigenesis and that the inhibition of STING function in this pathway may be selectively suppressed during cancer development. However, we have found that defects in STING signaling renders cancer cells more susceptible to oncolytic viral infection. Therefore, the examination of STING activity in cancers may lead to development of assays that will shed light into the outcome of select cancer therapies.
Results
Defective STING signaling in colorectal adenocarcinoma cells
STING-deficient mice have been reported to be prone to AOM/DSS-associated CAC (Ahn et al., 2015). However, whether STING function is deregulated to any extent in human colorectal adenocarcinoma (CA) is unknown. To start to evaluate this, we examined STING expression by immunoblot in a variety CA cells, generated from cancers diagnosed at various stages as described by Duke’s system (Wu, 2007). Results indicated that STING was expressed in 10 out of 11 cell lines examined, albeit at varying levels (Figure 1A). To correlate expression levels with STING function, we transfected cells with dsDNA to activate STING signaling, or with dsRNA (polyI:C) to activate the RIG-I like pathway (Ishikawa and Barber, 2008). We then measured type I IFN expression by ELISA, which is known to be STING-inducible. We noted that all 11 CA cells responded poorly to dsDNA-triggered type I IFN production. We confirmed that all cells were transfected adequately using FITC-labelled dsDNA activator and immunofluorescence analysis (Figure S1). This was in contrast to control hTERT cells or normal colon epithelial cells (FHC), which when transfected with dsDNA did express IFNβ (Figure 1B). In contrast, 8 of the 11 CA cells were able to produce type I IFN, in various amounts, in response to dsRNA, indicating that the RIG-I-Like pathway retained function in the majority of cases examined (Figure 1B and Figure S2A). A similar finding was noted upon examination of CXCL10 mRNA production by RT-PCR, although some CXCL10 was detected, albeit in low levels, in LoVo and HT29 in response to STING-dependent dsDNA transfection (Figure 1C), whereas polyI:C induced CXCL10 production in majority of CA cells at varying degrees (Figure S2B). To extend these findings further, we measured IL-1β production in the CA cells since we have previously noted that carcinogen triggered DNA damage can induce IL-1β through STING-signaling (Ahn et al., 2012). Loss of IL-1β has been shown to render mice susceptible to CAC due to wound healing responses being impaired (Elinav et al., 2013). This study indicated that IL-1β was produced in the normal hTERT and FHC cells by dsDNA, indicating the importance of STING-activity in this process. However, only 3 out of the 11 CA cells appeared able to produce IL-1β in response to dsDNA treatment, again suggesting that STING function is defective in the majority of CA cells examined (Figure 1D). SW48, which lacked STING expression, did not appear responsive to dsDNA transfection in any capacity. RNAi treatment confirmed that the upregulation of these cytokines was STING-dependent (Figure S3A–C). Given this data, we performed a more detailed analysis of dsDNA-dependent STING signaling in CA cells, by microarray analysis. CA cells were selected based on their ability to exhibit some STING function or not. For example, data from Figure 1C, indicated that HT29 and LoVo cells were partially able to produce CXCL10 in response to dsDNA. In contrast, SW480 and HT116 were noted to be unable to produce CXCL10 to any significant level. Microarray analysis revealed that all the CA cells examined did not respond to dsDNA signaling as efficiently as control FHC cells, and confirmed our RT-PCR analysis (Figure 1E, F). For instance, the level of CXCL10 was significantly higher in the control FHC cells compared to the CA cells analyzed. However, HT29 cells did appear able to retain some response to cytosolic dsDNA, more than any of the other CA cells examined, especially when compared to SW480 or HT116 (Figure 1E, F). While HT29 was able to produce IFNβ moderately as determined by microarray analysis, IFNβ protein production was not readily evident by ELISA, perhaps due to low level expression, which was similarly observed even in the FHC controls (Figure 1B). Nevertheless, taken together, our data indicates that a majority of CA cells exhibit defective STING-dependent signaling with only SW1116, LS123, LoVo and HT29 exhibiting some low level STING activity.
Figure 1. STING mediated dsDNA induced innate immune activation is impaired in majority of human colon cancer cell lines.

(A) Immunoblot of STING in hTERT fibroblasts, normal human colon epithelial (FHC) and a series of human colon cancer cell lines. (B) ELISA analysis of human Interferon β production in the media of cells (same as A) transfected with 3μg/ml polyIC or dsDNA90 or mock transfected for 16 hours. (C) qPCR analysis of human CXCL10 expression in cells (same as A) transfected with 3μg/ml dsDNA90 or mock transfected for 3 hours. (D) qPCR analysis of human IL1B expression in cells same as C. Data is representative of at least two independent experiments. Error bars indicate s.d. *, p<0.05; **, p<0.01; ***, p<0.001; Student’s t-test. (E) Microarray analysis of gene expression in indicated normal and colon cancer cells mock transfected or transfected with 3μg/ml dsDNA90 for 3 hours. Highest variable genes are shown. Rows represent individual genes; columns represent individual samples. Pseudo-colors indicate transcript levels below (green), equal to (black), or above (red) the mean. Scale represents the intensity of gene expression (log10 scale ranges between −3 and 3). (F) Fold change values of highest variable genes shown in E. See also Figure S1 and S2.
Loss of IRF3 function in CA cells
To examine the extent of defective STING signaling in CA cells, we performed immunofluorescence and immunoblot analysis to evaluate NF-κB and IRF3 function. In the presence of dsDNA, STING rapidly undergoes trafficking from the ER, along with TBK1, to perinuclear-associated endosomal regions, containing NF-kB and IRF3, in a process resembling autophagy (Ishikawa and Barber, 2008; Konno et al., 2013). This event accompanies STING phosphorylation and degradation, likely to avoid sustained STING-activated cytokine production which can manifest inflammation (Ahn and Barber, 2014). This approach confirmed that STING could traffic and undergo phosphorylation and degradation in the control hTERT and FHC cells, following treatment with dsDNA (Figure 2A and D, left panel). In these cells, TBK1 became phosphorylated as well as its cognate target IRF3 and the p65 subunit of NF-κB (Figure 2D, left panel). IRF3 and p65 were also noted to translocate into the nucleus, as expected (Figure 2B, C). A comparable effect was observed using SW1116 and LS123 CA cells which exhibited modest dsDNA-dependent IL-1β induction, confirming that the STING pathway retained some function in these two cells (Figure 2A–D and Figure 1C, D). However, while HT29 and LoVo displayed similar IRF3 translocation, these cells lacked p65 translocation. This likely helped to explain that the defect in dsDNA-mediated innate immune gene induction rested in the inability of STING to trigger p65 function (Figure 2A–D and Figure 1E, F). In addition, we noted that the other CA cells, such as SW480, SW1417, SW48 and HT116, exhibited very little STING activity or trafficking (Figure 2A, D right panel). Similarly, little evidence of TBK1 or IRF3 phosphorylation/translocation was noted (highlighted by red boxes). Some indication of p65 phosphorylation was revealed, for example in SW480, but translocation of this transcription factor was not evident in any of the LoVo, HT29, SW480, SW48, SW1417 or HT116 cells. In contrast, dsRNA induced IRF3 translocation in majority of CA cells although p65 translocation seemed to be impaired to a larger extent (Figure S2C–D). STING expression was not observed in SW48 cells as previously described (Figure 1A, 2A, D). This data indicates that dsDNA-signaling is affected at various points of the STING pathway. For example, STING retains some activity and ability to traffic and escort TBK1 to IRF3, as in HT29 or LoVo cells, but NF-kB signaling is affected. In contrast, STING does not appear to undergo any phosphorylation or trafficking in SW480, SW1417, SW48 or HT116 cells, suggesting that STING function is impeded upstream of IRF3/NF-kB interaction.
Figure 2. dsDNA induced STING signaling pathway is defective in majority of human colon cancer cell lines.

(A) Immunofluorescence Microscopy analysis of STING translocation in normal and human colon cancer cell lines transfected with 3μg/ml dsDNA90 or mock transfected for 3 hours. (B) Immunofluorescence Microscopy analysis of IRF3 translocation in cells same as A. (C) Immunofluorescence Microscopy analysis of p65 translocation in cells same as A. Representative images are shown at original magnification, 1260X. Bar size, 1μm. (D) Immunoblot analysis of STING signal activation in cells (same as above) transfected with 3μg/ml dsDNA90 for indicated time periods.
CA cells exhibit defective cGAS expression
Loss of STING trafficking in SW480, SW1417, SW48 or HT116 cells could indicate a problem with STING function in the ER, perhaps involving a mutation that would render STING unable to interact with CDNs. Conversely, the breakdown in STING-signaling could occur upstream and involve the synthase cGAS, which can generate CDNs following association with dsDNA, to augment STING function (Konno et al., 2013). To evaluate this, we sequenced the entire STING genome within all 11 CA cells (introns and exons comprise approximately 7.2 kb on chromosome 5q31.2). Sequence analysis indicated that 2 of the 11 CA cells (LoVo and SW480) exhibited a previously reported HAQ STING variant (Jin et al., 2011; Yi et al., 2013), which occurs in approximately 20% of the population, and which has been reported to be partially defective when overexpressed in 293T cells, yet is able to function normally in the presence of CDNs (Table S1). The remainder of the STING genes analyzed represented the R272 encoded product, which has not been reported to exert any defects in function and which represent approximately 85% of the population. Collectively, these findings do not suggest the existence of a major mutation in the STING gene contained within the CA cells and suggest that a defect upstream of STING, for example at the level of cGAS could plausibly be prevalent. We thus started to examine the expression and activity of cGAS in CA cells. We developed an RT-PCR assay and principally measured cGAS mRNA levels. Our results indicated that, of the 11 CA cells examined, cGAS expression was absent in 5 (45%) of them (LS174T, SW480, SW1417, SW48 and HT116) (Figure 3A). This data was confirmed via immunoblot and immunohistochemistry analysis using an antibody to cGAS (Figure 3A). A qPCR examination of 48 human colon adenocarcinoma samples similarly indicated low to undetectable level of cGAS in 15 of 48 samples (31%) (Figure S4). Our findings could be explained through loss of the cGAS gene. However, sequencing analysis similarly indicated that no major mutations or deletions existed within the genome encoding the cGAS gene (Table S2). In view of this, we examined whether cGAS expression was suppressed by epigenetic phenomena, such as by hypermethylation of the cGAS promoter region (Lao and Grady, 2011; Mitchell et al., 2014). Indeed, databank analysis indicated the presence of considerable CpG islands within the cGAS promoter region (Figure S5A). Control hTERT, or cGAS-defective LS174T, SW480, SW1417, SW48 or HT116 cells were thus treated with the de-methylating agent 5-Aza-2′-deoxycytidine (5AZADC) for 5 days, and cGAS mRNA levels again evaluated. Our study indicated that cGAS expression was rescued in 2 of the 5 cells examined (SW480 and HT116) (Figure 3B). The sequencing of bisulfite converted genomic DNA retrieved from normal and CA cells confirmed significant hypermethylation within the cGAS promoter region of CA cells where cGAS expression is suppressed (Figure S5B). It is not yet clear why expression levels of cGAS are muted in the remainder of the CA cells (LS174T, SW1417, SW48) but suppression could speculatively involve other epigenetic modifications such as histone modifications (Jin and Robertson, 2013). Accordingly, treatment of these cells with histone deacetylase or histone-lysine methyltransferases inhibitors partially rescued cGAS mRNA expression in CA cells examined (LS174T, SW1417, SW48) (Figure S5C). It may also be apparent that alternate mechanisms of cGAS suppression exist, such as those involving miRNAs (Yarbrough et al., 2014). To determine if reconstitution of cGAS expression rescued STING-dependent dsDNA signaling, we examined control hTERT or SW480, HT116 (cGAS rescued by 5AZADC) or LS174T (cGAS not rescued by 5AZADC) CA cells. We observed that the 5AZADC-treated cGAS-rescued SW480, HT116 CA cells, but not LS174T cells regained phosphorylation of TBK1 and IRF3, with concomitant phospho-IRF3 translocation (Figure 3C, D). These effects were reflected in modest expression of type I IFN and IL-1β in the 5AZADC treated SW480 and HT116 CA cells (Figure S5D, E). Thus, demethylating agents may be able to partially rescue STING-dependent innate immune gene induction in select CAs.
Figure 3. cGAS expression is suppressed in many human colon cancer cell lines and can be partially recapitulated through DNA demethylation.

(A) Immunoblot (upper) and qPCR analysis (lower) of cGAS expression in normal and human colon cancer cells. (B) qPCR analysis of cGAS expression in cGAS negative colon cell lines mock treated or treated with 1 μM 5-Azacytidine (5AZADC) for 5 days. (C) Immunoblot analysis of STING signal activation in cells (selected from B) mock treated or treated with 1 μM 5-Azacytidine (5AZADC) for 5 days, followed by dsDNA90 transfection at 3μg/ml for indicated time periods. Data is representative of at least two independent experiments. Error bars indicate s.d. *, p<0.05; **, p<0.01; ***, p<0.001; Student’s t-test. (D) Immunofluorescence Microscopy analysis of IRF3 translocation in SW480 and HT116 cells treated with 5AZADC same as above followed by dsDNA transfection at 3μg/ml dsDNA90 for 3 hours. Representative images are shown at original magnification, 1260X. Bar size, 500 nm. (E) Normal and colon cancer cells were treated with AOM or DMH at 3 mM for 20 hours. IFNB induction was analyzed by qPCR analysis. See also Figure S3, S4.
The question arises as to why the STING-signaling pathway may be inhibited in colon adenocarcinoma. Recently, we have shown that STING-deficient cells and mice are sensitive to AOM-induced DNA damage (Ahn et al., 2015). In this situation, the STING pathway may play a role in the DNA-damage response pathway, to induce the production of cytokines which facilitate tissue repair or damaged cell removal (Chatzinikolaou et al., 2014; Kidane et al., 2014; Lord and Ashworth, 2012). We thus examined innate immune induction of CA cells in response to DNA damaging agents. As shown in Figure 3E, the carcinogens AOM and DMH were able to induce the production of type I IFN in normal colon epithelial (CCD841) and in LS123 (which exhibited partial STING activity; Figure 1C and D). However, CA cells which exhibited defective STING activated IRF3 or NF-kB activity were unable to generate type I IFN in response to AOM or DMH (Figure 3E). Thus, the inhibition of the STING pathway may enable DNA damaged cells, harboring mutations, to escape part of the DNA damage response and the immune surveillance machinery to progress into a tumorigenic state.
Tumors with defective STING-Signaling are sensitive to viral Oncolysis
We have previously shown that loss of STING signaling in vitro or in vivo renders cells or mice, respectively, extremely sensitive to Herpes simplex virus (HSV) infection (Ishikawa and Barber, 2008; Ishikawa et al., 2009). HSV, containing a dsDNA genome of 375 kb is presently being evaluated in clinical trials as a therapeutic agent for the treatment of cancer (Kolodkin-Gal et al., 2009). However, the mechanisms of oncolysis remain to be fully determined and there is no evaluation, presently, for determining the efficacy of HSV antitumor treatment. Given that we have previously shown that STING signaling plays a critical role in host defense responses to HSV infection, and that STING activity is defective in numerous CA cells, we postulated that the ability of STING to signal may affect outcome to HSV oncoviral therapy. To start to address this we infected the CA cells or control hTERT and FHC with HSV1 lacking the γ34.5 encoding product that is presently being evaluated as an oncolytic agent, including against colon cancer as well as melanoma. The γ34.5 viral protein has been proposed to suppress host defense responses, although the mechanisms remain to be fully clarified. Thus, HSV1γ34.5 does not robustly repress innate immune signaling events and potently triggers STING-dependent innate immune gene induction, including type I IFNs (Ishikawa et al., 2009). This analysis indicated that similar to our dsDNA transfection results, HSV1γ34.5 induced the production of IFNβ mRNA significantly in control hTERT and FHC cells, as well as SW1116 and LS123 CA cells (Figure 4A). However, little type I IFN was induced in the remainder of the CA cells, including SW480 and HT116, deficient in cGAS expression. The ability to induce type I IFN inversely correlated with HSV1γ34.5 replication, due to the induced anti-viral effects (Figure 4B). Furthermore, cells such as SW480 and HT116 underwent rapid cell death, likely due to robust viral replication, while control cells and cells with partial STING function (SW1116 and HT29) were significantly more refractory (Figure 4C). We followed up by infecting CA cells with HSV expressing the luciferase gene that contained γ34.5 (HSV-Luc). This data confirmed that CA cells exhibiting defective STING-signaling such as SW480 and HT116 enabled more viral-induced luciferase expression (Figure 4D). siRNA treatment further confirmed that the IFNβ responses induced by HSV1γ34.5 in normal and STING functional CA cells are STING dependent (Figure S3D). Of note is that HSV1 is not the only DNA virus to be considered as an oncolytic therapeutic agent to treat cancer. Other candidate viruses under consideration, including as a therapeutic against colon cancer, comprise Vaccinia Virus (VV), a dsDNA virus with 190 kb genome that replicates in the cytoplasm of infected cells (Rowe and Cen, 2014). To evaluate whether VV can trigger host innate immune response in the absence of functional STING signaling, we infected CA cells with partial STING signaling capacity (SW116 and HT29) or completely defective STING signaling (SW480, HT116) with VV. Similar to the situation using HSV1γ34.5, VV triggered type I IFN and CXCL10 production only in the control cells or CA cells with partial STING signaling ability and not in cells with loss of STING function (SW480 and HT116) (Figure 4E, F). We further analyzed CA cell susceptibility of VV infection. As shown in Figure S6A–D, increased VV replication and cell death were observed in all cells exhibiting loss of cGAS or STING. Our data indicates that CA cells with defective STING-signaling are highly susceptible to HSV1 and VV oncolytic activity. Thus, it is plausible that being able to measure the presence or absence of STING/cGAS expression may help predict the response of patients with certain cancers to selected viral oncolytic therapy.
Figure 4. STING signal defect leads colon cancer cells more susceptible to DNA virus infection.

(A) Cells (same as in figure 1) were infected with HSV1γ34.5 at M.O.I. 5 for 1 hour and human IFNB induction was analyzed by qPCR 3 hours post infection. (B) Normal human hTERT cells and selected human colon cancer cell lines (cGAS positive: SW1116, HT29; cGAS negative: SW480, HT116) were infected with HSV1γ34.5 at indicated M.O.I. for 1 hour, and titration of HSV1γ34.5 was analyzed by standard plaque assay in Vero cells 24 hours later. (C) Cells (same as in B) were infected with HSV1γ34.5 at M.O.I. 1 for 1 hour, and cell viability was analyzed by trypan blue staining 24 hours and 48 hours later. (D) Cells (same as in A) were infected with HSV1-Luc at indicated M.O.I. for 1 hour, and luciferase activity was analyzed 24 hours later. (E) Colon Cancer cells were infected with Vaccinia Virus at M.O.I. 100 and analyzed by qPCR for IFNB expression 3 hours post infection. (F) Cells same as E were analyzed by qPCR for CXCL10 expression. Data is representative of at least two independent experiments. Error bars indicate s.d. *, p<0.05; **, p<0.01; Student’s t-test. See also Figure S2.
Predicting outcome to viral oncolytic therapy
Our data indicates that the outcome of oncolytic virotherapy involving DNA-based viruses such as HSV1 may be predicted by the presence or absence of STING signaling. Since the STING pathway naturally requires the presence of STING and cGAS to function, and since we have observed that STING and/or cGAS may be absent in 30–55% of colon cancer, being able to measure the presence of these two gene products may therefore indicate the effectiveness of DNA-viral oncotherapy. This could be achieved using RT-PCR methodology but biopsied tissue may contain infiltrating hematopoietic cells that contain normal STING/cGAS expression (Ishikawa et al., 2009). Thus, analysis of STING and/or cGAS protein or RNA expression within the cancer cell itself would provide more accurate information into the status of STING function. First, we designed a RNA in situ hybridization assay using RNAscope technology that can detect the single levels copies of an mRNA within individual cells. By labelling the STING probe green (FITC), and the cGAS probe red (Cy5), we were able to use both probes in the same assay and effectively quantitate the mRNA levels of STING and cGAS within the identical cell. To test the assay, we incubated control cells or cGAS positive (SW1116 or HT29) or negative (SW480 and HT116) CA cells with RNA probes recognizing cGAS (red) or STING (green) mRNA. This study indicated that STING and cGAS expression could be detected and quantitated in the control (hTERT and FHC) and STING/cGAS positive (SW1116 or HT29) CA cells using the RNAscope (Figure 5A, C). However, only STING was observed in the cGAS negative (SW480 and HT116) CA cells (Figure 5A, C). STING was not detectable in SW48 cells, as expected, using this assay (Figure 1A and 5A, C). This data also correlated with our previous expression analysis of cGAS in these cells by RT-PCR (Figure 3A). Moreover, we were able to observe cGAS expression by RNAscope in those CA cells where cGAS mRNA production was rescued following treatment with 5AZADC (SW480 and HT116) (Figure 5B, D). Thus, fluorescence in situ hybridization analysis may be able to predict the outcome to oncolytic viral therapy depending on the presence or absence of cGAS or STING.
Figure 5. RNA in situ hybridization and Immunohistochemistry analysis of STING and cGAS in human colon cancer cell lines.

(A) RNA fluorescence in situ hybridization (RNA FISH) analysis of STING and cGAS expression in normal and human colon cancer cell lines. Representative images are shown at 1260X. Bar size, 500 nm. (B) RNA FISH analysis of STING and cGAS expression in SW480 and HT116 mock treated or treated with 1 μM 5 AZADC for 5 days. Representative images are shown at 1260X. Bar size, 500 nm. (C) Quantitation of STING and cGAS RNA copy number in A. (D) Quantitation of cGAS RNA copy number in B. (E) STING and cGAS expression in formalin-fixed paraffin-embedded (FFPE) normal and human colon cancer cell lines were analyzed by Chromogenic RNA in situ hybridization (RNA CISH). Quantitation of STING and cGAS RNA copy number are shown in bar graph. Data is representative of at least two independent experiments. Error bars indicate s.d. *, p<0.05; **, p<0.01; ***, p<0.001; Student’s t-test. (F) Representative images of STING and cGAS RNA CISH analysis are shown at 600X. Bar size, 1μm. (G) Immunohistochemistry analysis of cGAS and STING expression in colon cancer cells. Images were shown at 400X. Bar size, 20μm.
To further follow up on this assay, we paraffin embedded normal hTERT, or cGAS positive (SW1116 or HT29) or negative (SW480 and HT116) CA cells. We also analyzed SW48 which had both cGAS and STING expression missing. This situation may mimic situations where biopsied and paraffin embedded patient derived material required analysis. We were again readily able to detect using the RNA probes both STING and cGAS expression in control, SW1116 and HT29 cells, as before, and only STING in the cGAS negative SW480 and HT116 CA cells (Figure 5E, F). Neither cGAS nor STING was observed in the double negative SW48 line (Figure 1A and 5E, F). Using antibody to cGAS and STING, we also performed immunohistochemistry (IHC) analysis on paraffin embedded cells and confirmed cGAS and STING expression, or not, in accord with our immunoblot and RNAscope studies (Figure 1A, 3A, 5A, F and G). Subsequently, we further tested our RNAscope assay on 12 normal and 80 CA samples in paraffin embedded tissue microarrays (TMA) and observed substantial loss of STING and cGAS RNA expression starting from stage II. Overall, 36% (29 out of 80) cancer samples had undetectable STING and/or cGAS RNA in stage II–IV colon cancer (Figure 6A, B). Furthermore, IHC analysis of a human colon cancer TMA corroborated our RNA analysis and similarly showed substantial loss of STING and/or cGAS protein expression starting predominantly in stage II samples, generating an overall (stage II–IV) of 54% loss (21 out of 39) (Figure 6C, D). Statistical analysis of STING and cGAS expression in different stages of colon cancer using the H-score method showed significant loss of cGAS expression by IHC as early as Stage II, whereas loss of STING is more profound in later stages (Table S3). Given this data, it is plausible that STING/cGAS expression is also lost in other tumor types. Subsequently, we indeed noted that STING expression and/or function was absent in a variety of other tumor types, indicating that suppression of this pathway may be widespread in human cancer (Figure S7). In summary, RNAscope and IHC procedures may be useful for the analysis of cGAS and STING expression in FFPE preserved samples.
Figure 6. RNA in situ hybridization analysis in colon cancer tissue microarray.

(A) RNA CISH analysis of STING and cGAS expression in a human colon cancer TMA. STING and/or cGAS expression status in each tissue is summarized in the table. (B) Representative images of A are shown at 400X. Bar size, 20μm. (C) IHC analysis of STING or cGAS expression in a human colon cancer TMA. Expression status is summarized in the table. (D) Representative images of C are shown at 200X. Bar size, 50μm. See Also Figure S5.
In vivo analysis of CA cells with defective STING signaling to HSV1γ34.5 therapy
It is possible that loss of STING signaling may affect the outcome to select oncoviral therapy (Figure 4A–D) (Kolodkin-Gal et al., 2009; Rowe and Cen, 2014). To start to evaluate this, in vivo, we correlated the in vitro oncolytic effect of HSV1 by subcutaneously inoculating nude mice with CA cells harboring partial (SW1116 or HT29) or defective (SW480 and HT116) STING signaling. HSV1γ34.5 was then administered intratumorally and tumor growth monitored (Figure 7A). This study indicated that tumors exhibiting partial STING-signaling (SW1116 and HT29) were refractory to viral oncolytic treatment (Figure 7B, C). While these tumors had different growth rates in vivo, they did not respond to viral therapy at all and the mice were sacked after the tumors burden became significant. In contrast, tumors derived from CA cells with defective STING-signaling (SW480 and HT116) were noted to be extremely susceptible to virus treatment (Figure 7D, E). In mice implanted with SW480, tumor size decreased rapidly 3 days after HSV1γ34.5 treatment, and 4 out of 7 tumors diminished 2–3 weeks after treatment (Figure 7D). In addition, we examined the oncolytic effect of vaccinia virus in CA cells exhibiting partial STING signaling (HT29) or completely defective in STING signaling (HT116), and observed that vaccinia virus reduced the growth of HT116 in nude mice significantly (p=0.012) when compared to HT29 (p=0.297) (Figure S6E–F). Our data thus correlates with our in vitro findings and indicates that the activity of the STING pathway may predict the outcome of DNA virus-related oncolytic therapy against colon and perhaps other cancers.
Figure 7. Increased HSV1γ34.5 oncolytic effect was observed in colon cancer cells with impaired STING signal in vivo.

(A) Scheme of HSV1γ34.5 treatment on xenograft tumor in nude mice. (B–D)The indicated xenograft tumors were generated in the right flank of nude Balb/c mice. When tumors had reached approximately 0.5 cm in diameter, tumors were injected every other day a total of three times (arrows) with 1E7 PFU HSV1γ34.5 in 50 μl PBS (N=7) or 50 μl PBS only (N=3) and tumor growth measured every other day. Data is representative of two independent experiments. Statistical analysis was carried out comparing the two treatment groups at the last time point using the unpaired Student’s t-test. P values are as indicated.
Discussion
The STING controlled signaling pathway is essential for facilitating innate immune gene transcription in response to the recognition of cytosolic DNA species (Ishikawa and Barber, 2008). STING activity can be triggered by CDNs such as cyclic-di-AMP or cyclic-di-GMP produced from intracellular bacteria such as Listeria monocytogenes or by cyclic-di-GMP-AMP (cGAMP) manufactured by the synthase cGAS following association with cytosolic dsDNA species (Burdette et al., 2011; Cai et al., 2014). Such DNA can represent the genome of DNA pathogens, such as HSV-1 or bacteria such as mycobacterium tuberculosis, as well as self DNA leaked from the nucleus of DNA damaged cells. STING-deficient mice, while viable, are extremely sensitive to lethal infection by a variety of pathogens (Ishikawa et al., 2009). However, chronic STING activity has been shown to cause a diversity of autoinflammatory disease, through the overproduction of pro-inflammatory cytokines (Ahn and Barber, 2014). Indeed, inappropriate overstimulation of STING has even been shown to aggravate inflammation driven skin cancer (Ahn et al., 2014b). Recently, transient STING activity has been shown to be essential for mediating the generation of anti-tumor T-cell responses (Woo et al., 2015). Data suggests that STING, in professional antigen presenting cells (CD8+ dendritic cells) becomes extrinsically activated by the DNA of engulfed dying tumor cells which results in the triggering of cytokines such as type I IFN, which facilitates cross-presentation and CTL priming (Woo et al., 2014). Correspondingly, the therapeutic administration of CDNs, intratumorally, has been shown to repress tumor growth, presumably by facilitating DC-dependent CTL production (Corrales et al., 2015; Woo et al., 2014). STING may also play a role in influencing the anti-tumor effects of checkpoint inhibitors such as PD1, although the mechanisms remain to be determined (Fu et al., 2015).
We have also recently demonstrated that STING-deficient mice are susceptible to carcinogen-aggravated CAC, however STING-deficient cells are not highly susceptible to oncogenic transformation (Ahn et al., 2015). In this situation, evidence indicates that damaged DNA can trigger STING intrinsic activity, perhaps by leaking out of the nucleus or through other mechanisms that remain to be clarified. Presumably, this event would augment cytokine production that would attract the immune system to the damaged cells (Ahn et al., 2014a; Ahn et al., 2014b; Chatzinikolaou et al., 2014; Deng et al., 2014; Kidane et al., 2014; Zhu et al., 2014). Eradication of such cells may ensue, as well as the stimulation of cytokine and growth factor dependent tissue repair. Data suggests that STING can trigger the production of cytokines that facilitate wound repair in the gut, such as IL-1β. Such cytokines are processed by nucleotide-binding oligomerization-domain protein like receptors (NLRs) such as NLRP3 and NLRP6, which interact with inflammasome-associated ASC and caspase-1 to process IL-1β and IL-18 (Elinav et al., 2013). These pro-inflammatory cytokines are secreted and bind to receptors mainly requiring MyD88 for signaling. IL-18 production can suppress IL-22BP, which is responsible for inhibiting the wound repair activity of IL-22 (Huber et al., 2012). Loss of ASC, caspase-I, MyD88 or IL22BP can increase tumorigenesis in colitis-associated colon cancer models, similar to loss of STING (Barker et al., 2011; Elinav et al., 2013; Huber et al., 2012; Salcedo et al., 2010). STING may therefore work in concert with inflammasome processing.
Thus, loss of STING suppresses tissue healing and damaged mucosal lining may enable the invasion and expansion of bacteria with enhanced genotoxic ability which would aggravate STING-independent inflammatory responses (Ahn et al., 2015; Arthur et al., 2012). The generation of ROS by overactive, infiltrating immune cells may enhance DNA damaging processes and facilitate mutational inactivation of TSGs or the mutational activation of growth stimulatory proteins such as k-ras (Fearon, 2011; Sosa et al., 2013). Thus, intrinsic STING-signaling may play a key role in preventing the development of cancer through responding to DNA damage and alerting the immune surveillance machinery (Chatzinikolaou et al., 2014; Kondo et al., 2013). In addition, extrinsic STING activity in DCs is also required for the generation of anti-tumor CTLs (Corrales et al., 2015; Woo et al., 2015). This places STING in a pivotal role in the host anti-cancer defense arsenal that intrinsic STING activity facilitates clearance of pre-cancerous cells by alerting the immune system and through extrinsic STING stimulation of anti-tumor T cell activity.
Given this, we analyzed the expression and regulation of STING signaling in colon cancer and found frequent suppression of STING activity. These events inhibited the production of DNA-damage dependent cytokine production, which may enable the damaged cell to escape the attention of the immune surveillance system. Such cells may evade eradication and further genetic mutation events may accrue to enhance the tumorigenic process (Elinav et al., 2013; Kidane et al., 2014). The inhibition of STING signaling was observed to mainly involve the suppression of STING expression, or of the synthase cGAS. We did not observe significant mutation or deletion events involving the STING or cGAS genes, but rather observed frequent transcriptional suppression involving hypermethylation of the promoter regions. We were able to partially rescue cytosolic DNA signaling using demethylating agents which regained cGAS expression in some but not other CA cells. However, it remains unclear whether the rescue of STING signaling in cancer cells may afford better responses to anti-cancer agents. Further, that cGAS and in some cases STING expression was not rescued by demethylating agents may indicate other forms of epigenetic silencing that requires additional characterization. In other CA types, we observed that the ability of STING to activate the transcription factors NF-κB or IRF3 was impaired, by molecular mechanisms that also remain to be determined. It is noteworthy that a number of other genes involved in DNA repair, such as the mismatch repair proteins MHS2 and MLH1 are also reported to be frequently silenced in colon cancer (Chatzinikolaou et al., 2014; Le et al., 2015; Lord and Ashworth, 2012). Thus targeting the DNA repair machinery maybe a common requirement in cancer development. Collectively, we observed that STING-dependent signaling was defective in numerous colon related tumors examined. This may indicate that suppression of STING function is also a key obligation for the tumorigenic process.
Since loss of STING may be common in tumors and may even predict outcome to anti-cancer therapy, we also developed assays to evaluate the expression levels of both STING and cGAS. Loss of either of these two proteins appears to repress cytosolic DNA mediated innate immune signaling. Our ability to measure STING and cGAS mRNA expression in situ, and STING protein expression using antibody enabled us to develop a screen that indicated loss of one or other of these proteins in over 40% of CAC. Such assays may be useful in predicting the effective response rates of cancers to select therapeutic interventions. Further, recapitulating STING signaling in tumors, via novel antitumor gene therapy approaches, might enable such cells to reactivate host antitumor immunity.
Accordingly, we noticed that loss of STING signaling in CA cells enabled the robust replication of DNA-based viruses such as HSV1. Viruses, such as HSV1 and vaccinia virus, are presently being used as oncolytic agents for the treatment of cancer (Kolodkin-Gal et al., 2009; Rowe and Cen, 2014). Such viruses may directly destroy the tumor cell by lysis as well as create a tumor antigen source for engulfment by APCs for the generation of CTLs (Woo et al., 2015). Data indicates that STING plays a key role in both these processes (Figure S8). However, the efficacy of successful oncoviral therapy remains low, for reasons that remain unclear. Mainly, assays based on molecular insight, that may help predict treatment outcome have not been developed. This is because the molecular mechanisms those explain oncolysis in cancer cells rather than normal cells remains to be fully appraised. Evidence suggests that innate immune signaling pathways that exert anti-viral activity may be defective in cancer cells (Heiber and Barber, 2012). In a tumor cell, functional intrinsic STING signaling would prevent efficient virus replication and oncolysis. We propose that this impedes the effectiveness of oncolytic activity. However, in cells lacking STING, greater oncolytic activity is expected and indeed observed and greater cytolysis occurs as our data shows. This would provide a larger amount of tumor cell lysate that would be engulfed and cross-presented for T-cell priming (which requires STING). Our data presented here is amongst the first clear indication that loss of an innate signaling pathway can predict outcome to oncoviral therapy. Thus, utilization of molecular biomarker assays similar to the ones portrayed here may enable a more predictive response to the use of microbes for the treatment of cancer. Such assays may also shed insight into whether other STING-dependent anti-tumor therapies based on CDNs, or even DNA-adduct based chemotherapeutic regimes, may work or not (Zitvogel et al., 2013). In this light, we have recently described that the immunological benefits of using chemotherapeutic agents such as cisplatin and etoposide significantly involved the STING-signaling pathway (Ahn et al., 2014b). Thus, further studies on the regulation and function of STING in cancer may provide acumen into the molecular mechanisms of tumorigenesis as well as provide a therapeutic target that may help in the treatment of cancer.
Experimental Procedures
Materials
All reagents were from Invitrogen, Thermo Scientific or Sigma unless specified.
Cell culture
Cells were purchased from Lozna and ATCC and cultured in their appropriate growth media. hTERT-BJ1 Telomerase Fibroblasts (hTERT) were originally from Clontech and cultured in 4:1 ratio of DMEM:Medium 199 supplement with 10%FBS, 4 mM L-Glutamine and 1 mM sodium pyruvate at 37°C in a 5% CO2-humidified atmosphere.
Immunoblot analysis
Equal amounts of proteins were resolved on sodium dodecyl sulfate (SDS)-Polyacrylamide gels and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore). After blocking with 5% Blocking Reagent, membranes were incubated with various primary antibodies (and appropriate secondary antibodies). The image was resolved using an enhanced chemiluminescence system ECL (Thermo Scientific) and detected by autoradiography (Kodak). Antibodies: rabbit anti STING polyclonal antibody was developed in our laboratory as described previously in Ishikawa et al, 2008; other antibodies were obtained from following sources: β-actin (Sigma Aldrich), p-IRF3 (Cell Signaling), IRF3 (Santa Cruz Biotechnology), p-p65 (Cell Signaling), p65 (Cell Signaling), p-TBK1 (Cell Signaling), TBK1 (Abcam), cGAS (Cell Signaling).
Interferon β Elisa analysis
Interferon β Elisa was performed using either the IFNβ human ELISA Kit from Invitrogen or the Human IFNβ ELISA Kit from PBL InterferonSource following the manufacturer’s protocol.
Immunofluorescence Microscopy
Cell were fixed with 4% parsformaldehyde for 15 minutes in at 37°C and permeabilized with 0.05% Triton X-100 for 5 minutes at room temperature. Immunostaining was performed with STING, IRF3 or p65 antibody followed by fluorescence conjugated secondary antibodies (FITC-goat-anti-rabbit) (Invitrogen). Images were taken with Leika LSM confocal microscope at the Image Core Facility, University of Miami.
Microarray Analysis
Total RNA was isolated from cells or tissues with RNeasy Mini kit (Qiagen). RNA quality was analyzed by Bionalyzer RNA 6000 Nano (Agilent Technologies). Gene array analysis was examined by Illumina Sentrix BeadChip Array (Human HT-12_V4_Bead Chip) at the Oncogenomics Core Facility, University of Miami. Gene expression profiles were processed and statistical analysis was performed at the Bioinformatics Core Facility, University of Miami. Briefly, raw intensity values from Illumina array are uploaded on GeneSpring™ software from Agilent. Values are Quantile normalized and log2 transformed to the median of all samples. Significantly differential expressed genes from a two-class comparison are computed using the Student’s t-test and selected using threshold of P-value ≤ 0.05. Hierarchical Clustering and visualization of selected differentially expressed genes is performed on GeneSpring using Pearson Correlation distance method and linkage was computed using the Ward method. Fold Change analysis was performed between two groups and differentially expressed genes were selected based on threshold of Fold Changes.
Quantitative Real-Time PCR (qPCR)
Total RNA was reverse transcribed using QuantiTect Reverse Transcription Kit (Qiagen). Real-time PCR was performed with the TaqMan gene Expression Assay (Applied Biosystems).
Immunohistochemistry and Histological Analysis
Tissue Microarray was purchased from Origene. Immunohistochemistry staining was performed with cGAS or STING antibody following standard protocol. The score for the extent of the IHC-stained area was set as 0 for no IHC signal at all, 1 for <10%, 2 for 10%to 50%, and 3 for >50% of tumor cells stained. The score for IHC intensity was also scaled as 0 for no IHC signal, 1 for weak, 2 for moderate, and 3 for strong IHC signals. The final score used in the analysis was calculated by multiplying the extent score and intensity score, with a maximum score of 9. Staining was considered position if scored ≥ 3.
Virus Amplification, Purification, Titration and Infection
HSV-1 γ34.5 was kindly provided by Bernard Roizman. HSV-1 luc was kindly provided by David Leib. Vaccinia virus (vTF7-3) was kindly provided by John Rose. Virus was amplified in Vero cells and purified by sucrose gradient ultracentrifugation following standard protocol. Plague assay using serial diluted virus was performed in Vero cells following standard protocol. Cells were infected with virus at specific M.O.I. for 1 hour, washed and then incubated for designated period for specific assay examination.
RNA in situ Hybridization
STING and cGAS RNA probed was custom designed by ACD and RNA in situ Hybridization was performed using RNAscope® Multiplex Fluorescent Reagent Kit for cultured cells and 2-plex RNAscope® Reagent Kit for FFPE cells and tissue following the manufacturer’s instruction. Staining quantification followed the score guideline which was categorized into five grades: 0, No staining or less than 1 dot in every ten cells (40X magnification); 1, 1–3 dots/cell (visible at 20–40X magnification); 2, 4–10 dots/cell, and very few dot clusters (visible at 20–40X magnification); 3, >10 dots/cell and less than 10% positive cells have dot clusters (visible at 20X magnification); and 4, >10 dots/cell and more than 10% positive cells have dot clusters (visible at 20X magnification). Staining was considered positive if scored ≥ 1.
Mouse Treatment
Balb/C nu/nu mice were purchased from Charles River and maintained in the institutional Division of Veterinary Resources (DVR). All experiments were performed with institutional animal care and use committee (IACUC) approval and in compliance with IACUC guidelines. Tumor cells were introduced in the flanks of Balb/c nude mice by subcutaneous injection of 2E6 of the appropriate tumor cells and tumors allowed to develop to an average diameter of approximately 0.5 cm. HSV1γ34.5 was then be injected into the tumors every other day for a total of three times at appropriate dosage (i.e. 50 μl at 1E7 PFU/ml). PBS was used as vehicle control. Effects on tumor growth was monitored. Mice was euthanized when tumor diameter exceeds 10mm.
C57/BL6 wild type (WT) or Sting knock out (SKO) mice were injected subcutaneously with B16-OVA tumor cells (5E105 cells/mouse) on day 0. On days 7, 9, 11, mice were injected intratumorally with HSV1γ34.5 (5E5 PFU/mouse). PBS was used as control. Tumor volume was evaluated every other day.
Interferon γ assay
Splenocytes were isolated from WT or SKO mice inoculated with HSV1γ34.5 (5E5 PFU/mouse) on day 13 after B16-OVA cell inoculation. The cells were plated at 1E6 cells per well and stimulated with OVA peptide (SIINFEKL). After 48 hours, IFNγ levels were determined from the supernatant using mouse IFNγ ELISA kit (R&D system) following manufacturer’s instruction.
Bisulfite Sequencing Analysis
Bisulfite conversion of genomic DNA was performed using EZDNA Methylation Kit from Zymo Research followed by PCR amplification. PCR products were then gel purified and sanger sequenced.
Genomic DNA Sequencing
Genomic DNA were extracted from CA as well as normal cells using Qiagen DNeasy Kit and specific locus was sequenced by Polymorphic DNA Technologies.
Statistical Analysis
All statistical analysis was performed by Student’s t test unless specified. The data were considered to be significantly different when P < 0.05.
Supplementary Material
Acknowledgments
We thank Dr. Biju Issac of the Sylvester Comprehensive Cancer Center Bioinformatics Core Facility for Gene expression array analysis; Ms. Delia Gutman and Ms. Auristela Rivera for mice breeding; Dr. Phillip Ruiz of Department of Surgery for IHC interpretation.
Footnotes
Accession Number
The GEO accession number for the microarray data reported in this paper is GSE75205.
Author Contribution
T.X. carried out most of the experiments; K.H. performed signaling immunoblot analysis; J.A. performed DNA damage signaling analysis; G.N.B wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Current opinion in immunology. 2014;31:121–126. doi: 10.1016/j.coi.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:19386–19391. doi: 10.1073/pnas.1215006109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Konno H, Barber GN. Diverse roles of STING-dependent signaling on the development of cancer. Oncogene. 2015 doi: 10.1038/onc.2014.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN. Intrinsic self-DNA triggers inflammatory disease dependent on STING. Journal of immunology. 2014a;193:4634–4642. doi: 10.4049/jimmunol.1401337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Konno H, Konno K, Ruiz P, Barber GN. Inflammation-driven carcinogenesis is mediated through STING. Nature communications. 2014b;5:5166. doi: 10.1038/ncomms6166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Gharaibeh RZ, Muhlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nature communications. 2014;5:4724. doi: 10.1038/ncomms5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Muhlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BR, Taxman DJ, Ting JP. Cross-regulation between the IL-1beta/IL-18 processing inflammasome and other inflammatory cytokines. Current opinion in immunology. 2011;23:591–597. doi: 10.1016/j.coi.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Molecular cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Chatzinikolaou G, Karakasilioti I, Garinis GA. DNA damage and innate immunity: links and trade-offs. Trends in immunology. 2014;35:429–435. doi: 10.1016/j.it.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Chen J, Huang XF. The signal pathways in azoxymethane-induced colon cancer and preventive implications. Cancer biology & therapy. 2009;8:1313–1317. doi: 10.4161/cbt.8.14.8983. [DOI] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell reports. 2015;11:1018–1030. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity. 2014;41:843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS, Jemal A. Cancer treatment and survivorship statistics, 2014. CA: a cancer journal for clinicians. 2014;64:252–271. doi: 10.3322/caac.21235. [DOI] [PubMed] [Google Scholar]
- Dzutsev A, Goldszmid RS, Viaud S, Zitvogel L, Trinchieri G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. European journal of immunology. 2015;45:17–31. doi: 10.1002/eji.201444972. [DOI] [PubMed] [Google Scholar]
- Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nature reviews Cancer. 2013;13:759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Fearon ER. Molecular genetics of colorectal cancer. Annual review of pathology. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, Mechette K, Leong JJ, Lauer P, Liu W, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Science translational medicine. 2015;7:283ra252. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Cairns BR, Jones DA. Epigenetic regulation of colon cancer and intestinal stem cells. Current opinion in cell biology. 2013;25:177–183. doi: 10.1016/j.ceb.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiber JF, Barber GN. Evaluation of innate immune signaling pathways in transformed cells. Methods in molecular biology. 2012;797:217–238. doi: 10.1007/978-1-61779-340-0_15. [DOI] [PubMed] [Google Scholar]
- Hornung V, Hartmann R, Ablasser A, Hopfner KP. OAS proteins and cGAS: unifying concepts in sensing and responding to cytosolic nucleic acids. Nature reviews Immunology. 2014;14:521–528. doi: 10.1038/nri3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber S, Gagliani N, Zenewicz LA, Huber FJ, Bosurgi L, Hu B, Hedl M, Zhang W, O’Connor W, Jr, Murphy AJ, et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Advances in experimental medicine and biology. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Xu LG, Yang IV, Davidson EJ, Schwartz DA, Wurfel MM, Cambier JC. Identification and characterization of a loss-of-function human MPYS variant. Genes and immunity. 2011;12:263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidane D, Chae WJ, Czochor J, Eckert KA, Glazer PM, Bothwell AL, Sweasy JB. Interplay between DNA repair and inflammation, and the link to cancer. Critical reviews in biochemistry and molecular biology. 2014;49:116–139. doi: 10.3109/10409238.2013.875514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World journal of gastroenterology: WJG. 2014;20:9872–9881. doi: 10.3748/wjg.v20.i29.9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodkin-Gal D, Edden Y, Hartshtark Z, Ilan L, Khalaileh A, Pikarsky AJ, Pikarsky E, Rabkin SD, Panet A, Zamir G. Herpes simplex virus delivery to orthotopic rectal carcinoma results in an efficient and selective antitumor effect. Gene therapy. 2009;16:905–915. doi: 10.1038/gt.2009.44. [DOI] [PubMed] [Google Scholar]
- Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, Komatsu K, Akira S, Kawai T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2969–2974. doi: 10.1073/pnas.1222694110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao VV, Grady WM. Epigenetics and colorectal cancer. Nature reviews Gastroenterology & hepatology. 2011;8:686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015 doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–294. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nature reviews Microbiology. 2014;12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- Mitchell SM, Ross JP, Drew HR, Ho T, Brown GS, Saunders NF, Duesing KR, Buckley MJ, Dunne R, Beetson I, et al. A panel of genes methylated with high frequency in colorectal cancer. BMC cancer. 2014;14:54. doi: 10.1186/1471-2407-14-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe J, Cen P. TroVax in colorectal cancer. Human vaccines & immunotherapeutics. 2014;10:3196–3200. doi: 10.4161/21645515.2014.973323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R, Cataisson C, Hasan U, Yuspa SH, Trinchieri G. MyD88 and its divergent toll in carcinogenesis. Trends in immunology. 2013;34:379–389. doi: 10.1016/j.it.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo R, Worschech A, Cardone M, Jones Y, Gyulai Z, Dai RM, Wang E, Ma W, Haines D, O’HUigin C, et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. The Journal of experimental medicine. 2010;207:1625–1636. doi: 10.1084/jem.20100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sosa V, Moline T, Somoza R, Paciucci R, Kondoh H, MELL Oxidative stress and cancer: an overview. Ageing research reviews. 2013;12:376–390. doi: 10.1016/j.arr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends in immunology. 2015;36:250–256. doi: 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JS. Rectal cancer staging. Clinics in colon and rectal surgery. 2007;20:148–157. doi: 10.1055/s-2007-984859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarbrough ML, Zhang K, Sakthivel R, Forst CV, Posner BA, Barber GN, White MA, Fontoura BM. Primate-specific miR-576-3p sets host defense signalling threshold. Nature communications. 2014;5:4963. doi: 10.1038/ncomms5963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single nucleotide polymorphisms of human STING can affect innate immune response to cyclic dinucleotides. PloS one. 2013;8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Man SM, Gurung P, Liu Z, Vogel P, Lamkanfi M, Kanneganti TD. Cutting edge: STING mediates protection against colorectal tumorigenesis by governing the magnitude of intestinal inflammation. Journal of immunology. 2014;193:4779–4782. doi: 10.4049/jimmunol.1402051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G. Mechanism of action of conventional and targeted anticancer therapies: reinstating immunosurveillance. Immunity. 2013;39:74–88. doi: 10.1016/j.immuni.2013.06.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.