

Abstract
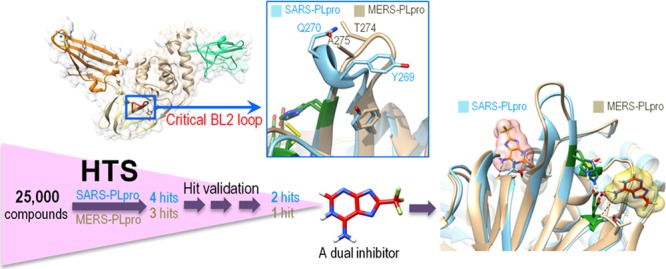
The Middle East Respiratory Syndrome coronavirus (MERS-CoV) papain-like protease (PLpro) blocking loop 2 (BL2) structure differs significantly from that of SARS-CoV PLpro, where it has been proven to play a crucial role in SARS-CoV PLpro inhibitor binding. Four SARS-CoV PLpro lead inhibitors were tested against MERS-CoV PLpro, none of which were effective against MERS-CoV PLpro. Structure and sequence alignments revealed that two residues, Y269 and Q270, responsible for inhibitor binding to SARS-CoV PLpro, were replaced by T274 and A275 in MERS-CoV PLpro, making critical binding interactions difficult to form for similar types of inhibitors. High-throughput screening (HTS) of 25 000 compounds against both PLpro enzymes identified a small fragment-like noncovalent dual inhibitor. Mode of inhibition studies by enzyme kinetics and competition surface plasmon resonance (SPR) analyses suggested that this compound acts as a competitive inhibitor with an IC50 of 6 μM against MERS-CoV PLpro, indicating that it binds to the active site, whereas it acts as an allosteric inhibitor against SARS-CoV PLpro with an IC50 of 11 μM. These results raised the possibility that inhibitor recognition specificity of MERS-CoV PLpro may differ from that of SARS-CoV PLpro. In addition, inhibitory activity of this compound was selective for SARS-CoV and MERS-CoV PLpro enzymes over two human homologues, the ubiquitin C-terminal hydrolases 1 and 3 (hUCH-L1 and hUCH-L3).
Middle East Respiratory Syndrome coronavirus (MERS-CoV), previously called human coronavirus-Erasmus Medical Center (HCoV-EMC), was first reported in Saudi Arabia in 2012 and spread to 20 different countries,1−4 resulting in 853 infections with 301 deaths as of October 2, 2014.5 The unusually high case-fatality rate (CFR) of MERS-CoV infections (∼35%) is alarming as it far exceeds that of all other known human coronaviruses, including the human severe acute respiratory syndrome coronavirus (SARS-CoV). SARS-CoV caused a fatal global outbreak in 2003, resulting in 800 deaths (∼10% CFR).6 There are over 20 known coronaviruses (CoV), six of which are identified as human coronaviruses (HCoV; Supplementary Figure S1). Coronaviruses are classified into four genera (α, β, γ, and δ), and each genus can be divided into lineage subgroups. Of the six HCoVs, two (NL63 and 229E) belong to genus α, and the remaining four (HKU1, OC43, SARS-CoV, and MERS-CoV) belong to genus β. Within the betacoronavirus genus, SARS-CoV is classified as lineage group B, while MERS-CoV is categorized into lineage group C based on their genomes. Two bat CoVs from lineage group C, BtCoV-HKU4 and BtCoV-HKU5, are the most closely related to the MERS-CoV.2,7−9 MERS-CoV and SARS-CoV are highly pathogenic, with evidence of person-to-person transmission via either household or hospital contacts.10,11 MERS-CoV and SARS-CoV use different receptors, dipeptidyl peptidase 4 (DPP4 or CD26) and angiotensin-converting enzyme 2 (ACE2), respectively,12,13 and the epidemiology of MERS-CoV is still being investigated. Both MERS-CoV and SARS-CoV exhibit as a severe respiratory infection, while MERS-CoV exhibits an additional unique symptom of renal failure.2 Even though the MERS-CoV transmission rate is slower than that of SARS-CoV, the number of MERS-CoV infections continues to grow.11,14,15 Due to the recent emergence of this new coronavirus and the potential of SARS-CoV retransmission from zoonotic reservoirs to humans,16−18 the possibility of another deadly pandemic has been seriously raised. However, there is still no effective therapeutic available against either coronavirus. Therefore, developing treatments against both coronaviruses is important.
Both MERS-CoV and SARS-CoV are single-stranded positive-sense RNA viruses with approximately 30 kb genome sizes. Each of their genes encodes two polyproteins called pp1a and pp1b (Figure 1A) that are processed by two proteases, a 3-C-like protease (3CLpro) and a papain-like protease (PLpro). Many coronaviruses contain two PLpro enzymes (PLP1 and PLP2), but MERS-CoV and SARS-CoV have only one PLpro enzyme.19,20 PLpro enzymes are part of a large nonstructural protein 3 (nsp3) that contains four other domains, a ubiquitin-like fold (UB1), an ADP-ribose-1d-phosphatase (ADRP) domain, a SARS-unique domain (SUD), and a transmembrane (TM) domain (Figure 1A). PLpro is responsible for cleavage of the first three positions of its polyprotein, while 3CLpro cleaves the remaining 11 locations, releasing a total of 16 nonstructural proteins (nsp) in both MERS-CoV and SARS-CoV. Sequence motifs recognized by MERS-CoV PLpro (MERS-PLpro) and SARS-CoV PLpro (SARS-PLpro) are (L/I)XGG↓(A/D)X and LXGG↓(A/K)X, respectively (Figure 1B). Unlike 3CLpro, SARS-PLpro has been shown to be a multifunctional protein involved in de-ISGylation, deubiquitination, and viral evasion of the innate immune response in addition to viral peptide cleavage as a protease.16,21 Researchers have discovered that the MERS-PLpro also exhibits deubiquitination and de-ISGylation functions, blocking the interferon regulatory factor 3 (IRF3) pathway.22,23 Both 3CLpro and PLpro are known to be essential for viral replication, making them attractive targets in antiviral drug discovery.20,24 In this work we investigated four known SARS-PLpro lead inhibitors against MERS-PLpro. In addition, high-throughput screening (HTS) of a 25 000-compound antimicrobial focused library against both MERS-CoV and SARS-PLpro enzymes identified a low molecular weight compound that showed activity against both PLpro enzymes via two different modes of inhibition.
Figure 1.

Schematics of SARS-CoV and MERS-CoV polyproteins. (A) Cleavage positions of PLpro (pink) and 3CLpro (cyan) are shown by different colored arrows in their polyproteins. (B) Cleavage site comparison between SARS and MERS PLpro enzymes. Sequence motifs recognized by SARS-CoV PLpro (SARS-PLpro) and MERS-CoV PLpro (MERS-PLpro) are LXGG↓(A/K)X and (L/I)XGG↓(A/D)X, respectively.
Results and Discussion
SARS-PLpro Lead Inhibitors Do Not Inhibit MERS-PLpro
We and others have previously identified and developed a series of noncovalent SARS-PLpro inhibitors using high-throughput screening (HTS) and structure-based drug design that can be classified into two distinct scaffolds.25−28 Four inhibitor structures from these two scaffolds are shown in Figure 2A, with I-1 and I-2 representing scaffold 1 and I-3 and I-4 representing scaffold 2. Inhibitors I-2 and I-3 exhibited excellent inhibitory activities with IC50 values of 0.34 μM and 0.6 μM against SARS-PLpro, with SARS antiviral activities of 2 μM and 15 μM, respectively.25,28 The four SARS-PLpro lead inhibitors (I-1–I-4) were tested against MERS-PLpro to determine whether these two PLpro enzymes behave similarly or not. Surprisingly, none of them showed any inhibitory activity against MERS-PLpro. This result led us to further analyze what determined the interaction between SARS-PLpro and its inhibitors.
Figure 2.

SARS-PLpro lead inhibitors and structures. (A) Structures of four SARS-PLpro lead inhibitors (I-1–I-4).25,26 (B) X-ray crystal structure of inhibitor I-2 bound to SARS-PLpro (PDB: 3E9S). The amide group of inhibitor I-2 forms two hydrogen bonds with D165 and Q270 in the BL2 loop. (C) X-ray crystal structure of inhibitor I-3 bound to SARS-PLpro (PDB: 3MJ5). The amide group of inhibitor I-3 forms a hydrogen bond with Q270 in the BL2 loop. The aromatic ring of Y269 forms a hydrophobic interaction with the naphthyl rings of both I-2 and I-3. The three catalytic site residues are shown in green. (D) Overlay of the SARS-PLpro blocking loop 2 (BL2) and the corresponding loop of MERS-PLpro.
There are seven SARS-PLpro crystal structures available to date, four of which are complexes with an inhibitor and two are substrate-bound.21,25−28 In the case of MERS-PLpro, one apo and two substrate-bound complexes have been recently published.29,30 We also determined the X-ray crystal structure of unbound MERS-PLpro (PDB code: 4RNA) at 1.8 Å (Supporting Information Table S1) in addition to our previously released apo structure at lower resolution (PDB code: 4PT5). Both SARS-PLpro and MERS-PLpro contain two blocking loops named BL1 and BL2 that could be structurally important. The two corresponding loops of human ubiquitin-specific protease 14 (USP14) have been proven to be crucial in blocking accessibility to the active site.31 Indeed, two SARS-PLpro complex crystal structures, with lead inhibitors from each scaffold (I-2 or I-3) revealed that inhibitors bind not to the catalytic site of the PLpro enzyme but to the BL2 loop, blocking the entrance of the active site. This appears to prevent substrate access to the catalytic site, inhibiting PLpro enzyme activity. The amide group of inhibitor I-2 forms two hydrogen bonds with D165 and Q270 (Figure 2B). The amide group of inhibitor I-3 also forms a hydrogen bond with Q270 in the BL2 loop (Figure 2C). The aromatic ring of Y269 forms a hydrophobic interaction with the naphthyl ring of both I-2 and I-3. Therefore, residues Q270 and Y269 form common key interactions in both scaffold 1 and 2 lead inhibitors of SARS-CoV PLpro. However, neither Q270 nor Y269 residues exist in MERS-PLpro (Figure 2D). In MERS-PLpro, A275 exists in place of Q270 of SARS-PLpro, eliminating potential hydrogen bonding with inhibitors. Additionally, the second key interaction is also impossible because T274 of the MERS-PLpro does not have the aromatic ring of Y269. Apparently due to the lack of these two key residues, none of the SARS-PLpro lead inhibitors had any inhibitory efficacy against MERS-PLpro.
In order to further analyze the interaction between SARS-PLpro and its inhibitors, we aligned all four available SARS-PLpro–inhibitor complexes, one SARS-PLpro in complex with a ubiquitin aldehyde substrate, one MERS-PLpro complex with a ubiquitin substrate, and the apo structures of both SARS-PLpro and MERS-PLpro (Figure 3).21,25−28,30 In addition, all residues involved in inhibitor binding by SARS-PLpro and their corresponding residues in MERS-PLpro are compared to three bat coronaviruses because of the similarities of MERS-CoV with bat coronaviruses BtCoV-HKU4 and BtCoV-HKU5. The residues compared include the active site, catalytic triad, and BL2 loop residues (Figure 3E). All eight structures aligned well with each other, except for several distinct locations, including the zinc-binding motif and BL2 regions. The zinc atoms were shifted similar distances and locations in all five SARS-PLpro complex structures as compared to the apo structure, as noted with a red arrow pointed to the right in the bottom insert in Figure 3A. The equivalent zinc atom in MERS-PLpro was located in a different position and was shifted in a different direction from those in SARS-PLpro (dark brown arrow in Figure 3A). Residues involved in the SARS-PLpro inhibitor binding are shown in Figure 3B, along with inhibitor I-3, in order to illustrate the orientation of the inhibitor, inhibitor-interacting SARS-PLpro residues, and the flexible BL2 loop. Figure 3C and D show the binding locations of inhibitors and ubiquitin substrates and the flexible BL2 loop in exactly the same orientation. The SARS-PLpro flexible BL2 loop blocks the entrance of the tunnel to the active site when it is unbound and becomes well-ordered upon binding of either an inhibitor or a substrate (Figure 3C and D) through conformational changes.21,26−28,32,33 For MERS-PLpro, the flexible BL2 loop is positioned much further from its active site than that of SARS-PLpro when it is unbound. Upon substrate binding to MERS-PLpro, its BL2 loop moves to an orientation similar to that of the substrate bound SARS-PLpro loop, as noted with red and dark brown arrows in Figure 3D.30 Both BL1 and BL2 loops of USP14 appear to play a regulatory role in its deubiquitinating activity,31 while mainly BL2 seems to serve this role in SARS-PLpro. The question of whether the BL2 loop of MERS-PLpro plays a regulatory role in its deubiquitinating activity remains to be answered.
Figure 3.

Structure comparison of SARS-PLpro complexes and MERS-PLpro. (A) Overlay of five SARS-PLpro complex structures with an inhibitor or a substrate, apo SARS-PLpro, apo MERS-PLpro, and MERS-CoV-PLpro complex with a ubiquitin. The PDB codes of aligned structures are Apo MERS-CoV-PLpro (PDB: 4RNA), MERS-CoV-PLpro complex with a ubiquitin (PDB: 4RF1), Apo SARS-CoV-PLpro (PDB: 2FE8), SARS-CoV-PLpro inhibitor complex with inhibitor I-2 (PDB: 3E9S), SARS-CoV-PLpro complex with inhibitor I-3 (PDB: 3MJ5), SARS-CoV-PLpro complex with inhibitor 3k (PDB: 4OVZ), SARS-CoV-PLpro complex with inhibitor 3j (PDB: 4OW0), and SARS-CoV-PLpro complex with ubiquitin aldehyde substrate (PDB: 4MM3). (B) Expanded overlaid structures of BL2 and surrounding residues involved with inhibitor binding. (C) Different orientation of Figure 4B, showing catalytic residues and relative BL2 orientations. (D) Expanded overlaid structures of BL2 loops and a ubiquitin aldehyde (blue) and ubiquitin (orange) substrates for SARS-PLpro and MERS-PLpro, respectively. Ubiquitin is hidden, and only part of the each substrate is shown in this figure due to space constraints. (E) The active site, catalytic triad (CT), and two blocking loop (BL1 and BL2) residues of MERS-PLpro and their corresponding aligned residues in the active sites of SARS-PLpro and three bat coronaviral PLpro enzymes.
Prescreen Assay Optimization for High-Throughput Screening
Prescreen assay optimization prior to a large scale HTS is crucial to achieving a high quality outcome. Essential factors for consideration include the enzyme and substrate concentrations, additives (detergent and reducing agent), the DMSO tolerance, and enzyme stability. The most important factor is the substrate concentration, and it is recommended to use a substrate concentration near or slightly lower than the Michaelis constant (KM) value in order to select for both competitive and noncompetitive inhibitors.34 The KM was determined with the ubiquitin-derived peptide substrate, RLRGG-AMC, for both SARS-PLpro and MERS-PLpro enzymes side by side for comparison. The substrate KM value of MERS-PLpro was ∼2-fold larger at 142 μM and 75.9 μM for MERS-PLpro and SARS-CoV, respectively (Supporting Information Table S2). Therefore, a higher substrate concentration was used for MERS-PLpro than for the SARS-PLpro HTS screen. The kcat (turnover number) value of SARS-CoV was ∼25-fold larger than that of MERS-PLpro, which made the catalytic efficiency (kcat/KM) of SARS-PLpro for this substrate ∼45-fold higher than that of MERS-PLpro. Accordingly, a 20-fold higher MERS-PLpro enzyme concentration was necessary to yield an enzyme activity signal similar to that of SARS-PLpro. DMSO tolerance, reducing agent effect, two additives (BSA and Triton X-100) effect, and enzyme stability at RT were determined in addition to optimal substrate and enzyme concentrations for HTS.
High-Throughput Screening (HTS) and Hit Validation
To search for MERS-PLpro inhibitors, HTS was performed with a 25 000-compound Life Chemicals antimicrobial/antiviral focused library against both SARS-PLpro and MERS-PLpro. The overall screening and hit validation process are described in Figure 4A. The primary HTS screen against SARS-PLpro was performed in duplicate, generating average Z′-factors of 0.64 ± 0.08. The MERS-PLpro primary screen was done in a single pass with Z′-factors of 0.65 ± 0.11. HTS hits with over 50% inhibition at 50 μM compound concentration were cherry picked and reanalyzed by a continuous kinetic assay to filter out false positives. The enzyme omission assay with exactly the same assay conditions, but without the PLpro enzyme, was performed to remove fluorescence signal interfering compounds. Confirmed hits were repurchased, and their inhibitory activities (IC50 values) were determined from full inhibition curves.
Figure 4.

HTS results from Life Chemicals antimicrobial/antiviral focused library and hit validation. (A) Schematic of HTS with 25 000 Life Chemicals compounds and hit validation process. (B) Bar graphs of IC50 values and the dissociation equilibrium constants (KD) of six hit compounds determined by fluorescence-based enzymatic assay and Surface Plasmon Resonance (SPR), respectively. All data were normalized for immobilization levels of target proteins and reference. Bars that reach the top of the graph represent either IC50 or KD values of over 200 μM (no inhibition or no binding).
Of ∼25 000 compounds, four (compounds 1–4 in Figure 4B) and three (compounds 4–6 in Figure 4B) exhibited inhibitory activity with IC50 values below 50 μM for SARS-PLpro and MERS-PLpro, respectively. Surface plasmon resonance (SPR) was used as a secondary orthogonal binding assay to eliminate false positives from the primary hits since our primary screen was done by a fluorescence-based enzymatic assay. Binding affinity of each hit compound can be determined by measuring the dissociation equilibrium constant (KD) using SPR, and IC50 and KD values for the six hit compounds are compared in Figure 4B. Compound 2 from the four SARS-PLpro hits did not bind to the enzyme, indicating that it is a false positive, while the remaining three were confirmed to be binders with KD values below 50 μM. The binding affinities of these validated hits varied from 26.3 μM to 39.9 μM, and their corresponding IC50 values varied from 10.9–31.4 μM. The IC50 and KD values of 3 are 31.4 μM and 39.9 μM, respectively, which are similar. The KD value of 4 (26.3 μM) is approximately 2.4-fold greater than its IC50 value (10.9 μM). Of the three MERS-PLpro hits, only 4 showed specific binding to the enzyme, with 18.4 μM binding affinity, while 5 and 6 were false positives. Compound 5 did not bind to MERS-PLpro at all, whereas compound 6 bound nonspecifically. These three distinct binding patterns determined by SPR, specific, no binding, and nonspecific interactions, are shown in Supporting Information Figure S3A–C. Interestingly, compound 4 showed similar strength of inhibitory activities and binding affinity against both SARS-PLpro and MERS-PLpro enzymes.
Mechanism of Inhibition
There have been two types of noncovalent small molecule inhibitor scaffolds against SARS-PLpro previously discovered by our research group. Mechanism of inhibition studies revealed that these compounds are mixed-type inhibitors with α values greater than 1, indicating that they bind to an allosteric site other than its catalytic site but behave as if they are competitive inhibitors.35 As noted above, these lead inhibitors bind to the flexible BL2 region and induce conformational changes to block substrate access to the catalytic site of the enzyme.26,28 There have been no MERS-PLpro inhibitors published to date; we have identified one compound (4) that inhibits both SARS-PLpro and MERS-PLpro. Our mode of inhibition studies with compound 4 were done with a series of increasing substrate concentrations and enzyme-compound complexes (Figure 5A and B). The kinetic data were fit to four different enzyme inhibition models (competitive, noncompetitive, uncompetitive, and mixed-type) using the Sigmaplot Enzyme Kinetics Module. The best fit equation was selected based on Akaike Information Criterion-corrected (AICc) values.36 The equation with the lowest AICc value corresponds to the best fit, and a minimum of a two AICc unit difference from the next lowest is required to be considered statistically significant. Interestingly, compound 4 exhibited mixed-type inhibition for SARS-PLpro but competitive inhibition for MERS-PLpro for the same substrate. The second best fit equation of compound 4 with SARS-PLpro was noncompetitive inhibition with 7.1 AICc values lower than the best fit (mixed-type) inhibition, which was also 38.6 AICc values lower than competitive inhibition, clearly indicating that compound 4 is an allosteric inhibitor of SARS-PLpro. For MERS-PLpro, the AICc value of competitive inhibition was 19.4 units lower than the next lowest, noncompetitive inhibition. Because of the large AICc value differences from the next best fit equations for both SARS and MERS-PLpro, it is clear that the same compound acts as an allosteric inhibitor for SARS-PLpro and acts as a competitive inhibitor for MERS-PLpro. Therefore, this compound inhibits the two PLpro enzymes via two different inhibitory mechanisms even though the inhibitory activities are similar, with IC50 values of 10.9 μM (SARS-PLpro) and 6.2 μM (MERS-PLpro). The Ki values of compound 4 against SARS-PLpro and MERS-PLpro are 11.5 μM and 7.6 μM, respectively (Figure 5C).
Figure 5.

Mechanism of inhibition. Dixon plots of compound 4 against SARS-PLpro (A) and MERS-PLpro (B). (C) Summary table of kinetic mode of inhibition of compound 4. Mechanism of enzyme inhibition of compound 4 was determined to be a mixed inhibition for SARS-PLpro and a competitive inhibition for MERS-PLpro. Determined Ki values of compound 4 were 11.5 μM and 7.5 μM for SARS-PLpro and MERS-PLpro, respectively. (D) IC50 value comparison of four SARS-PLpro lead inhibitors in combination with the newly identified compound 4 to determine if they inhibit synergistically. (E) Bar graphs of the dissociation equilibrium constants (KD) of compound 4 in the absence (solid bars) and in the presence (striped bars) of substrate determined by Surface Plasmon Resonance (SPR).
Compound 4 appears to interact with MERS-PLpro by binding to the catalytic site since it is a competitive inhibitor with respect to the substrate. However, we were uncertain where 4 might bind to SARS-PLpro since it is an allosteric inhibitor. Our first hypothesis was that it may bind to the BL2 loop where two other SARS-PLpro lead inhibitors (I-2 and I-3) bound. We thus evaluated inhibition by 4 in the presence of each of the four SARS-PLpro lead inhibitors (I-1–I-4) in order to see if it has an additive effect (Figure 5D). If 4 binds to the same location as I-2 and I-3, their inhibitory activities (IC50 values) would not be improved by the addition of compound 4. But their IC50 values should be enhanced if compound 4 binds elsewhere. Inhibitory activities of the four lead inhibitors alone varied from 0.23 μM to 2.26 μM, and their IC50 values were enhanced up to almost 7-fold (0.034–0.67 μM) in the presence of 4 at a concentration of 10 μM, slightly lower than its IC50 value. This led us to conclude that 4 binds to an allosteric site of the SARS-PLpro other than the BL2 loop. In addition to enzymatic mode of inhibition analysis, competition SPR studies of compound 4 with SARS-PLpro and MERS-PLpro were each performed in the presence and in the absence of the substrate. Binding affinity of compound 4 to SARS-PLpro was the same regardless of substrate presence, whereas that of compound 4 to MERS-PLpro was 4.5-fold weaker in the presence of the substrate than 4 alone. These results indicate that the substrate is competing with 4 for the same binding site in MERS-PLpro (Figure 5E). Therefore, both the enzymatic mechanism of inhibition and SPR studies support compound 4 being an allosteric inhibitor for SARS-PLpro, while 4 is a competitive inhibitor for MERS-PLpro. In order to further clarify this, cocrystallizations of both SARS-PLpro and MERS-PLpro with compound 4 are currently in process.
Inhibitor Selectivity
A concern of potential nonspecificity was raised due to the fact that our newly identified dual inhibitor is a small fragment-like compound and also exhibited inhibitory activity against SARS-CoV 3CLpro with an IC50 value of 13.9 μM.37 X-ray crystallography and mode of inhibition studies showed that 4 binds to the dimer interface of the SARS-CoV 3CLpro, inhibiting its enzyme activity by breaking the dimer since SARS-CoV 3CLpro is a functional dimer (unpublished data). Therefore, compound 4 acts as an allosteric inhibitor against both SARS-CoV proteases (3CLpro and PLpro), whereas it acts as a competitive inhibitor against MERS-PLpro. We further investigated the specificity of 4 and a lead inhibitor I-3 (control) against two human cysteine proteases also called human ubiquitin C-terminal hydrolases (hUCH-L1 and hUCH-L3) and two unrelated enzymes (Hepatitis C Virus NS3 serine protease and Bacillus anthracis dihydroorotase). The hUCH-L1 is one of the human homologues most closely related to PLpro, which makes it an excellent control to test the selectivity of a newly identified inhibitor. Structural alignment of these two human homologues revealed that their catalytic triads are very similar (Figure 6A). Compound 4 was selective for SARS-PLpro and MERS-PLpro proteases over the two human cysteine proteases and both unrelated enzymes (Figure 6B).
Figure 6.
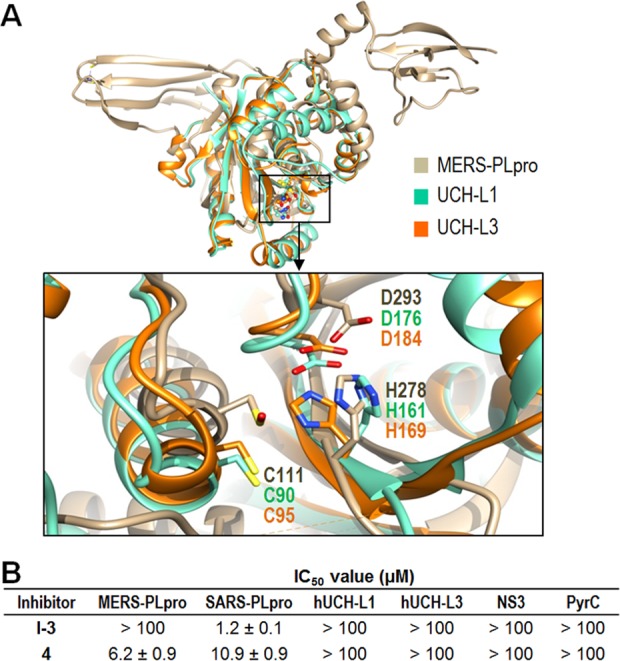
Selectivity of compound 4. (A) Structural alignment of MERS-PLpro with two human deubiquitinating enzymes. The aligned catalytic triads of two human ubiquitin C-terminal hydrolases, hUCH-L1 (green, PDB: 2ETL)38 and hUCH-L3 (orange, PDB: 1UCH),39 are shown with that of MERS-PLpro (tan, PDB: 4RNA) in the expanded box. (B) Selectivity of the confirmed hit compound 4. In addition to two human cysteine proteases (hUCH-L1 and hUCH-L3), two unrelated enzymes, Hepatitis C Virus NS3 serine protease (NS3) and Bacillus anthracis dihydroorotase (PyrC), were also tested along with both PLpro enzymes.
Active-Site Comparison and Oxyanion Hole Stabilization
The cysteine/serine protease mechanism involves tetrahedral intermediate formation, following nucleophilic attack by the cysteine/serine side chain. Generally, there is an oxyanion close to the catalytic site, which interacts with a negatively charged tetrahedral intermediate to stabilize it. Ratia et al. demonstrated that the SARS-PLpro W107 located below the catalytic cysteine plays this vital role in forming a hydrogen bond (H-bond) with an intermediate as an H-bond donor in the active site by showing that the SARS-PLpro W107A mutant completely lost catalytic activity.21 However, in the MERS-PLpro active site, the equivalent position is occupied by L106, which is not capable of being an H-bond donor (Figure 7A). Lei et al. recently demonstrated that the L106W mutation resulted in catalytic activity enhancement of MERS-PLpro,29 indicating that the MERS-PLpro oxyanion hole may not be complete in comparison to that of SARS-PLpro. Interestingly, the leucine residue at this position is highly conserved in three bat coronaviruses (BtCoV-HKU4, BtCoV-HKU5, and BtCoV-133) that belong to the same lineage group C as MERS-CoV (Figure 7B). On the other hand, two human coronavirus (NL63 and 229E) PLpro enzymes have a residue (Q or T) that can be an H-bond donor similar to SARS-PLpro. Therefore, another residue must play this intermediate-stabilizing function in MERS-PLpro. Asparagine (N110) in SARS-PLpro is highly conserved among various coronavirus PLpro enzymes, and Ratia et al. suggested that this residue could be another residue contributing to the oxyanion hole stabilization in addition to W107.21 From the structural alignment of the active site, we noted that the N109 of MERS-PLpro overlaps with N110 of SARS-PLpro. This suggested that N109, located above the catalytic cysteine, might be the residue that plays this critical role in MERS-PLpro. We hypothesized two potential mechanisms: First, the side chain amine group of N109 could form an H-bond with the intermediate’s oxyanion as an H-bond donor (Figure 7C). Alternatively, the carbonyl group of N109 could bind to a water molecule, followed by the water forming another H-bond with the negatively charged intermediate (Figure 7D). The positions of N109 in these two scenarios could differ. We generated two MERS-PLpro mutants, N109A and N109D, to investigate these two hypotheses. Enzyme activity of the N109A mutant was completely abolished, while the N109D mutant exhibited only ∼13.8% of the wild-type MERS-PLpro activity (Figure 7E). This indicates that the N109 residue is indeed crucial for stabilizing the intermediate for the enzyme to perform its catalytic function. If N109 stabilized the intermediate via the second hypothesis, the side chain of N109D could still form an H-bond with a water molecule through the carbonyl group of aspartic acid, rescuing the MERS-PLpro enzyme activity. However, the N109D mutant also showed very low enzyme activity as compared to the wild-type, suggesting that the second hypothesis is not likely to be the main stabilization mechanism. This result suggests that N109 is a critical residue for intermediate stabilization, most likely through an H-bond formation with the side chain amine group of N109.
Figure 7.

Active site analysis of the MERS-PLpro. (A) Active site alignment of MERS-PLpro (tan) and SARS-PLpro (cyan). The three catalytic triad residues (C111, H278, and D293) of MERS-PLpro are aligned with the SARS-PLpro catalytic triad (C112, H273 and D287). (B) Sequence alignment of important residues near the catalytic triad between various CoV. Residue numbers are shown for MERS-PLpro. (C) Potential mechanism 1 for oxyanion hole stabilization via N109. Active site and substrate residues are shown in green and pink, respectively. (D) Potential mechanism 2 for oxyanion hole stabilization via N109. (E) Enzyme activity comparison between wild-type and two mutant MERS-PLpro enzymes.
In addition to containing a crucial residue that stabilizes the oxyanion, the small loop (residues 101–108) next to the active site in SARS-PLpro is important for its catalytic activity via controlling active site access. The hydrogen bond between D109 from this loop and W94 restrains the loop conformation, preventing it from moving to block active site access.21 These two residues (D108 and W93 in MERS-PLpro) are conserved in MERS-PLpro, playing the same role as that of SARS-PLpro (Figure 7B).
Conclusion
SARS-CoV and MERS-CoV cause contagious and highly virulent infectious diseases in humans, threatening public health.10,27 Both coronaviruses apparently originated from animal reservoirs such as bats or camels but surprisingly have rapidly evolved to human-to-human transmission, although limited cases have been reported for MERS-CoV.11 SARS-CoV has been contained by public health measures since 2003, but MERS-CoV has spread into 12 different countries so far, and the numbers of infections continue to rise. There is currently no specific treatment or vaccine available.
In this study, we determined that none of the tested SARS-PLpro lead inhibitors were effective against MERS-PLpro. Thorough structural comparison between these two PLpro enzymes using all available structures revealed crucial structural differences, providing insights for developing inhibitors against PLpro. The overall MERS-PLpro structure is similar to that of SARS-PLpro including the N-terminal Ubl-domain. However, the flexible BL2 loop of MERS-PLpro differs significantly from that of SARS-PLpro, which raised the possibility of differing roles in inhibitor binding. This may explain the observation that all of the tested SARS-PLpro lead inhibitors were ineffective against MERS-PLpro. It was surprising to discover that SARS-PLpro has a deubiquitinating function,21 and now it has been shown that MERS-PLpro also exhibits the same function.22,23 We determined the catalytic activity of MERS-PLpro in direct comparison with that of SARS-PLpro. The catalytic efficiency (kcat/KM) of SARS-PLpro was ∼45-fold higher (8.2 × 105 M–1 s–1) than that of MERS-PLpro (1.9 × 104 M–1 s–1). Although the deubiquitinating activity of MERS-PLpro is lower than SARS-PLpro, it is still much more active than two closely related human homologues of PLpro, herpes-associated ubiquitin-specific protease (HAUSP) and ubiquitin-specific protease 14 (USP14), which exhibit catalytic efficiencies of 2.2 × 103 M–1 s–1 and 107 M–1 s–1, respectively.40
We performed HTS of 25 000 compounds against both PLpro enzymes, and identified a dual noncovalent inhibitor that was active against both PLpro enzymes. Interestingly, this inhibitor was determined to be a competitive inhibitor against MERS-PLpro, whereas it was an allosteric inhibitor against SARS-PLpro. These results suggest that inhibitor recognition specificity of MERS-PLpro may differ from that of SARS-PLpro even though the overall structures of the whole protein and the catalytic sites are very similar. The most probable contributing factor for inhibitor selectivity of these two PLpro enzymes could be attributed to the structural differences of the BL2 loop.
Materials and Methods
Details about cloning, expression, and purification; crystallization, confirmation assay, and IC50 value determination by dose response curve; and reversibility of inhibition are provided in the SI Materials and Methods.
Primary High-Throughput Screening
The 25 000-compound Life Chemicals library was screened against the two PLpro cysteine proteases from SARS-CoV and MERS-CoV. All assays against SARS-PLpro were done in duplicate, and those against MERS-PLpro were done in a single pass in black 384-well plates (Matrix Technologies). The SARS-PLpro enzyme (20 nM final concentration) was prepared in an assay buffer (50 mM HEPES, pH 7.5, 0.01% Triton X-100 (v/v), 0.1 mg mL–1 BSA, and 2 mM GSH). The MERS-PLpro enzyme (400 nM final concentration) was prepared in the same assay buffer with 5 mM DTT in place of 2 mM GSH. A total of 30 μL of enzyme solution was dispensed into wells, and then 200 nL of 10 mM compounds (50 μM final concentrations) was added and incubated for 5 min. Enzyme reactions were initiated with 10 μL of substrate Z-Arg-Leu-Arg-Gly-Gly-AMC (Bachem Bioscience; 50 μM and 75 μM for SARS- and MERS-PLpro, respectively) dissolved in the assay buffer and incubated for 6 min, followed by adding 10 μL of 10% SDS (w/v) as a stop solution. Fluorescence intensity was monitored at 360 nm (excitation) and 450 nm (emission).
Determination of Dissociation Equilibrium Constant (KD) by SPR
Compound solutions with a series of increasing concentrations (0–200 μM at 1.5-fold dilution) were applied to all four channels at a 30 μL/min flow rate. Sensorgrams were analyzed using the Biacore T200 evaluation software 2.0, and response units were measured during the equilibration phase at each concentration. Each PLpro enzyme was immobilized on a CM5 sensor chip using standard amine-coupling with running buffer HBS-P (10 mM HEPES, 150 mM NaCl, 0.05% surfactant P-20, pH 7.4) using a Biacore T200 instrument. The MERS-PLpro enzyme was immobilized to flow channels 2 and 3, and immobilization levels of flow channels 2 and 3 were ∼16 900 RU and ∼16 700 RU, respectively. SARS-PLpro was immobilized to flow channel 4 at the immobilization level of ∼14 600 RU to be compared with MERS-PLpro. Data were referenced with blank (enthanolamine) RU values. SigmaPlot 12.0 was used to fit the data to a single rectangular hyperbolic curve to determine KD values. The hyperbolic, y = ymax·x/(KD + x), was used to plot response units and corresponding concentration, where y is the response, ymax is the maximum response, and x is the compound concentration.
Mechanism of Inhibition
Enzyme activities of both MERS-PLpro and SARS-PLpro were monitored in the same way as the primary screen with varying concentrations of inhibitors and substrates (0–300 μM). The concentration of compounds was varied from 0 to at least 10× the IC50 value of each compound. The data were fit to four equations (shown in SI Materials and Methods) using SigmaPlot Enzyme Kinetics Module 1.3 in order to determine the best fit inhibition mechanism and kinetic parameters for each compound.
Inhibitor Selectivity Assay
To test for selectivity, two human ubiquitin C-terminal hydrolases (UCH-L1 and UCH-L3) and two unrelated enzymes (Hepatitis C Virus NS3 serine protease and B. anthracis dihydroorotase) were tested with the top hit compound from HTS and a lead SARS-PLpro inhibitor (I-1) using a fluorometric assay. The fluorogenic substrate used in this study was ubiquitin-AMC (Boston Biochem). All assays were performed in 384-well black plates (Corning) in a total volume of 24 μL of the assay buffer containing 50 mM HEPES (pH 7.5), 5 mM DTT, 0.1 mg mL–1 BSA, and 0.01% Triton X-100 (v/v) in triplicate. A series of compound concentrations (0 to 200 μM final concentration at 2-fold serial dilution) in 100% DMSO was prepared in a 384-well plate. Then 3× compound solutions were prepared in the assay buffer prior to assays. A total of 8 μL of each enzyme solution was distributed into wells, and 8 μL of varying concentrations of compounds was added and incubated for 10 min. The enzyme reaction was initiated by adding 8 μL of the substrate (50 μM final concentration), and fluorescence intensity was continuously monitored at excitation/emission wavelengths of 350 nm/460 nm for 10 min.
X-ray Data Collection, Processing, and Structure Solvation
Data were collected at the LS-CAT end station 21-ID-F at the Advanced Photon Source, Argonne National Laboratory, using a wavelength of λ = 0.97872 Å, and the crystal at 100 K under a dry liquid nitrogen stream. Data were recorded by a MAR CCD 225 mm detector with an oscillation angle of 1.0° using a total of 190 frames. Data were processed and scaled by XDS.41 The crystal space group belonged to C2, containing one monomer in the asymmetric unit. The Matthews coefficient (VM) was calculated as 2.5, and solvent content was estimated to be 50%. Molecular replacement was carried out using Phaser42 from the CCP4 package. The SARS-PLpro crystal structure (2FE8)21 was used as a search model. The zinc binding domain in the initial model was truncated and manually rebuilt by Coot.43 Structural refinement was conducted using Refmac5.5.44
Acknowledgments
This work was supported in part by National Institutes of Health Grants R56 AI089535. We thank K. Ratia for performing HTS and primary screening data analysis and J. Ren for computational assistance. A.J.R. was supported during a portion of this work by NIDCR T32-DE018381, UIC College of Dentistry, MOST program. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation, grant number OCI-1053575. Use of the APS, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. We thank ChemAxon for a free academic license of their cheminformatics suite including JChem and JChem for excel for HTS data analysis.
Glossary
Abbreviations
- MERS-CoV
Middle East Respiratory Syndrome coronavirus
- HCoV-EMC
human coronavirus-Erasmus Medical Center
- SARS-CoV
Severe Acute Respiratory Syndrome
- 3CLpro
3C-like protease
- PLpro
papain-like protease
- nsp
nonstructural protein
- CFR
case-fatality rate
- BL2
blocking loop 2
- HTS
high-throughput screening
- SPR
surface plasmon resonance
- hUCH
human ubiquitin C-terminal hydrolases
Supporting Information Available
Additional Materials and Methods, Tables S1 and S2, Figures S1–S3, and NMR Spectra and MS analysis. This material is available free of charge via the Internet.
Accession Codes
4PT5 (unbound MERS-PLpro at 2.5 Å resolution), 4RNA (unbound MERS-PLpro at 1.8 Å resolution)
Author Contributions
§ Equal contributors.
Author Contributions
H. Lee performed all experiments with the assistance of J.L.G., K.P., M.Z.S., and I.O. H. Lei and B.D.S. solved the MERS-PLpro structure. S.C. performed computational studies. A.K.G. synthesized current SARS-PLpro lead compounds. H. Lee, H. Lei, and M.E.J. designed the experiments, and H. Lee, H. Lei, S.C., A.J.R., and M.E.J. wrote the manuscript.
The authors declare no competing financial interest.
This article is made available for a limited time sponsored by ACS under the ACS Free to Read License, which permits copying and redistribution of the article for non-commercial scholarly purposes.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Anderson L. J.; Baric R. S. (2012) Emerging human coronaviruses--disease potential and preparedness. N. Engl. J. Med. 367, 1850–1852. [DOI] [PubMed] [Google Scholar]
- Chan J. F.; Li K. S.; To K. K.; Cheng V. C.; Chen H.; Yuen K. Y. (2012) Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic?. J. Infect. 65, 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki A. M.; van Boheemen S.; Bestebroer T. M.; Osterhaus A. D.; Fouchier R. A. (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820. [DOI] [PubMed] [Google Scholar]
- de Groot R. J.; Baker S. C.; Baric R. S.; Brown C. S.; Drosten C.; Enjuanes L.; Fouchier R. A.; Galiano M.; Gorbalenya A. E.; Memish Z. A.; Perlman S.; Poon L. L.; Snijder E. J.; Stephens G. M.; Woo P. C.; Zaki A. M.; Zambon M.; Ziebuhr J. (2013) Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 87, 7790–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2014). Middle East respiratory syndrome coronavirus (MERS-CoV) – update. http://www.who.int/csr/don/2014_07_23_mers/en/.
- Ziebuhr J. (2004) Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 7, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K.; Li K. S.; Tsang A. K.; Lam C. S.; Ahmed S.; Chen H.; Chan K. H.; Woo P. C.; Yuen K. Y. (2013) Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 87, 8638–8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boheemen S.; de Graaf M.; Lauber C.; Bestebroer T. M.; Raj V. S.; Zaki A. M.; Osterhaus A. D.; Haagmans B. L.; Gorbalenya A. E.; Snijder E. J.; Fouchier R. A. (2012) Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 3, e00473-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan G. (2013) A novel coronavirus capable of lethal human infections: an emerging picture. Virol. J. 10, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rha B. (2013) Update: Severe Respiratory Illness Associated with a Novel Coronavirus - Worldwide, 2012–2013. MMWR Morbid Mortal W 62, 194–195. [PMC free article] [PubMed] [Google Scholar]
- Assiri A.; McGeer A.; Perl T. M.; Price C. S.; Al Rabeeah A. A.; Cummings D. A.; Alabdullatif Z. N.; Assad M.; Almulhim A.; Makhdoom H.; Madani H.; Alhakeem R.; Al-Tawfiq J. A.; Cotten M.; Watson S. J.; Kellam P.; Zumla A. I.; Memish Z. A. (2013) Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 369, 407–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Moore M. J.; Vasilieva N.; Sui J.; Wong S. K.; Berne M. A.; Somasundaran M.; Sullivan J. L.; Luzuriaga K.; Greenough T. C.; Choe H.; Farzan M. (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M. A.; Raj V. S.; Muth D.; Meyer B.; Kallies S.; Smits S. L.; Wollny R.; Bestebroer T. M.; Specht S.; Suliman T.; Zimmermann K.; Binger T.; Eckerle I.; Tschapka M.; Zaki A. M.; Osterhaus A. D.; Fouchier R. A.; Haagmans B. L.; Drosten C. (2012) Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. MBio 3, e00515-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guery B.; Poissy J.; el Mansouf L.; Sejourne C.; Ettahar N.; Lemaire X.; Vuotto F.; Goffard A.; Behillil S.; Enouf V.; Caro V.; Mailles A.; Che D.; Manuguerra J. C.; Mathieu D.; Fontanet A.; van der Werf S. (2013) Clinical features and viral diagnosis of two cases of infection with Middle East Respiratory Syndrome coronavirus: a report of nosocomial transmission. Lancet 381, 2265–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailles A.; Blanckaert K.; Chaud P.; van der Werf S.; Lina B.; Caro V.; Campese C.; Guery B.; Prouvost H.; Lemaire X.; Paty M. C.; Haeghebaert S.; Antoine D.; Ettahar N.; Noel H.; Behillil S.; Hendricx S.; Manuguerra J. C.; Enouf V.; La Ruche G.; Semaille C.; Coignard B.; Levy-Bruhl D.; Weber F.; Saura C.; Che D. (2013) First cases of Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infections in France, investigations and implications for the prevention of human-to-human transmission, France, May 2013. Eurosurveillance 18, 2–6. [PubMed] [Google Scholar]
- Barretto N.; Jukneliene D.; Ratia K.; Chen Z.; Mesecar A. D.; Baker S. C. (2005) The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79, 15189–15198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K.; Woo P. C.; Li K. S.; Huang Y.; Tsoi H. W.; Wong B. H.; Wong S. S.; Leung S. Y.; Chan K. H.; Yuen K. Y. (2005) Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A. 102, 14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Shi Z.; Yu M.; Ren W.; Smith C.; Epstein J. H.; Wang H.; Crameri G.; Hu Z.; Zhang H.; Zhang J.; McEachern J.; Field H.; Daszak P.; Eaton B. T.; Zhang S.; Wang L. F. (2005) Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679. [DOI] [PubMed] [Google Scholar]
- Thiel V.; Ivanov K. A.; Putics A.; Hertzig T.; Schelle B.; Bayer S.; Weissbrich B.; Snijder E. J.; Rabenau H.; Doerr H. W.; Gorbalenya A. E.; Ziebuhr J. (2003) Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 84, 2305–2315. [DOI] [PubMed] [Google Scholar]
- Harcourt B. H.; Jukneliene D.; Kanjanahaluethai A.; Bechill J.; Severson K. M.; Smith C. M.; Rota P. A.; Baker S. C. (2004) Identification of severe acute respiratory syndrome coronavirus replicase products and characterization of papain-like protease activity. J. Virol. 78, 13600–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K.; Saikatendu K. S.; Santarsiero B. D.; Barretto N.; Baker S. C.; Stevens R. C.; Mesecar A. D. (2006) Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A 103, 5717–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielech A. M.; Kilianski A.; Baez-Santos Y. M.; Mesecar A. D.; Baker S. C. (2014) MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 450–451, 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Chen X.; Bian G.; Tu J.; Xing Y.; Wang Y.; Chen Z. (2014) Proteolytic processing, deubiquitinase and interferon antagonist activities of Middle East respiratory syndrome coronavirus papain-like protease. J. Gen. Virol. 95, 614–626. [DOI] [PubMed] [Google Scholar]
- Yang H.; Bartlam M.; Rao Z. (2006) Drug design targeting the main protease, the Achilles’ heel of coronaviruses. Curr. Pharm. Des 12, 4573–4590. [DOI] [PubMed] [Google Scholar]
- Ghosh A. K.; Takayama J.; Aubin Y.; Ratia K.; Chaudhuri R.; Baez Y.; Sleeman K.; Coughlin M.; Nichols D.; Mulhearn D. C.; Prabhakar B. S.; Baker S. C.; Johnson M. E.; Mesecar A. D. (2009) Structure-Based Design, Synthesis, and Biological Evaluation of a Series of Novel and Reversible Inhibitors for the Severe Acute Respiratory Syndrome-Coronavirus Papain-Like Protease. J. Med. Chem. 52, 5228–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A. K.; Takayama J.; Rao K. V.; Ratia K.; Chaudhuri R.; Mulhearn D. C.; Lee H.; Nichols D. B.; Baliji S.; Baker S. C.; Johnson M. E.; Mesecar A. D. (2010) Severe acute respiratory syndrome coronavirus papain-like novel protease inhibitors: design, synthesis, protein-ligand X-ray structure and biological evaluation. J. Med. Chem. 53, 4968–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baez-Santos Y. M.; Barraza S. J.; Wilson M. W.; Agius M. P.; Mielech A. M.; Davis N. M.; Baker S. C.; Larsen S. D.; Mesecar A. D. (2014) X-ray Structural and Biological Evaluation of a Series of Potent and Highly Selective Inhibitors of Human Coronavirus Papain-like Proteases. J. Med. Chem. 57, 2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K.; Pegan S.; Takayama J.; Sleeman K.; Coughlin M.; Baliji S.; Chaudhuri R.; Fu W.; Prabhakar B. S.; Johnson M. E.; Baker S. C.; Ghosh A. K.; Mesecar A. D. (2008) A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. U. S. A. 105, 16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei J.; Mesters J. R.; Drosten C.; Anemuller S.; Ma Q.; Hilgenfeld R. (2014) Crystal structure of the papain-like protease of MERS coronavirus reveals unusual, potentially druggable active-site features. Antiviral Res. 109C, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Elkin B. A.; Knaap R. C.; Johnson G. G.; Dalebout T. J.; Ninaber D. K.; van Kasteren P. B.; Bredenbeek P. J.; Snijder E. J.; Kikkert M.; Mark B. L. (2014) Crystal Structure of the MERS Coronavirus Papain-Like Protease Bound to Ubiquitin Facilitates Targeted Disruption of Deubiquitinating Activity to Demonstrate its Role in Innate Immune Suppression. J. Biol. Chem. 289, 34667–34682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M.; Li P.; Song L.; Jeffrey P. D.; Chenova T. A.; Wilkinson K. D.; Cohen R. E.; Shi Y. (2005) Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 24, 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratia K.; Kilianski A.; Baez-Santos Y. M.; Baker S. C.; Mesecar A. (2014) Structural Basis for the Ubiquitin-Linkage Specificity and deISGylating activity of SARS-CoV papain-like protease. PLoS Pathog. 10, e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C. Y.; Lai H. Y.; Chen H. Y.; Cheng S. C.; Cheng K. W.; Chou Y. W. (2014) Structural basis for catalysis and ubiquitin recognition by the severe acute respiratory syndrome coronavirus papain-like protease. Acta Crystallogr., Sect. D: Biol. Crystallogr. 70, 572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2005) Evaluation of enzyme inhibitors in drug discovery. A guide for medicinal chemists and pharmacologists. Methods Biochem. Anal. 46, 1–265. [PubMed] [Google Scholar]
- Lee H.; Cao S.; Hevener K. E.; Truong L.; Gatuz J. L.; Patel K.; Ghosh A. K.; Johnson M. E. (2013) Synergistic Inhibitor Binding to the Papain-Like Protease of Human SARS Coronavirus: Mechanistic and Inhibitor Design Implications. ChemMedChem. 8, 1361–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnham K. P., Anderson D. R. (1998) Model Selection and Inference, Springer-Verlag. [Google Scholar]
- Lee H.; Mittal A.; Patel K.; Gatuz J. L.; Truong L.; Torres J.; Mulhearn D. C.; Johnson M. E. (2014) Identification of novel drug scaffolds for inhibition of SARS-CoV 3-Chymotrypsin-like protease using virtual and high-throughput screenings. Bioorg. Med. Chem. 22, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C.; Hoang Q. Q.; Kreinbring C. A.; Luchansky S. J.; Meray R. K.; Ray S. S.; Lansbury P. T.; Ringe D.; Petsko G. A. (2006) Structural basis for conformational plasticity of the Parkinson’s disease-associated ubiquitin hydrolase UCH-L1. Proc. Natl. Acad. Sci. U. S. A. 103, 4675–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S. C.; Larsen C. N.; Cook W. J.; Wilkinson K. D.; Hill C. P. (1997) Crystal structure of a deubiquitinating enzyme (human UCH-L3) at 1.8 A resolution. EMBO J. 16, 3787–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernova T. A.; Allen K. D.; Wesoloski L. M.; Shanks J. R.; Chernoff Y. O.; Wilkinson K. D. (2003) Pleiotropic effects of Ubp6 loss on drug sensitivities and yeast prion are due to depletion of the free ubiquitin pool. J. Biol. Chem. 278, 52102–52115. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A. A.; Steiner R. A.; Lebedev A. A.; Potterton L.; McNicholas S.; Long F.; Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2184–2195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.