

Abstract
Background
While aspirin is a well‐established and generally effective anti‐platelet agent, considerable inter‐individual variation in drug response exists, for which mechanisms are not completely understood. Metabolomics allows for extensive measurement of small molecules in biological samples, enabling detailed mapping of pathways involved in drug response.
Methods and Results
We used a mass‐spectrometry‐based metabolomics platform to investigate the changes in the serum oxylipid metabolome induced by an aspirin intervention (14 days, 81 mg/day) in healthy subjects (n=156). We observed a global decrease in serum oxylipids in response to aspirin (25 metabolites decreased out of 30 measured) regardless of sex. This decrease was concomitant with a significant decrease in serum linoleic acid levels (−19%, P=1.3×10−5), one of the main precursors for oxylipid synthesis. Interestingly, several linoleic acid‐derived oxylipids were not significantly associated with arachidonic‐induced ex vivo platelet aggregation, a widely accepted marker of aspirin response, but were significantly correlated with platelet reactivity in response to collagen.
Conclusions
Together, these results suggest that linoleic acid‐derived oxylipids may contribute to the non‐COX1 mediated variability in response to aspirin. Pharmacometabolomics allowed for more comprehensive interrogation of mechanisms of action of low dose aspirin and of variation in aspirin response.
Keywords: aspirin, drugs, fatty acids, lipids, pharmacology, platelets
Introduction
Acetyl salicylic acid, or aspirin, was first marketed in 1899 for reducing fever, pain, and inflammation. It was not until the 1960s that aspirin's antiplatelet effects were realized, and aspirin is now taken by over 50 million people in the United States alone to prevent primary and secondary cardiovascular disease. The antiplatelet effects of aspirin are mediated through its ability to irreversibly inhibit cyclooxygenase‐1 (COX‐1), which subsequently prevents the conversion of arachidonic acid (AA) to the potent platelet activator thromboxane A2. Specifically, at low dose, aspirin acetylates platelet COX‐1 in the presystemic circulation before its metabolism by the liver.1, 2 Therefore, platelet function inhibition occurring at low aspirin doses is thought to result in little to no systemic effects. However, the non‐COX‐mediated effects of aspirin are increasingly apparent. For example, low‐dose (81 mg/day) but not high‐dose (650 mg/day) aspirin initiated the biosynthesis of anti‐inflammatory mediators, the 15‐epi‐lipoxins.3 Similarly, our group demonstrated that several amino acids, purine metabolites, and nonesterified fatty acids, which are not directly related to COX, were significantly altered in healthy volunteers after 81 mg/day aspirin treatment for 14 days.4, 5 Understanding these non‐COX‐1‐mediated mechanisms is critical in order to better understand the wide interindividual variability observed in aspirin response. Though aspirin use significantly reduces the risk of cardiovascular death, still ≈25% of high‐risk patients on aspirin therapy show persistent platelet reactivity6, 7 (ie, laboratory aspirin resistance) and atherothrombotic events (ie, clinical aspirin resistance) remain relatively common8 in patients on aspirin therapy. The mechanisms underlying variability in aspirin response are poorly understood. Incomplete COX‐1 inhibition has been observed in several settings;9, 10 however, poor response despite complete COX‐1 inhibition has also been reported.9, 11
Omega‐6 fatty acids such as linoleic acid (LA) (C18:2), dihomo‐γ‐linolenic acid (C20:3), or AA (C20:4) and omega‐3 fatty acids such as α‐linolenic acid (C18:3), eicosapentaenoic acid (C20:5), or docosahexaenoic acid (C22:6) are essential polyunsaturated fatty acids. The oxidation products of polyunsaturated fatty acids, so‐called oxylipids, are highly potent mediators of both pro‐ and anti‐inflammatory processes.12 Oxylipids can be formed via COXs, lipoxygenases (LOXs), cytochrome P‐450 monooxygenases (CYPs), and by nonenzymatic oxidation. Oxylipids have myriad functions that are still being elucidated. Aberrant oxylipid signaling has been shown to lead to a number of pathologies important to cardiovascular disease including hyperlipidemia, hypertension, thrombosis, and hemostasis.13 For example, coronary artery disease patients have higher plasma levels of CYP‐mediated AA metabolites (namely, the oxylipids epoxyeicosatrienoic acid).14 While some studies have described the effects of aspirin on individual oxylipids,15, 16, 17 to our knowledge, the systematic effect of aspirin on oxylipid as a class has not been described. Given the complexity of the oxylipid metabolic network, a systematic approach will offer insight into how the entire oxylipid network responds to drug exposure.
Pharmacometabolomics is an emerging field that aims to use metabolomics tools to define the mechanisms of action for drugs and the biochemical basis for variation in response to treatment.18, 19 Metabolic profiles integrate genetic and environmental influences and provide unique information that can help explain the drug–response phenotype.20, 21 In the present investigation, we have used a robust, broad‐spectrum mass spectroscopy (MS)‐based metabolomics platform to measure the concentrations of multiple oxylipids in serum samples from 156 healthy volunteers of the Heredity and Phenotype Intervention (HAPI) Heart Study before and after 14 days of low‐dose (81 mg/day) aspirin treatment. We aim to characterize the effect of low‐dose aspirin therapy on the oxylipid metabolic pathways, to investigate sex differences in aspirin‐induced oxylipid changes, and to determine whether oxylipids are associated with aspirin‐induced inhibition of platelet aggregation.
Methods
HAPI Heart Study Design and Platelet Aggregation Measures
Samples for metabolomic profiling were obtained from subjects enrolled in the HAPI Heart Study, which has been described previously.22 Briefly, participants were relatively healthy adult members of the Old Order Amish population from Lancaster County, PA. Subjects (N=745) participated in a short‐term aspirin intervention where they were given 81 mg of aspirin for 14 consecutive days.23 Medication adherence was high and was monitored by pill counts. Blood samples were obtained after an overnight fast, and ex vivo platelet aggregometry was performed before aspirin therapy and again the morning after the last dose by the same technician. Aggregation was induced with collagen (2 μg/mL) or AA (0.5 mmol/L). More details on sample collection and ex vivo platelet testing are provided in Data S1. Blood samples for serum preparation were allowed to clot at room temperature for 15 minutes, centrifuged at 2025 g for 10 minutes then immediately frozen at −80°C.
All study procedures were in accordance with the Declaration of Helsinki. The study was approved by the institutional review board of the University of Maryland, Baltimore and was monitored by an external data safety and monitoring board. Participants provided informed consent, including permission to contact relatives, before participation.
Metabolomics
In this metabolomics study, samples originated from 156 HAPI participants. Figure 1 describes the procedure used to select this subset. The 745 original HAPI participants were grouped into sex‐specific quartiles of aspirin response as measured by post aspirin collagen‐stimulated platelet aggregation adjusted for participant age and preaspirin collagen‐stimulated platelet aggregation. We used collagen‐induced platelet aggregation to select our samples because we sought to probe non‐COX‐1‐mediated pathways implicated in aspirin's mechanism of action and in variation in aspirin response. Twenty‐five non–first‐degree relatives from each sex‐specific drug–response quartile were selected for metabolic profiling. Given sample availability, samples from 156 subjects were successfully profiled both pre‐ and post aspirin and were included for analysis.
Figure 1.
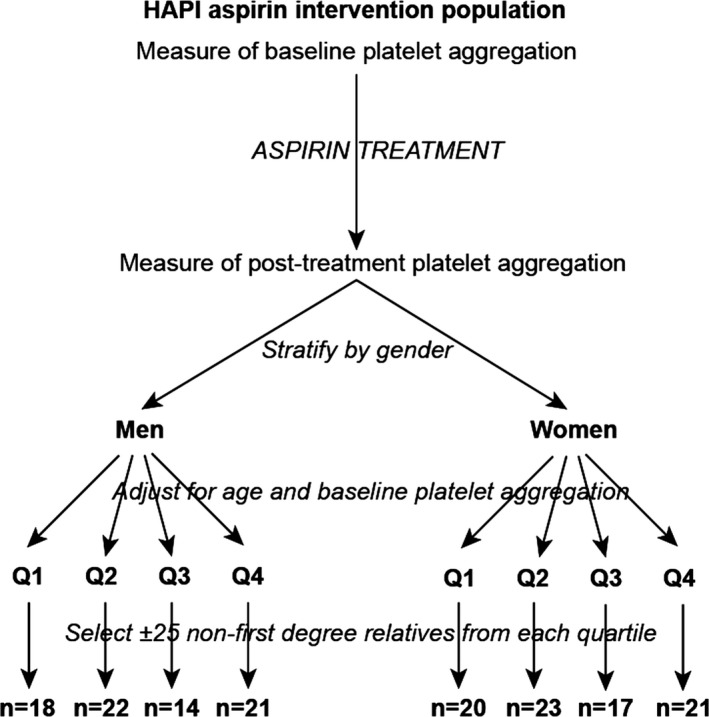
Procedures for sample selection. Flow diagram shows procedure for sample selection for the 156 metabolomics substudy participants from the overall Heredity and Phenotype Intervention (HAPI) Heart Study (n=745).
Oxylipids were extracted from 250 μL serum according to the procedure described previously.24 Samples were analyzed by liquid chromatography (Agilent 1260; San Jose, CA) coupled to electrospray ionization on a triple quadrupole mass spectrometer (Agilent 6460; San Jose, CA). Quantitation of oxylipid response was calculated as the peak area ratios of the target analyte to the respective internal standard. To obtain actual concentrations, calibration samples with spiked oxylipid levels were included in the measurement series. Relative response ratios of oxylipids were converted into actual concentrations (nmol/L) using the chemCal package in R.25 Twenty‐eight metabolites were successfully quantified and 2 metabolites (12‐HETE and 12‐HHTrE) were semiquantified because calibration curves failed. All metabolites measured are listed in Table 1. AA and LA were measured using gas chromatography–mass spectrometry as described previously.4
Table 1.
Metabolites Detected
| Name | Precursor FA | Omega 6/3 | Enzyme | Lipid Map ID | HMDB ID |
|---|---|---|---|---|---|
| 12‐HETE | AA | n‐6 | 12‐LOX | LMFA03060088 | HMDB06111 |
| 15‐HETE | AA | n‐6 | 15‐LOX | LMFA03060001 | HMDB03876 |
| 8‐HETE | AA | n‐6 | 15‐LOX | LMFA03060006 | HMDB04679 |
| LTB4 | AA | n‐6 | 5‐LOX | LMFA03020001 | HMDB01085 |
| 5‐HETE | AA | n‐6 | 5‐LOX | LMFA03060002 | HMDB11134 |
| TXB2 | AA | n‐6 | COX | LMFA03030002 | HMDB03252 |
| PGF2α | AA | n‐6 | COX | LMFA03010002 | HMDB01139 |
| 13,14‐dihydro‐PGF2α | AA | n‐6 | COX | LMFA03010079 | HMDB04239 |
| 12S‐HHTrE | AA | n‐6 | COX | LMFA03050002 | HMDB12535 |
| 11‐HETE | AA | n‐6 | COX | LMFA03060028 | HMDB04682 |
| 14,15‐DiHETrE | AA | n‐6 | CYP | LMFA03050010 | HMDB02265 |
| 11,12‐DiHETrE | AA | n‐6 | CYP | LMFA03050008 | HMDB02314 |
| 8,9‐DiHETrE | AA | n‐6 | CYP | LMFA03050006 | HMDB02311 |
| 20‐HETE | AA | n‐6 | CYP | LMFA03060009 | HMDB05998 |
| 5,6‐DiHETrE | AA | n‐6 | CYP | LMFA03050004 | HMDB02343 |
| 9‐HOTrE | ALA | n‐3 | 5‐LOX | LMFA02000024 | HMDB10224 |
| 15(S)‐HETrE | DGLA | n‐6 | 15‐LOX | LMFA03050007 | HMDB05045 |
| PGF1α | DGLA | n‐6 | COX | LMFA03010137 | HMDB02685 |
| 19,20‐DiHDPA | DHA | n‐3 | CYP | LMFA04000043 | HMDB10214 |
| 12(S)‐HEPE | EPA | n‐3 | 12‐LOX | LMFA03070008 | HMDB10202 |
| 15(S)‐HEPE | EPA | n‐3 | 15‐LOX | LMFA03070009 | HMDB10209 |
| 5(S)‐HEPE | EPA | n‐3 | 5‐LOX | LMFA03070010 | HMDB05081 |
| 13‐HODE | LA | n‐6 | 15‐LOX | LMFA02000228 | HMDB06939 |
| 9‐HODE | LA | n‐6 | 5‐LOX | LMFA02000151 | HMDB10223 |
| 9‐KODE | LA | n‐6 | 5‐LOX | LMFA02000274 | HMDB04669 |
| 9,12,13‐TriHOME | LA | n‐6 | 5‐/15‐LOX | LMFA02000014 | HMDB04708 |
| 12,13‐DiHOME | LA | n‐6 | CYP | LMFA02000230 | HMDB04705 |
| 9,10‐DiHOME | LA | n‐6 | CYP | LMFA02000229 | HMDB04704 |
| 12(13)‐EpOME | LA | n‐6 | CYP | LMFA02000038 | HMDB04702 |
| 9(10)‐EpOME | LA | n‐6 | CYP | LMFA02000037 | HMDB04701 |
AA indicates arachidonic acid; ALA, α‐linolenic acid; COX, cyclooxygenase; CYP, cytochrome P450; DGLA, dihomo‐γ‐linolenic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; FA, fatty acid; LA, linoleic acid; LOX, lipoxygenase.
Statistical Analysis
Missing ion intensity values were assumed to result from areas falling below the limits of detection and were imputed with values randomly drawn from a normal distribution with mean close to the limit of detection. For the 2 metabolites for which absolute concentrations were not obtained (12‐HETE and 12‐HHTrE), we report metabolite response values divided by the mean metabolite response value for the metabolite, a scaling method that does not affect our conclusions. Metabolite response values were log‐transformed before statistical analysis and back‐transformed for presentation. We assessed the significance of the effects of aspirin exposure and sex on metabolite level using linear models and linear mixed models in GenStat 14th edition (VSN International, Hemel Hempstead, UK) as described in details in Data S1.
All other statistical analyses were performed using Matlab version R2009a (MathWorks Inc, Natick, MA) or R.25 Correlations were performed using Spearman's rank correlation. The modulated modularity clustering algorithm26 was used to cluster oxylipids based on their correlation coefficients. Significant changes in correlation (post‐ versus pre‐aspirin) were assessed comparing the differences in correlation with a null distribution of correlation differences. The null distribution was generated by randomly permuting the post‐ and pre‐aspirin results of individuals, so that on average half of the data are permuted. Repeating this random permutation multiple times generated the null‐distribution. For all statistical tests, each set of P values was corrected for multiple comparisons using the procedure described by Benjamini and Yekutieli27 in R package multtest.28 This function computes the false discovery rate (q value) to control the expected proportion of rejected null hypotheses that were incorrect rejections (false discoveries) when conducting multiple comparisons. Significance was achieved when both P<0.05 and q<0.05.
Results
Participants’ Characteristics
Healthy volunteers enrolled in the HAPI Heart Study were selected for metabolic profiling based on their ex‐vivo response to low‐dose aspirin treatment. Characteristics and platelet aggregation measures of the 156 subjects selected for this metabolomics investigation are shown in Table 2. Samples originated from 81 women and 75 men and sex‐specific ex‐vivo response quartiles were comparable for age, body mass index and pre‐aspirin measures of collagen‐induced platelet aggregation. Women were slightly older (P=0.02) and had a higher body mass index (P=0.02) than men, as well as higher collagen‐induced platelet aggregation pre‐ and post aspirin (P=6.0×10−3 and 1.0×10−4, respectively). However, change in collagen‐induced platelet aggregation upon aspirin exposure was not significantly different between men and women (P=0.06). We also compared the subset of 156 subjects with metabolomics profiling to the full HAPI sample (N=745) and did not detect differences in sex (P=0.18), age (P=0.26), body mass index (P=0.36), presence of diabetes (P=0.59), AA‐induced platelet aggregation pre‐ and postaspirin (P=0.054 and 0.07, respectively) or collagen‐induced platelet aggregation pre‐ and post aspirin (P=0.58 and 0.76, respectively).
Table 2.
Subject Characteristics
| Response Quartile | Women | Men | P valueb | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q1 | Q2 | Q3 | Q4 | P Valuea | Total | Q1 | Q2 | Q3 | Q4 | P valuea | Total | ||
| N | 20 | 23 | 17 | 21 | 81 | 18 | 22 | 14 | 21 | 75 | |||
| Age, y | 43±12 | 49±14 | 43±15 | 42±11 | 0.2 | 45±13 | 43±15 | 39±11 | 40±11 | 38±10 | 0.8 | 40±12 | 0.02 |
| BMI, m/kg2 | 30±6 | 29±6 | 26±5 | 26±4 | 0.07 | 28±6 | 25±3 | 27±3 | 25±3 | 26±3 | 0.4 | 26±3 | 0.02 |
| Whole blood indirect COX‐1 pathway: collagen‐induced platelet aggregation | |||||||||||||
| Pre‐aspirin, Ω | 14±3 | 14±2 | 14±3 | 14±2 | 0.9 | 14±2 | 13±2 | 13±2 | 13±2 | 13±3 | 0.7 | 13±2 | 6×10−3 |
| Post aspirin, Ω | 6±2 | 11±2 | 12±1 | 15±1 | 1×10−14 | 11±3 | 5±2 | 8±1 | 10±1 | 13±2 | 1×10−14 | 9±3 | 1×10−4 |
| Change | −8±3 | −4±1 | −2±2 | 1±2 | 3×10−13 | −3±4 | −8±3 | −5±1 | −3±2 | 0±3 | 2×10−11 | −4±4 | 0.06 |
| Whole blood direct COX‐1 pathway: arachidonic acid–induced platelet aggregation | |||||||||||||
| Pre‐aspirin, Ω | 10±2 | 11±6 | 11±2 | 11±2 | 0.6 | 11±2 | 8±4 | 8±2 | 9±3 | 9±4 | 0.72 | 8±3 | 2×10−7 |
| Post aspirin, Ω | 0±0 | 1±3 | 2±3 | 2±3 | 0.12 | 1±3 | 0±0 | 0±1 | 1±1 | 2±3 | 8×10−3 | 1±2 | 0.96 |
BMI indicates body mass index; COX‐1, cyclooxygenase‐1.
P values for comparing mean values of gender‐specific quartiles (using Kruskal–Wallis tests).
P values for comparing mean values in all women vs all men (using Wilcoxon tests).
Metabolic Profiling
The metabolomic platform successfully detected 30 unique oxylipids in our samples. Besides the well‐described AA metabolites, oxylipids derived from LA, dihomo‐γ‐linolenic acid, α‐linolenic acid, eicosapentaenoic acid, and docosahexaenoic acid were also measured. Figure S1 illustrates the biochemical coverage of the platform in a pathway‐specific context.
Effect of Sex
Given that sex differences in the metabolome and in aspirin response have been reported previously,1, 2, 23, 29 we first assessed whether the effect of aspirin on oxylipids was sex dependent. We compared oxylipid levels pre‐ and post aspirin in men versus women. Prior to aspirin exposure, 8 metabolites were significantly higher in women and 1 (5,6‐DiHETrE) was significantly higher in men (Table 3). After aspirin exposure, 6 of these metabolites remained significantly different between men and women (Table 3). When linear mixed models were fitted to test the interaction between aspirin treatment and sex on oxylipid levels, no interaction remained significantly different after correction for multiple testing (8×10−3<P<0.92, 0.22<q<0.92), demonstrating that aspirin affected oxylipid levels similarly in men and women. Therefore, all subsequent analyses were performed in the entire cohort.
Table 3.
Significant Oxylipid Differences in Women Versus Men
| Metabolites | Pre‐aspirin | Postaspirin | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Precursor | Enzyme | Women | Men | P Value | Q | Women | Men | P value | Q |
| 5‐HEPE | EPA | 5‐LOX | 5.3±3.4 | 3.4±2.9 | 2×10−8 | 4×10−7 | 3.6±2.3 | 2.3±1.2 | 1×10−5 | 1.8×10−4 |
| 13‐HODE | LA | 15‐LOX | 24±10.5 | 18.3±8.7 | 3×10−5 | 3×10−4 | 17.2±7.3 | 14.2±7.8 | 2×10−5 | 1.8×10−4 |
| 9‐HODE | LA | 5‐LOX | 22.4±10.6 | 16.9±7.2 | 7×10−5 | 4×10−4 | 14.8±5.6 | 13.1±7 | 2×10−3 | 0.007 |
| 5,6‐DiHETrE | AA | CYP | 3.4±1.5 | 4.4±2 | 3×10−4 | 1.2×10−3 | 2.5±1.1 | 3.2±1.4 | 4×10−4 | 0.002 |
| 5‐HETE | AA | 5‐LOX | 15±6.6 | 12±5.4 | 2×10−3 | 6×10−3 | 11.5±5.7 | 9.2±4.5 | 3×10−3 | 0.008 |
| 9‐HOTrE | ALA | 5‐LOX | 1.1±0.6 | 0.8±0.4 | 4×10−3 | 0.01 | 0.7±0.4 | 0.6±0.4 | 0.25 | 0.2 |
| 9,10‐EpOME | LA | CYP | 3.3±1.6 | 2.7±2.6 | 2×10−4 | 0.001 | 2.6±2 | 2.3±2 | 0.2 | 0.2 |
| 12;13‐DiHOME | LA | CYP | 12.7±5.6 | 10.7±6.3 | 3×10−3 | 0.008 | 11.3±6.2 | 9.3±4.9 | 3×10−3 | 0.008 |
| 11,12‐DiHETrE | AA | CYP | 3.7±1.3 | 3.2±1 | 0.01 | 0.02 | 3±0.9 | 2.8±0.9 | 0.04 | 0.06 |
Data represent mean±SD of metabolite concentrations (nmol/L) in men (n=75) and women (n=81). AA indicates arachidonic acid; ALA, α‐linolenic acid; CYP, cytochrome P450; EPA, eicosapentaenoic acid; LA, linoleic acid; LOX, lipoxygenase.
Oxylipid Signature of Aspirin Exposure
Investigating the effect of aspirin exposure on oxylipid levels in our entire cohort revealed that the levels of 26 metabolites were significantly decreased post‐ compared to pre‐aspirin exposure, while only 1 metabolite (13,14‐dihydroPGF2) was increased (Table 4). As expected, the metabolites with the largest magnitude of decrease were known COX products, namely, TXB2 (−97%) 12‐HHTrE (−87%), and 11‐HETE (−85%). However, it was quite surprising to observe that aspirin also significantly decreased the levels of almost all metabolites measured, independently of their fatty acid precursor or the enzyme used for their synthesis (Figure 2). For example, aspirin strongly decreased LOX‐products derived from AA (5‐HETE, 12‐HETE, 8‐HETE, and 15‐HETE), LA (9‐HODE and 13‐HODE), dihomo‐γ‐linolenic acid (15‐HETrE), eicosapentaenoic acid (5‐HEPE, 12‐HEPE, and 15‐HEPE), and α‐linolenic acid (9‐HOTrE). Although to a lesser extent, aspirin also significantly decreased levels of CYP‐produced metabolites derived from LA (EpOMEs and DiHOMEs) and AA (DiHETrEs and 20‐HETE).
Table 4.
Effect of Aspirin on Oxylipids
| Name | Precursor | Enzyme | Pre‐aspirin | Postaspirin | % Change | P value | Q value |
|---|---|---|---|---|---|---|---|
| TXB2 | AA | COX | 241.3±245.8 | 7±12.9 | −97 | 1.9×10−26 | 3.2×10−25 |
| 12‐HHTrE* | AA | COX | 1.8±1.8 | 0.2±0.1 | −87 | 2.3×10−26 | 3.2×10−25 |
| 11‐HETE | AA | COX | 13.7±11.9 | 1.8±1.2 | −85 | 3.3×10−26 | 3.2×10−25 |
| 15‐HETE | AA | 15‐LOX | 16.2±12.9 | 5.2±3.6 | −67 | 1.7×10−24 | 1.2×10−23 |
| 12‐HEPE | EPA | 12‐LOX | 26.2±34.5 | 11.1±17.4 | −60 | 3.3×10−13 | 8.2×10−13 |
| 12‐HETE* | AA | 12‐LOX | 1.3±1.4 | 0.7±1 | −53 | 8×10−13 | 1.2×10−12 |
| 15‐HETrE | DGLA | 15‐LOX | 3.2±1.5 | 1.8±0.9 | −45 | 9×10−22 | 5.8×10−21 |
| 15‐HEPE | EPA | 15‐LOX | 1.7±1.1 | 1±0.7 | −44 | 5×10−16 | 2.1×10−15 |
| 8‐HETE | AA | 15‐LOX | 5.5±3.6 | 3.4±2.6 | −43 | 1.4×10−12 | 3×10−12 |
| PGF2α | AA | COX | 5.9±3.4 | 3.9±2.3 | −31 | 7.4×10−20 | 3.7×10−19 |
| 9‐HOTrE | ALA | 5‐LOX | 1±0.5 | 0.7±0.4 | −31 | 9.2×10−10 | 1.5×10−9 |
| 9‐HODE | LA | 5‐LOX | 19.7±9.5 | 14±6.4 | −29 | 7.3×10−14 | 2.4×10−13 |
| 5‐HEPE | EPA | 5‐LOX | 4.4±3.3 | 3±2 | −28 | 2.5×10−13 | 6.7×10−13 |
| 5,6‐DiHETrE | AA | CYP | 3.9±1.8 | 2.8±1.3 | −27 | 3.6×10−14 | 1.3×10−13 |
| 13‐HODE | LA | 15‐LOX | 21.3±10 | 15.8±7.7 | −27 | 6.5×10−12 | 1.3×10−11 |
| 5‐HETE | AA | 5‐LOX | 13.6±6.2 | 10.4±5.3 | −26 | 2.5×10−13 | 6.7×10−13 |
| 12,13‐DiHOME | LA | CYP | 11.7±6 | 10.3±5.7 | −19 | 1.3×10−4 | 1.6×10−4 |
| 20‐HETE | AA | CYP | 7.2±2.8 | 5.9±2.2 | −19 | 1.9×10−8 | 2.8×10−8 |
| 11,12‐DiHETrE | AA | CYP | 3.5±1.2 | 2.9±0.9 | −16 | 9.2×10−10 | 1.5×10−9 |
| 9‐KODE | LA | 5‐LOX | 2.6±1.8 | 2.2±1.6 | −15 | 1.1×10−7 | 1.5×10−7 |
| 9,10‐DiHOME | LA | CYP | 6.3±4 | 5.5±3.9 | −15 | 1×10−4 | 1.3×10−8 |
| 8,9‐DiHETrE | AA | CYP | 2.3±0.7 | 2±0.6 | −14 | 1.8×10−9 | 2.8×10−8 |
| PGF1α | DGLA | COX | 2.6±0.5 | 2.2±0.4 | −11 | 1.5×10−11 | 2.8×10−11 |
| 9,10‐EpOME | LA | CYP | 3±2.1 | 2.4±2 | −11 | 1.3×10−6 | 1.7×10−6 |
| 12,13‐EpOME | LA | CYP | 5±3.3 | 4.6±3 | −11 | 8.5×10−3 | 9.4×10−3 |
| 14,15‐DiHETrE | AA | CYP | 4.8±1.2 | 4.3±1.1 | −9 | 7.8×10−8 | 1.1×10−7 |
| 9,12,13‐TriHOME | LA | 5/15‐LOX | 1.5±0.6 | 1.4±1 | −3 | 0.036 | 0.038 |
| 13,14‐dihydro‐PGF2α | AA | COX | 39.9±6.5 | 41.9±6.2 | 3 | 0.004 | 0.005 |
| 19,20‐DiHDPA | DHA | CYP | 7.3±3.2 | 7.1±3 | −2 | 0.56 | 0.56 |
| LTB4 | AA | 5‐LOX | 3.1±3.7 | 2.6±3 | 0 | 0.06 | 0.06 |
Data represent mean±SD of metabolite concentrations (nmol/L) in all subjects (n=156) pre‐ and postaspirin treatment and median % change. Absolute concentrations could not be determined for metabolites with an asterisk, so data represent mean±SD of metabolite levels divided by the mean metabolite level detected in this study. AA indicates arachidonic acid; ALA, α‐linolenic acid; COX, cyclooxygenase; CYP, cytochrome P450; DGLA, dihomo‐γ‐linolenic acid; DHA, docosahexaenoic acid; EPA, eicosapentaenoic acid; LA, linoleic acid; LOX, lipoxygenase.
Figure 2.
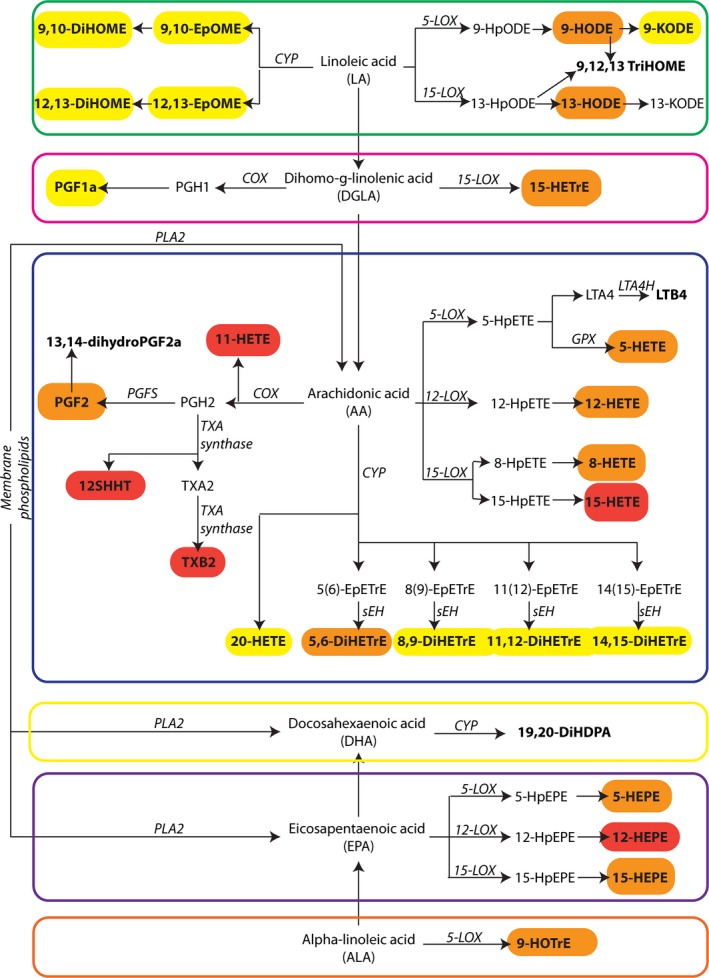
Effect of aspirin on oxylipids. Metabolites detected in our study samples are in bold. Metabolites significantly changed upon aspirin treatment are circled and color coded according to their percentage change upon aspirin treatment: red: change >−60%; orange: −60%> change >−25%; yellow: −25%> change >0%. COX indicates cyclooxygenase; CYP, cytochrome P‐450 monooxygenases; EpETrE, epoxyeicosatrienoic acid; LOX, lipoxygenases; TXA, thromboxane A2; PGH2: prostaglandin H2, PGF2: prostaglandin F2, PGFS: prostaglandin F synthase, PLA2: phospholipase A2.
Changes in Correlation Between Oxylipids Upon Aspirin Exposure
To further evaluate the impact of aspirin exposure on the oxylipid pathways, we conducted cluster plot analyses in order to observe correlations among oxylipid metabolites. Figure 3A displays correlations between metabolite levels pre‐aspirin. Three clusters reflecting the main oxylipid biosynthetic pathways emerge from this unsupervised clustering analysis. Cluster 1 comprises AA‐derived oxylipids formed through LOX or COX. Cluster 2 consists of LA‐derived metabolites synthesized through LOX or CYP. Finally, AA‐ and docosahexaenoic acid–derived CYP products correlate with each other in cluster 3. Figure 3B displays these correlations post aspirin treatment. Significance of change in correlation between metabolites post‐ versus pre‐aspirin exposure was evaluated using permutation testing. Figure 3C displays the q‐values to evaluate whether correlations between metabolites changed significantly post‐ versus pre‐aspirin exposure. Most significant q values can be observed within Cluster 1, where correlations between COX‐generated metabolites (TXB2, 12‐HHTrE, PGF1a, and PGF2a) and other metabolites from Cluster 1 were close to zero after aspirin treatment. This finding was expected given the known direct COX inhibition of aspirin, which led to strong inhibition of all COX‐products in our samples post–aspirin administration. Surprisingly, however, in Clusters 2 and 3, correlations between metabolites were not changed post–aspirin exposure, although the drug significantly decreased most metabolite levels. This might result from a disruption of the metabolic pathways somewhere upstream, maybe at the level of the fatty acid precursors. To evaluate this hypothesis, we measured the levels of free AA and LA, the 2 main fatty acid precursors for the oxylipid measured. Free AA levels were not significantly affected by aspirin treatment (−8%, P=0.42), whereas those of LA were significantly decreased (−19%, P=1.3×10−5).
Figure 3.
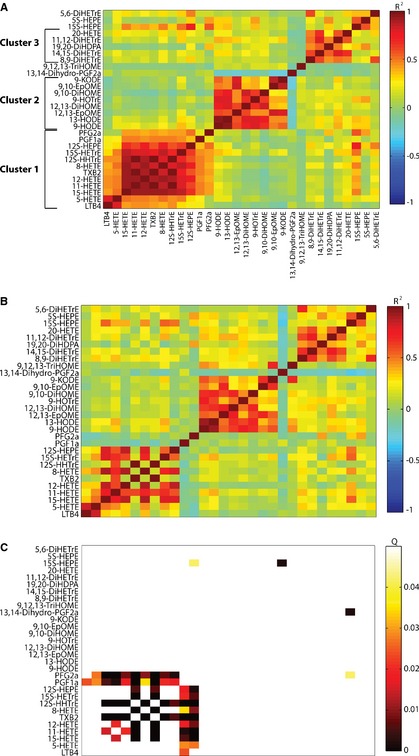
Correlations between oxylipids. A, Spearman correlation coefficients for correlation between metabolite levels pre‐aspirin in all 156 subjects are displayed. Metabolites were clustered based on Spearman correlation coefficients using modulated modularity clustering algorithm. B, Spearman correlation coefficients for correlation between metabolite levels post aspirin in all 156 subjects are displayed. The clustering order is the same as in Figure 3A. C, Q values evaluating the significance of the change in correlation between metabolites post‐ vs pre‐aspirin are displayed. The clustering order is the same as in Figure 3A.
Correlations Between Fatty Acid Precursors and Oxylipid Products
We further evaluated the causal relationship between free fatty acid precursor levels and the levels of their product oxylipids by computing the correlations between them prior to aspirin administration (Table 5 and Figure 4). We observed significant correlations between LA‐derived oxylipids and LA, with correlation coefficients (ρ) ranging from 0.27 to 0.5, whereas no significant correlation was found between AA and AA‐derived oxylipids (−0.13<ρ<0.17).
Table 5.
Correlations Between Serum Oxylipid and Free Fatty Acid Levels Pre‐aspirin
| Metabolite | Correlation | ||
|---|---|---|---|
| Name | Enzyme | ρ | P value |
| LA‐derived oxylipids | |||
| 9,10‐DiHOME | CYP | 0.39 | 1.2×10−6 |
| 9,10‐EpOME | CYP | 0.42 | 9×10−8 |
| 12,13‐DiHOME | CYP | 0.44 | 1.7×10−8 |
| 12,13‐EpOME | CYP | 0.36 | 6×10−6 |
| 9‐HODE | 5 LOX | 0.5 | 1×10−10 |
| 9‐KODE | 5 LOX | 0.27 | 8×10−4 |
| 13‐HODE | 15 LOX | 0.48 | 5×10−10 |
| AA‐derived oxylipids | |||
| 11‐HETE | COX | −0.1 | 0.35 |
| PGF2α | COX | −0.13 | 0.09 |
| 12S‐HHT | COX | −0.11 | 0.17 |
| TXB2 | COX | −0.09 | 0.27 |
| 20‐HETE | CYP | 0 | 0.86 |
| 5,6‐DiHETrE | CYP | 0 | 0.74 |
| 8,9‐DiHETrE | CYP | 0.11 | 0.17 |
| 11,12‐DiHETrE | CYP | 0.17 | 0.05 |
| 14,15‐DiHETrE | CYP | 0.13 | 0.12 |
| 5‐HETE | 5 LOX | 0.1 | 0.21 |
| 12‐HETE | 12 LOX | −0.1 | 0.36 |
| 8‐HETE | 15 LOX | 0 | 0.9 |
| 15‐HETE | 15 LOX | 0 | 0.5 |
AA indicates arachidonic acid; COX, cyclooxygenase; CYP, cytochrome P450; LA, linoleic acid; LOX, lipoxygenase.
Figure 4.
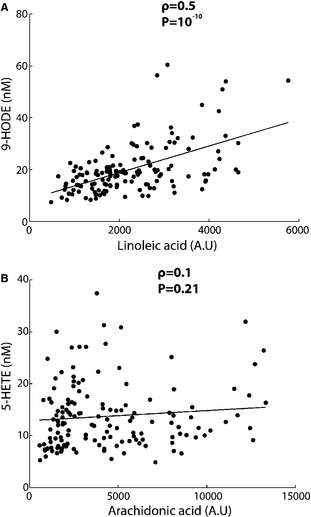
Typical correlations between oxylipid levels pre‐aspirin and their precursor fatty acid levels pre‐aspirin. Correlations between (A) linoleic acid (LA) and 9‐HODE (LA‐derived oxylipid) and (B) arachidonic acid (AA) and 5‐HETE (AA‐derived oxylipid).
Association Between Oxylipids and Aspirin‐Induced Inhibition of Platelet Aggregation
Finally, our study assessed whether oxylipid levels correlated with ex‐vivo platelet aggregation measures. We observed significant correlations between post aspirin levels of 13‐HODE, 9‐HODE, 12,13‐DiHOME, 9‐HOTrE, and 12,13‐EpOME and postaspirin collagen‐induced platelet aggregation (Table 6). LA and AA levels did not correlate with collagen‐induced platelet aggregation. Correlation analyses with AA‐induced platelet aggregation measures were not conducted since 75% (n=117) of the subjects had a complete inhibition of AA‐induced platelet aggregation postaspirin. Therefore, we evaluated differences in oxylipid, AA, and LA levels between the subjects with complete and those with incomplete AA‐induced platelet aggregation postaspirin. We observed a trend towards higher levels of TXB2 in the subjects with incomplete inhibition of AA‐induced platelet aggregation (P=0.008, q=0.1). No other significant difference was found.
Table 6.
Significant Correlations Between Serum Oxylipid Levels Postaspirin and Collagen‐Induced Platelet Aggregation Postaspirin
| Metabolite | ρ | P value | Q |
|---|---|---|---|
| 13‐HODE | 0.27 | 7×10−4 | 0.009 |
| 9‐HODE | 0.24 | 0.002 | 0.01 |
| 12,13‐DiHOME | 0.23 | 0.004 | 0.02 |
| 9‐HOTrE | 0.22 | 0.005 | 0.02 |
| 12,13‐EpOME | 0.19 | 0.005 | 0.02 |
Discussion
In this study, we investigated the metabolic signature of aspirin exposure in healthy volunteers using a quantitative mass spectrometry–based metabolomics platform targeted to the measurement of oxylipids. We aimed to evaluate the changes in the oxylipid metabolome in response to aspirin and to determine whether oxylipids contributed to variation in aspirin response.
The mechanisms underlying variation in response to aspirin as antiplatelet therapy are largely unknown. Several studies have reported less effective inhibition of platelet function in response to aspirin in women than in men,3, 30, 31 and whether women benefit equally from aspirin therapy as men is still in debate. In the present study, we therefore first evaluated constitutive and aspirin‐induced sex differences in oxylipid levels. Several previous metabolomics studies have shown considerable sex differences in the human metabolome,4, 5, 29, 32, 33, 34 none of them including the oxylipid metabolites. Here, we report for the first time that many oxylipid CYP products (the LA‐derived 9,10 EpOME and 12,13 DiHOME and the AA‐derived 5,6 DiHETrE and 11‐12 DiHETrE) differ between sexes. The CYP2J and 2C families are responsible for the synthesis of these molecules,6, 7, 35 and sex differences in human CYP activity has been extensively reported.8, 36 We also observed higher levels of 5‐LOX products (namely, the eicosapentaenoic acid–derived 5S‐HEPE, the LA‐derived 9‐HODE, the AA‐derived 5‐HETE, and the α‐linolenic acid–derived 9‐HOTrE) in women. This is in accordance with previous findings that androgens downregulate 5‐LOX product formation.9, 10, 37, 38 Most of these constitutive differences in oxylipid levels persisted after aspirin treatment and no sex‐specific effect of aspirin on oxylipid was observed, consistent with our previous findings in this population.4, 9, 11, 12, 39
Secondly, we observed that aspirin significantly decreased almost all of the oxylipids measured in our samples (26 out of 30), independently of their fatty acid precursors or their synthesizing enzymes. This finding may seem surprising, as one might have thought that inhibition of 1 enzymatic branch (COX) would divert fatty acid substrates towards other branches of the pathways, leading to higher levels of LOX‐ or CYP‐derived products. However, oxylipid metabolism does not seem to follow a simple mass‐action rule, at least when measured globally in serum, and our findings are in agreement with a previous report. In a murine inflammatory model, a single high dose of aspirin significantly lowered plasma levels of COX‐, LOX‐, and CYP‐derived AA metabolites.13, 40 The authors concluded that the COX, LOX, and CYP pathways “do not proceed in a parallel way but communicate in a dynamic manner.” In contrast, Shinde et al14, 41 reported that aspirin significantly decreased TXB2 levels in plasma of healthy volunteers, but not the levels of CYP‐, LOX‐, or AA‐derived metabolites. However, they measured oxylipids 2 hours after a single high dose of aspirin, which might explain our differing results.
To unravel the key points at which metabolic regulation has changed in the oxylipid pathways, we next examined aspirin‐induced changes in pairwise correlations of metabolite levels. We observed that, except for the direct COX‐ metabolites, correlations between oxylipids did not change significantly upon aspirin administration, even though metabolite levels were altered. We postulated that this coordinated change in oxylipid levels could result from an effect of aspirin on the upstream parts of the biosynthetic pathways and found that LA levels were indeed significantly lowered by aspirin treatment. Further, we observed a high correlation between LA levels and its oxylipid products in serum. Therefore, based on our findings, we postulate that, for LA‐derived oxylipids, aspirin‐induced decrease in precursor availability might account for the observed decrease in circulating oxylipid levels. The correlations were poor between AA levels and its oxylipid products, suggesting that free AA is not a major source of synthesis for circulating oxylipids.
We show for the first time that low‐dose aspirin therapy, such as that routinely used for prevention of cardiovascular disease, broadly decreases circulating fatty acid levels in healthy humans. We previously reported in the same population that aspirin decreased the levels of many of the most abundant FAs,15, 16, 17, 42 including oleic (−28%), palmitic (−10%), palmitoleic (−31%), mystiric (−15%), and lauric (−15%) acids, as well as glycerol levels (−13%).5, 18, 19 The concomitant decrease of free fatty acids and glycerol indicates a potential inhibition of lipolysis by aspirin in our subjects. The metabolic effects of aspirin are one of the least appreciated pharmacological actions of this drug but have been described >125 years ago. A decrease in serum fatty acid levels has been reported following salicylate administration in rats20, 21, 43 and in healthy and diabetic humans.22, 44, 45, 46, 47 There is evidence that salicylates inhibit lipolysis and enhance fatty acid re‐esterification in animal adipose tissue ex‐vivo23, 48 and downregulate the expression and activity of adipose 11β‐hydroxysteroid dehydrogenase type 1, a lipolytic enzyme.24, 49 However, all previous studies have used high dose of salicylates (minimum 1 g per day for 4 days25, 44). The long‐term consequences of the broad decrease in fatty acid levels we present here in participants taking low‐dose aspirin are unknown. In the Women's Health Study, low‐dose aspirin (100 mg every other day) did not decrease the incidence of type 2 diabetes in healthy women.50 However, whether daily aspirin dosing would alter or delay progression to diabetes or benefit some subgroup of patients is unknown. Based on our findings, future studies evaluating various doses of aspirin in different patient populations (men and women of varying cardiovascular risk) for its potential role in preventing diabetes or insulin resistance may be warranted.
Serum AA levels were not significantly decreased postaspirin in our study samples. However, low correlation between adipose and circulating levels of AA have been reported in nondiabetic women under prolonged fasting, whereas LA adipose and serum levels were strongly correlated.4, 51 These findings suggest that circulating AA levels poorly reflect adipose tissue lipolysis and might explain why AA levels were not significantly decreased upon aspirin exposure in our study. Unfortunately, n‐3 fatty acid levels were not measured by our analytical platform, so whether those levels are also decreased upon low‐dose aspirin treatment remains to be determined.
Finally, we investigated whether oxylipids are involved in variation in response to aspirin by correlating oxylipid levels to ex‐vivo platelet aggregation measures postaspirin. Not surprisingly, we observed higher levels of TXB2 in individuals with incomplete inhibition of AA‐induced platelet aggregation. However, no other metabolite was correlated with AA‐induced platelet aggregation post–aspirin exposure, suggesting that differences in oxylipid levels do not contribute appreciably to variability in traditional, COX‐1‐mediated measures of aspirin response. In contrast, we observed significant correlations between 4 LA‐derived oxylipids (13‐HODE, 9‐HODE, 12,13‐DiHOME, and 12,13‐EpOME) and collagen‐induced platelet aggregation post aspirin. Endothelial,16, 25, 52, 53 epidermal,26, 54 polymorphonuclear cells,27, 55 and platelets28, 56 convert LA into 13‐HODE and 9‐HODE. HODEs are active mediators in hemostasis, inflammation, and cancer invasion. 13‐HODE modulates the adhesive properties of endothelium52, 57, 58 and inhibits both AA‐ and collagen‐induced platelet aggregation.59 Low 13‐HODE, 9‐HODE, 12,13‐DiHOME, and 12,13‐EpOME in serum correlating to better non‐COX‐1‐mediated aspirin response may result from a negative feedback mechanism, although these results deserve further confirmation in other extended cohorts of healthy volunteers and/or patients with cardiovascular diseases on aspirin.
A limitation of our study that should be mentioned is the lack of a control group with oxylipid measurements taken over time without the administration of aspirin. Therefore, we are unable to separate the natural time variation of oxylipid levels from the effects of aspirin. However, based on the relatively short time period of our intervention and previous studies in which we have demonstrated minimal changes in the metabolome in placebo‐treated participants, with 0 metabolites differing after 1 week of placebo and 6 of 348 measured metabolites differing significantly after 4 weeks of placebo treatment,60 we hypothesize that the changes in oxylipids (27 out of 30 measured) we observed after 2 weeks are likely aspirin‐driven rather than lifestyle‐driven.
In conclusion, our findings revealed a sex‐independent, global decrease in serum oxylipids in response to low‐dose aspirin treatment in healthy volunteers, as well as in oxylipids that are not synthesized by COX. We show that this global decrease in oxylipids might be related to aspirin‐induced decrease in precursor nonesterified fatty acids. Finally, we observed that several LA‐derived oxylipids were significantly correlated with non‐COX1‐mediated measures of aspirin response, thereby suggesting that these metabolites might contribute to the non‐COX1‐mediated variability in response to aspirin. Pharmacometabolomics provided a powerful tool to understand the mechanisms of action of aspirin and the metabolic determinants of variation of response to aspirin's antiplatelet effects.
Sources of Funding
The National Institutes of Health (NIH) supported this study through (RC2GM092729) as part of the American Recovery and Reinvestment Act (ARRA). Work was implemented by the Pharmacometabolomics Research Network. The effort of Dr Ellero‐Simatos was supported by the research programme of the Netherlands Metabolomics Centre (NMC), part of The Netherlands Genomics Initiative/Netherlands Organization for Scientific Research; the HAPI Heart Study was supported by grants (U01‐HL72515), the University of Maryland General Clinical Research Center (GCRC; M01‐RR‐16500), the Johns Hopkins University GCRC (M01‐RR‐000052), National Center for Research Resources, and the Clinical Nutrition Research Unit of Maryland (P30‐DK072488). The effort of Dr Beitelshees was supported by NIH grant K23‐HL091120, Dr Lewis was supported by NIH grant K23‐GM102678, and Dr Yerges‐Armstrong was supported by grant K01‐HL116770.
Disclosures
None.
Supporting information
Data S1. Supplementary Methods.
Figure S1. Oxylipid biosynthetic pathways (adapted from (1)). Metabolites detected in our samples are in bold.
Acknowledgments
We would like to thank Keith Tanner for his help with sample preparation.
(J Am Heart Assoc. 2015;4:e002203 doi: 10.1161/JAHA.115.002203)
Accompanying Data S1 and Figure S1 are available at http://jaha.ahajournals.org/content/4/10/e002203/suppl/DC1
References
- 1. Patrono C, Ciabattoni G, Patrignani P, Pugliese F, Filabozzi P, Catella F, Davì G, Forni L. Clinical pharmacology of platelet cyclooxygenase inhibition. Circulation. 1985;72:1177–1184. [DOI] [PubMed] [Google Scholar]
- 2. Pedersen AK, FitzGerald GA. Dose‐related kinetics of aspirin. Presystemic acetylation of platelet cyclooxygenase. N Engl J Med. 1984;311:1206–1211. [DOI] [PubMed] [Google Scholar]
- 3. Chiang N, Bermudez EA, Ridker PM, Hurwitz S, Serhan CN. Aspirin triggers antiinflammatory 15‐epi‐lipoxin A4 and inhibits thromboxane in a randomized human trial. Proc Natl Acad Sci USA. 2004;101:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yerges‐Armstrong LM, Ellero‐Simatos S, Georgiades A, Zhu H, Lewis JP, Horenstein RB, Beitelshees AL, Dane A, Reijmers T, Hankemeier T, Fiehn O, Shuldiner AR, Kaddurah‐Daouk R. Purine pathway implicated in mechanism of resistance to aspirin therapy: pharmacometabolomics‐informed pharmacogenomics. Clin Pharmacol Ther. 2013;94:525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lewis JP, Yerges‐Armstrong LM, Ellero‐Simatos S, Georgiades A, Kaddurah‐Daouk R, Hankemeier T. Integration of pharmacometabolomic and pharmacogenomic approaches reveals novel insights into antiplatelet therapy. Clin Pharmacol Ther. 2013;94:570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hovens MMC, Snoep JD, Eikenboom JCJ, van der Bom JG, Mertens BJA, Huisman MV. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175–181. [DOI] [PubMed] [Google Scholar]
- 7. Gum PA, Kottke‐Marchant K, Welsh PA, White J, Topol EJ. A prospective, blinded determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–965. [DOI] [PubMed] [Google Scholar]
- 8. Antithrombotic Trialists’ Collaboration . Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frelinger AL, Li Y, Linden MD, Barnard MR, Fox ML, Christie DJ, Furman MI, Michelson AD. Association of cyclooxygenase‐1‐dependent and ‐independent platelet function assays with adverse clinical outcomes in aspirin‐treated patients presenting for cardiac catheterization. Circulation. 2009;120:2586–2596. [DOI] [PubMed] [Google Scholar]
- 10. Eikelboom JW, Hankey GJ, Thom J, Bhatt DL, Steg PG, Montalescot G, Johnston SC, Steinhubl SR, Mak K‐H, Easton JD, Hamm C, Hu T, Fox KAA, Topol EJ; Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management and Avoidance (CHARISMA) Investigators . Incomplete inhibition of thromboxane biosynthesis by acetylsalicylic acid: determinants and effect on cardiovascular risk. Circulation. 2008;118:1705–1712. [DOI] [PubMed] [Google Scholar]
- 11. Ohmori T, Yatomi Y, Nonaka T, Kobayashi Y, Madoiwa S, Mimuro J, Ozaki Y, Sakata Y. Aspirin resistance detected with aggregometry cannot be explained by cyclooxygenase activity: involvement of other signaling pathway(s) in cardiovascular events of aspirin‐treated patients. J Thromb Haemost. 2006;4:1271–1278. [DOI] [PubMed] [Google Scholar]
- 12. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. 2005;6:1191–1197. [DOI] [PubMed] [Google Scholar]
- 13. Gleim S, Stitham J, Tang WH, Martin KA, Hwa J. An eicosanoid‐centric view of atherothrombotic risk factors. Cell Mol Life Sci. 2012;69:3361–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Theken KN, Schuck RN, Edin ML, Tran B, Ellis K, Bass A, Lih FB, Tomer KB, Poloyac SM, Wu MC, Hinderliter AL, Zeldin DC, Stouffer GA, Lee CR. Evaluation of cytochrome P450‐derived eicosanoids in humans with stable atherosclerotic cardiovascular disease. Atherosclerosis. 2012;222:530–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cortelazzo S, Marchetti M, Orlando E, Falanga A, Barbui T, Buchanan MR. Aspirin increases the bleeding side effects in essential thrombocythemia independent of the cyclooxygenase pathway: role of the lipoxygenase pathway. Am J Hematol. 1998;57:277–282. [DOI] [PubMed] [Google Scholar]
- 16. Kaduce TL, Figard PH, Leifur R, Spector AA. Formation of 9‐hydroxyoctadecadienoic acid from linoleic acid in endothelial cells. J Biol Chem. 1989;264:6823–6830. [PubMed] [Google Scholar]
- 17. Colli S, Caruso D, Tremoli E, Stragliotto E, Morazzoni G, Galli G. Effect of single oral administrations of non steroidal antiinflammatory drugs to healthy volunteers on arachidonic acid metabolism in peripheral polymorphonuclear and mononuclear leukocytes. Prostaglandins Leukot Essent Fatty Acids. 1988;34:167–174. [DOI] [PubMed] [Google Scholar]
- 18. Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost J‐P, Le Net J‐L, Baker D, Walley RJ, Everett JR, Nicholson JK. Pharmaco‐metabonomic phenotyping and personalized drug treatment. Nature. 2006;440:1073–1077. [DOI] [PubMed] [Google Scholar]
- 19. Kaddurah‐Daouk R, Weinshilboum RM; Pharmacometabolomics Research Network . Pharmacometabolomics: implications for clinical pharmacology and systems pharmacology. Clin Pharmacol Ther. 2014;95:154–167. [DOI] [PubMed] [Google Scholar]
- 20. Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK. Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA. 2009;106:14728–14733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaddurah‐Daouk R, Baillie RA, Zhu H, Zeng Z‐B, Wiest MM, Nguyen UT, Watkins SM, Krauss RM. Lipidomic analysis of variation in response to simvastatin in the Cholesterol and Pharmacogenetics Study. Metabolomics. 2010;6:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mitchell BD, McArdle PF, Shen H, Rampersaud E, Pollin TI, Bielak LF, Jaquish C, Douglas JA, Roy‐Gagnon M‐H, Sack P, Naglieri R, Hines S, Horenstein RB, Chang Y‐PC, Post W, Ryan KA, Brereton NH, Pakyz RE, Sorkin J, Damcott CM, O'Connell JR, Mangano C, Corretti M, Vogel R, Herzog W, Weir MR, Peyser PA, Shuldiner AR. The genetic response to short‐term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am Heart J. 2008;155:823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shen H, Herzog W, Drolet M, Pakyz R, Newcomer S, Sack P, Karon H, Ryan KA, Zhao Y, Shi X, Mitchell BD, Shuldiner AR. Aspirin resistance in healthy drug‐naive men versus women (from the Heredity and Phenotype Intervention Heart Study). Am J Cardiol. 2009;104:606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strassburg K, Huijbrechts AML, Kortekaas KA, Lindeman JH, Pedersen TL, Dane A, Berger R, Brenkman A, Hankemeier T, van Duynhoven J, Kalkhoven E, Newman JW, Vreeken RJ. Quantitative profiling of oxylipins through comprehensive LC‐MS/MS analysis: application in cardiac surgery. Anal Bioanal Chem. 2012;404:1413–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. RCoreTeam . R: A Language and Environment for Statistical Computing [Internet]. Available at: http://www.R-project.org. Accessed 1 March 2013. [Google Scholar]
- 26. Stone EA, Ayroles JF. Modulated modularity clustering as an exploratory tool for functional genomic inference. PLoS Genet. 2009;5:e1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29:1165–1188. [Google Scholar]
- 28. Pollard KS, Gilbert HN, Ge Y, Taylor S, Dudoit S. Multtest: resampling‐based multiple hypothesis testing. R package version 2.8.0.
- 29. Lawton KA, Berger A, Mitchell M, Milgram KE, Evans AM, Guo L, Hanson RW, Kalhan SC, Ryals JA, Milburn MV. Analysis of the adult human plasma metabolome. Pharmacogenomics. 2008;9:383–397. [DOI] [PubMed] [Google Scholar]
- 30. Gum PA, Kottke‐Marchant K, Poggio ED, Gurm H, Welsh PA, Brooks L, Sapp SK, Topol EJ. Profile and prevalence of aspirin resistance in patients with cardiovascular disease. Am J Cardiol. 2001;88:230–235. [DOI] [PubMed] [Google Scholar]
- 31. Yee DL, Sun CW, Bergeron AL, Dong J‐F, Bray PF. Aggregometry detects platelet hyperreactivity in healthy individuals. Blood. 2005;106:2723–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mittelstrass K, Ried JS, Yu Z, Krumsiek J, Gieger C, Prehn C, Roemisch‐Margl W, Polonikov A, Peters A, Theis FJ, Meitinger T, Kronenberg F, Weidinger S, Wichmann HE, Suhre K, Wang‐Sattler R, Adamski J, Illig T. Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genet. 2011;7:e1002215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu Z, Kastenmüller G, He Y, Belcredi P, Möller G, Prehn C, Mendes J, Wahl S, Roemisch‐Margl W, Ceglarek U, Polonikov A, Dahmen N, Prokisch H, Xie L, Li Y, Wichmann H‐E, Peters A, Kronenberg F, Suhre K, Adamski J, Illig T, Wang‐Sattler R. Differences between human plasma and serum metabolite profiles. PLoS One. 2011;6:e21230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saito K, Maekawa K, Pappan KL, Urata M, Ishikawa M. Differences in metabolite profiles between blood matrices, ages, and sexes among Caucasian individuals and their inter‐individual variations. Metabolomics. 2014;10:402–413. [Google Scholar]
- 35. Konkel A, Schunck W‐H. Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta. 2011;1814:210–222. [DOI] [PubMed] [Google Scholar]
- 36. Waxman DJ, Holloway MG. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol Pharmacol. 2009;76:215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pergola C, Rogge A, Dodt G, Northoff H, Weinigel C, Barz D, Rådmark O, Sautebin L, Werz O. Testosterone suppresses phospholipase D, causing sex differences in leukotriene biosynthesis in human monocytes. FASEB J. 2011;25:3377–3387. [DOI] [PubMed] [Google Scholar]
- 38. Pergola C, Dodt G, Rossi A, Neunhoeffer E, Lawrenz B, Northoff H, Samuelsson B, Rådmark O, Sautebin L, Werz O. ERK‐mediated regulation of leukotriene biosynthesis by androgens: a molecular basis for gender differences in inflammation and asthma. Proc Natl Acad Sci USA. 2008;105:19881–19886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ellero‐Simatos S, Lewis JP, Georgiades A, Yerges‐Armstrong LM, Beitelshees AL, Horenstein RB, Dane A, Harms AC, Ramaker R, Vreeken RJ, Perry CG, Zhu H, Sànchez CL, Kuhn C, Ortel TL, Shuldiner AR, Hankemeier T, Kaddurah‐Daouk R. Pharmacometabolomics reveals that serotonin is implicated in aspirin response variability. CPT Pharmacometrics Syst Pharmacol. 2014;3:e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu J‐Y, Yang J, Inceoglu B, Qiu H, Ulu A, Hwang S‐H, Chiamvimonvat N, Hammock BD. Inhibition of soluble epoxide hydrolase enhances the anti‐inflammatory effects of aspirin and 5‐lipoxygenase activation protein inhibitor in a murine model. Biochem Pharmacol. 2010;79:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shinde DDD, Kim K‐BK, Oh K‐SK, Abdalla NN, Liu K‐HK, Bae SKS, Shon J‐HJ, Kim H‐SH, Kim D‐HD, Shin JGJ. LC‐MS/MS for the simultaneous analysis of arachidonic acid and 32 related metabolites in human plasma: basal plasma concentrations and aspirin‐induced changes of eicosanoids. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;911:113–121. [DOI] [PubMed] [Google Scholar]
- 42. Halliwell KJ, Fielding BA, Samra JS, Humphreys SM, Frayn KN. Release of individual fatty acids from human adipose tissue in vivo after an overnight fast. J Lipid Res. 1996;37:1842–1848. [PubMed] [Google Scholar]
- 43. Bizzi A, Codegoni SA. Salicylate, a powerful inhibitor of free fatty acid release. Nature. 1964;204:1205. [DOI] [PubMed] [Google Scholar]
- 44. Micossi P, Pontiroli AE, Baron SH, Tamayo RC, Lengel F, Bevilacqua M, Raggi U, Norbiato G, Foà PP. Aspirin stimulates insulin and glucagon secretion and increases glucose tolerance in normal and diabetic subjects. Diabetes. 1978;27:1196–1204. [DOI] [PubMed] [Google Scholar]
- 45. Carlson LA, Osiman J. Effect of sancylates on plasma free fatty acid in normal and diabetic subjects. Metab Clin Exp. 1961;10:781–787. [PubMed] [Google Scholar]
- 46. Carlson LA, Ostman J. Inhibition of the mobilization of free fatty acids from adipose tissue in diabetes. II. Effect of nicotinic acid and acetylsalicylate on blood glucose in human diabetics. Acta Med Scand. 1965;178:71–79. [DOI] [PubMed] [Google Scholar]
- 47. Giugliano D, Luyckx AS, Lefebvre PJ. Effects of acetylsalicylic acid on blood glucose, plasma FFA, glycerol, 3‐hydroxybutyrate, alanine, C‐peptide, glucagon and growth hormone responses to arginine in insulin‐dependent diabetics. Diabetes Metab. 1980;6:39–46. [PubMed] [Google Scholar]
- 48. Vik‐Mo H, Mjøs OD. Mechanisms for inhibition of free fatty acid mobilization by nicotinic acid and sodium salicylate in canine subcutaneous adipose tissue in situ. Scand J Clin Lab Invest. 1978;38:209–216. [DOI] [PubMed] [Google Scholar]
- 49. Nixon M, Wake DJ, Livingstone DE, Stimson RH, Esteves CL, Seckl JR, Chapman KE, Andrew R, Walker BR. Salicylate downregulates 11β‐HSD1 expression in adipose tissue in obese mice and in humans, mediating insulin sensitization. Diabetes. 2012;61:790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pradhan AD, Cook NR, Manson JE, Ridker PM, Buring JE. A randomized trial of low‐dose aspirin in the prevention of clinical type 2 diabetes in women. Diabetes Care. 2009;32:3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hellmuth C, Demmelmair H, Schmitt I, Peissner W, Blüher M, Koletzko B. Association between plasma nonesterified fatty acids species and adipose tissue fatty acid composition. PLoS One. 2013;8:e74927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Buchanan MR, Haas TA, Lagarde M, Guichardant M. 13‐Hydroxyoctadecadienoic acid is the vessel wall chemorepellant factor, LOX. J Biol Chem. 1985;260:16056–16059. [PubMed] [Google Scholar]
- 53. Camacho M, Godessart N, Antón R, García M, Vila L. Interleukin‐1 enhances the ability of cultured human umbilical vein endothelial cells to oxidize linoleic acid. J Biol Chem. 1995;270:17279–17286. [DOI] [PubMed] [Google Scholar]
- 54. Nugteren DH, Kivits GA. Conversion of linoleic acid and arachidonic acid by skin epidermal lipoxygenases. Biochim Biophys Acta. 1987;921:135–141. [DOI] [PubMed] [Google Scholar]
- 55. Claeys M, Kivits GA, Christ‐Hazelhof E, Nugteren DH. Metabolic profile of linoleic acid in porcine leukocytes through the lipoxygenase pathway. Biochim Biophys Acta. 1985;837:35–51. [DOI] [PubMed] [Google Scholar]
- 56. Daret D, Blin P, Larrue J. Synthesis of hydroxy fatty acids from linoleic acid by human blood platelets. Prostaglandins. 1989;38:203–214. [DOI] [PubMed] [Google Scholar]
- 57. Buchanan MR, Butt RW, Magas Z, van Ryn J, Hirsh J, Nazir DJ. Endothelial cells produce a lipoxygenase derived chemo‐repellent which influences platelet/endothelial cell interactions–effect of aspirin and salicylate. Thromb Haemost. 1985;53:306–311. [PubMed] [Google Scholar]
- 58. Buchanan MR, Bertomeu MC, Haas TA, Orr FW, Eltringham‐Smith LL. Localization of 13‐hydroxyoctadecadienoic acid and the vitronectin receptor in human endothelial cells and endothelial cell/platelet interactions in vitro. Blood. 1993;81:3303–3312. [PubMed] [Google Scholar]
- 59. Truitt A, McNeill G, Vanderhoek JY. Antiplatelet effects of conjugated linoleic acid isomers. Biochim Biophys Acta. 1999;1438:239–246. [DOI] [PubMed] [Google Scholar]
- 60. Kaddurah‐Daouk R, Bogdanov MB, Wikoff WR, Zhu H, Boyle SH, Churchill E, Wang Z, Rush AJ, Krishnan RR, Pickering E, Delnomdedieu M, Fiehn O. Pharmacometabolomic mapping of early biochemical changes induced by sertraline and placebo. Transl Psychiatry. 2013;3:e223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary Methods.
Figure S1. Oxylipid biosynthetic pathways (adapted from (1)). Metabolites detected in our samples are in bold.