

Abstract
Background
Patients with dilated cardiomyopathy (DCM) may present with ventricular arrhythmias early in the disease course, unrelated to the severity of left ventricular dysfunction. These patients may be classified as having an arrhythmogenic DCM (AR‐DCM). We investigated the phenotype and natural history of patients with AR‐DCM.
Methods and Results
Two hundred eighty‐five patients with a recent diagnosis of DCM (median duration of the disease 1 month, range 0 to 7 months) and who had Holter monitoring at baseline were comprehensively evaluated and followed for 107 months (range 29 to 170 months). AR‐DCM was defined by the presence of ≥1 of the following: unexplained syncope, rapid nonsustained ventricular tachycardia (≥5 beats, ≥150 bpm), ≥1000 premature ventricular contractions/24 hours, and ≥50 ventricular couplets/24 hours, in the absence of overt heart failure. The primary end points were sudden cardiac death (SCD), sustained ventricular tachycardia (SVT), or ventricular fibrillation (VF). The secondary end points were death from congestive heart failure or heart transplantation. Of the 285 patients, 109 (38.2%) met criteria for AR‐DCM phenotype. AR‐DCM subjects had a higher incidence of SCD/SVT/VF compared with non–AR‐DCM patients (30.3% vs 17.6%, P=0.022), with no difference in the secondary end points. A family history of SCD/SVT/VF and the AR‐DCM phenotype were statistically significant and cumulative predictors of SCD/SVT/VF.
Conclusions
One‐third of DCM patients may have an arrhythmogenic phenotype associated with increased risk of arrhythmias during follow‐up. A family history of ventricular arrhythmias in DCM predicts a poor prognosis and increased risk of SCD.
Keywords: arrhythmia, cardiomyopathy, prognosis, sudden death
In patients with dilated cardiomyopathy (DCM), ventricular arrhythmias may occur in the absence of signs and symptoms of overt heart failure and may not be related to the severity of left ventricular (LV) dysfunction. An arrhythmogenic trait associated with increased risk of sudden cardiac death (SCD) is well known in cardiomyopathies such as arrhythmogenic right ventricular (RV) cardiomyopathy (ARVC) and left‐dominant arrhythmogenic cardiomyopathy,1, 2 as well as in hypertrophic cardiomyopathy3 and LV noncompaction,4, 5, 6, 7 and is used for the purpose of risk stratification. However, the specific criteria for “arrhythmogenic” DCM (AR‐DCM) are lacking. As a consequence, in DCM, clinicians often face challenges in the appropriate selection of candidates for an implantable cardioverter‐defibrillator (ICD) for the prevention of SCD in the presence of an arrhythmogenic profile or positive family history of SCD or rapid ventricular tachycardia in the absence of heart failure or significant LV dysfunction. We investigated the arrhythmogenic phenotype in a large cohort of DCM patients, based on specific diagnostic criteria, to better understand the prevalence, natural history, and prognostic implications of AR‐DCM.
Methods
Study Population
We analyzed 285 patients with a recent diagnosis of DCM (the median duration of the disease was 1 month [range 0 to 7 months]) who were prospectively enrolled in our Familial Cardiomyopathy Registry and who had 24‐hour Holter monitoring data within 12 months from enrollment. We excluded 183 patients for whom a baseline Holter monitoring was either never performed or performed >1 year from enrollment. The enrollment period was from 1991 to 2012. Informed consent was obtained from all enrolled subjects, and the respective institutional review committees approved the study. Enrolled patients had a physical examination, electrocardiogram, echocardiogram, and laboratory investigations performed according to published guidelines.8, 9, 10
None of the patients was treated with amiodarone at the time of Holter monitoring. Additional clinically indicated studies were performed, including right‐ and left‐side heart catheterization, ventriculography, coronary angiography, endomyocardial biopsy, and neuromuscular evaluation.
Clinical Definitions
Criteria for the diagnosis of DCM were the presence of LV fractional shortening <25% and/or LV ejection fraction (LVEF) <45%, and LV end‐diastolic dimensions (LVEDD) >117% of the predicted value based on the Henry formula.8, 11
Exclusion criteria included arterial hypertension (>140/90 mm Hg),12 coronary artery disease (obstruction >50% of the luminal diameter in a major branch by coronary angiography), history of chronic excess alcohol consumption (>100 g/d), tachycardia‐induced cardiomyopathy, systemic diseases affecting short‐term prognosis, pericardial disease, congenital heart disease, and cor pulmonale.8, 13
Familial DCM was defined by the presence of ≥2 family members affected by DCM.8, 9, 10 AR‐DCM phenotype was diagnosed by the presence of 1 of the following: (1) unexplained syncope (likely due to ventricular tachyarrhythmia),2, 14, 15, 16 (2) rapid nonsustained ventricular tachycardia (NSVT) defined as ≥5 consecutive ventricular beats,17 lasting <30 seconds, with a rate ≥150/min on 24‐hour Holter monitoring,18 (3) ≥1000 premature ventricular contractions (PVCs) in 24 hours1 or (4) ≥50 couplets in 24 hours.19 ICD implantation had been performed for primary prevention in selected patients with DCM considered at high risk for SCD (ie, persistent LV dysfunction with LVEF ≤35% and New York Heart Association class II or III while being treated with optimal medical therapy).
End points
The primary end point was the occurrence of SCD or rapid and sustained ventricular arrhythmias. Specifically, SCD was defined as witnessed sudden cardiac death with or without documented ventricular fibrillation (VF) or death within 1 hour of acute symptoms or nocturnal death with no antecedent history of immediate worsening symptoms. Ventricular arrhythmias were defined as sustained (≥30 seconds, hemodynamically symptomatic) ventricular tachycardia (SVT), VF, appropriate SVT or VF treatment with implantable cardioverter‐defibrillator (ICD) (shock or antitachycardia pacing for termination of SVT ≥185 bpm), cardiopulmonary resuscitation after cardiac arrest, or SCD.14 The secondary end point was death due to heart failure (excluding SCD) or heart transplantation. A history of familial SCD was considered to be present if at least 1 family member (up to third degree) died suddenly at age younger than 60 years.14
Statistical Analysis
Clinical and demographic characteristics were compared by using the χ2 test for discrete variables expressed as proportions (or the Fisher exact test when necessary), the ANOVA test for continuous parameters, and the Brown–Forsythe statistic when the assumption of equal variances did not hold. The nonparametric median test was used when appropriate. For the time‐to‐event analysis, we started from the date of birth until events or censoring.1, 20, 21, 22 Patients with no events were censored at the date of the last available follow‐up.
Cause‐specific event‐free survival curves for SCD/SVT/VF and heart failure death (excluding SCD)/heart transplantation were estimated and plotted by using the Kaplan–Meier method, and the log‐rank test was applied to investigate for differences in survival. Cox regression analysis was then used to identify predictors for the SCD or malignant ventricular arrhythmias end point.
To mark the family code as a cluster indicator in the Cox model, we used the “cluster” argument of the “coxph” function in R: this is a special function used in the context of survival models. It identifies correlated groups of observations and is used on the right hand side of the formula. Use of the argument “cluster” in the survival model implies that robust sandwich variance estimators are calculated (the resulting variance is what is known as the “working independence” variance in a GEE model). The proportional hazards assumption of the Cox model was verified by using the Grambsch–Therneau test.23 Hazard ratios and 95% CIs were calculated; variables reaching P<0.05 at univariable analysis were included in the multivariable model. Statistical analyses were performed with use of the IBM SPSS Statistical Package 20.0 and the R statistical package version 2.14.1. A P value of ≤0.05 was considered statistically significant.
Results
Prevalence and Phenotype of AR‐DCM
Of 285 DCM patients with Holter monitor data at baseline, 109 (38.2%) fulfilled the prespecified diagnostic criteria of AR‐DCM. One hundred thirty (45.6%) subjects had a familial history of DCM: 71 patients were clustered in 30 families, with an average of 2.4 individuals per family (median of 2), and 59 were the only family members studied. The remaining 155 patients were sporadic “nonfamilial” cases. Table 1 shows the arrhythmogenic profile of AR‐DCM patients. Table 2 shows the baseline characteristics of AR‐DCM versus non–AR‐DCM patients. No significant clinical, echocardiographic or electrocardiographic difference was found between the 2 groups. None of the patients had significant RV involvement or sufficient criteria to fulfill the 2010 revised criteria for ARVC.24
Table 1.
Arrhythmic Profile of 109 AR‐DCM Patients
| Criteria | AR‐DCM Patients, n (%) |
|---|---|
| NSVT (≥5 beats, ≥150 bpm) | 43 (39.4) |
| ≥1000 PVCs/24 h | 90 (82.6) |
| ≥50 Couplets/24 h | 40 (36.7) |
| Syncope | 8 (7.3) |
AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; NSVT, nonsustained ventricular tachycardia; PVCs, premature ventricular contractions.
Table 2.
Comparison of Clinical and Instrumental Characteristics at Enrollment of AR‐DCM and Non–AR‐DCM Patients
| Baseline Characteristics | AR‐DCM (n=109) | Non–AR‐DCM (n=176) | P Valuea |
|---|---|---|---|
| Male sex, n (%) | 76 (69.7) | 126 (71.6) | 0.736 |
| Age at diagnosis, y | 41±13 | 41±14 | 0.955 |
| LVEDD, mm | 66±10 | 65±11 | 0.476 |
| LVEF, % | 34±11 | 32±14 | 0.425 |
| Complete LBBB, n (%) | 22 (20.2) | 47 (26.7) | 0.222 |
| Complete RBBB, n (%) | 4 (3.7) | 7 (4) | 1.000 |
| Inverted T waves in leads V2 to V3, n (%) | 2 (1.8) | 7 (4) | 0.490 |
| QRS >110 ms in leads V1 to V3, n (%) | 36 (33) | 66 (37.5) | 0.468 |
| Family history of SCD/SVT/VF, n (%) | 11 (10.1) | 14 (7.9) | 0.535 |
| β‐Blocker therapy, n (%) | 99 (90.8) | 153 (86.9) | 0.527 |
| ACEI therapy, n (%) | 88 (80.7) | 138 (78.4) | 0.843 |
AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; LBBB, left bundle branch block; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; RBBB, right bundle branch block; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation; ACEI, angiotensin‐converting enzyme inhibitor.
Between groups.
Natural History of the AR‐DCM Phenotype
During long‐term follow‐up (median 107 months, range 29 to 170 months), a greater number of AR‐DCM patients had SCD/SVT/VF (33/109 cases, 30.3%) than did non–AR‐DCM patients (31/176, 17.6%). They also had worse event‐free long‐term survival when considering SCD/SVT/VF, as shown in Figure 1A (P=0.02). There was no significant difference between AR‐DCM (23/109, 21.1%) and non–AR‐DCM (37/176, 21%) patients regarding the secondary end points of heart failure death or heart transplantation (Figure 1B, P=0.948). When using the date of enrollment in the registry for survival analysis, the SCD/SVT/VF survival curves for AR‐DCM and non–AR‐DCM diverged and became significantly different during follow‐up (P=0.001 at 100 months), as shown in Figure 2.
Figure 1.
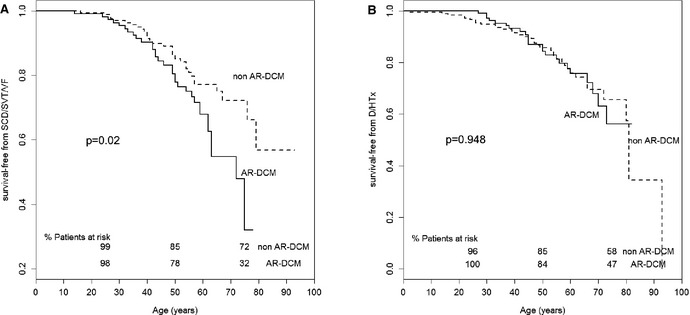
Kaplan–Meier event‐free survival. Comparison of (A) SCD/SVT/VF‐free survival (primary end point) and (B) D/HTx‐free survival (secondary end point) between AR‐DCM patients and non–AR‐DCM patients. AR‐DCM patients have a greater risk of life‐threatening arrhythmic events compared with the other DCM patients (P=0.02). Follow‐up from birth to end point/last follow‐up evaluation. Survival rates (as percentage of patients at risk) are provided at ages 25, 50, and 75 years. AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; D/HTx, heart failure death (excluding SCD)/heart transplant; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation.
Figure 2.
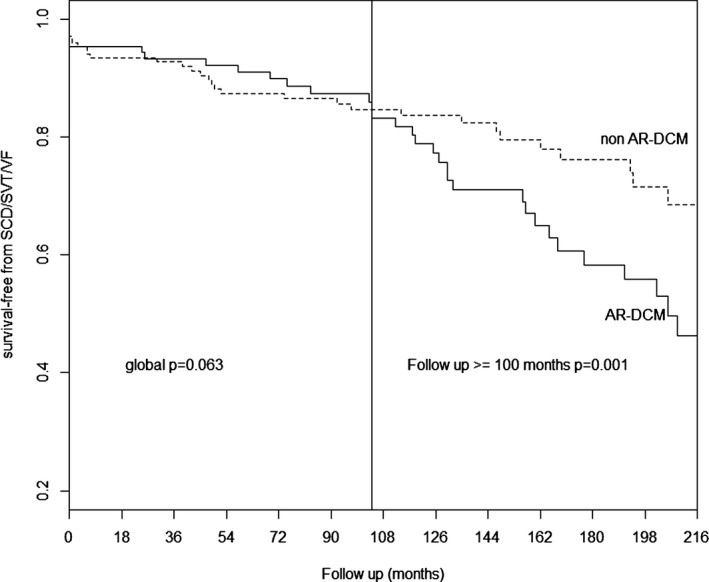
Kaplan–Meier event‐free survival. Comparison of SCD/SVT/VF ‐free survival (primary end point) between AR‐DCM patients and non–AR‐DCM patients. With the progress of follow‐up, AR‐DCM patients have a greater risk of ventricular arrhythmic events compared with to the other DCM patients (P=0.001 at 100 months). Follow‐up from enrollment/last follow‐up evaluation. AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation.
A total of 85 (29.8%) of 285 study patients―45 (41%) of 109 AR‐DCM and 40 (23%) of 176 non–AR‐DCM patients―received an ICD during follow‐up (P=0.001). Of these, 25 patients experienced ICD therapy during the follow‐up. There was a trend toward a higher number of patients who received ≥1 appropriate ICD therapy during follow‐up in the AR‐DCM cohort compared with non–AR‐DCM: 35.6% (n=16) versus 22.5% (n=9), respectively (P=0.19).
In the AR‐DCM cohort, there were no significant differences in the incidence of SCD/SVT/VF between patients with LVEF ≤35% and >35% at enrollment: 31.2% (20/64 events) and 28.9% (13/45 events), respectively; P=0.79. Of 285 subjects, 73 (25.6%)―36 AR‐DCM and 37 non–AR‐DCM―had an LVEF between 35% and ≤45% at enrollment. Of these, 15 subjects had SCD or SVT/VF: 30.1% (n=11) AR‐DCM and 10.8% (n=4) non–AR‐DCM (P=0.037).
Prognostic Impact of AR‐DCM Phenotype
Table 3 shows the results of the univariable and multivariable analysis of the association with SCD or SVT/VF in the overall DCM population. The AR‐DCM phenotype (hazard ratio 1.81, 95% CI 1.10 to 2.97, P=0.02) and a positive family history of SCD or SVT/VF (hazard ratio 2.21, 95% CI 1.04 to 4.66, P=0.038) were identified as the only early significant predictors for SCD or SVT/VF in the overall DCM population. Table 4 shows the cross‐tabulation of family history of SCD/SVT/VF versus AR‐DCM (χ2 P=0.535).
Table 3.
Cox Univariable and Multivariable Analysis for SCD/SVT/VF in the Entire Study Population (285 Patients)
| Univariable Analysis | Multivariable Analysis | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P Valuea | HR | 95% CI | P Valuea | |
| AR‐DCM | 1.77 | 1.08–2.91 | 0.024 | 1.81 | 1.10–2.97 | 0.020 |
| Family history of SCD/SVT/VF | 2.11 | 1.00–4.45 | 0.049 | 2.21 | 1.04–4.66 | 0.038 |
| Male sex | 1.64 | 0.91–2.97 | 0.102 | |||
| LVEF, % | 0.99 | 0.97–1.02 | 0.74 | |||
| LVEDD, mm | 1.02 | 0.99–1.04 | 0.20 | |||
| Inverted T waves in leads V2 to V3 | 0.51 | 0.70–3.78 | 0.50 | |||
| QRS >110 ms in leads V1 to V3 | 0.73 | 0.44–1.22 | 0.23 | |||
| Family history of DCM | 1.35 | 0.83–2.22 | 0.23 | |||
| Family history of AR‐DCM | 1.12 | 0.62–2.03 | 0.71 | |||
| Complete LBBB | 0.61 | 0.34–1.11 | 0.11 | |||
| Complete RBBB | 0.88 | 0.21–3.63 | 0.86 | |||
SCD/SVT/VF indicates sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation; HR, hazard ratio; AR‐DCM, arrhythmogenic dilated cardiomyopathy; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LBBB, left bundle branch block; RBBB, right bundle branch block;.
Between groups.
Table 4.
Cross‐Tabulation of Family History of SCD/Ventricular Arrhythmias Versus AR‐DCM
| Family History of SCD/SVT/VF | Total | |||
|---|---|---|---|---|
| Yes | No | |||
| AR‐DCM | ||||
| Yes | Number | 11 | 98 | 109 |
| % with family history of SCD/SVT/VF | 44% | 37.7% | 38.2% | |
| No | Number | 14 | 162 | 176 |
| % with family history of SCD/SVT/VF | 56% | 62.3% | 61.8% | |
| Total | Number | 25 | 260 | 285 |
| % with family history of SCD/SVT/VF | 100% | 100% | 100% | |
AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation.
These 2 variables showed an additive prognostic effect on the arrhythmic risk (Figure 3, Table 3) The P value of the proportional hazard test (PH) for the Cox model was 0.23. The P value for family history of SCD/SVT/VF was 0.65, and the P value for AR‐DCM was 0.09, indicating a trend of nonproportionality of the hazards for the AR‐DCM effect.
Figure 3.
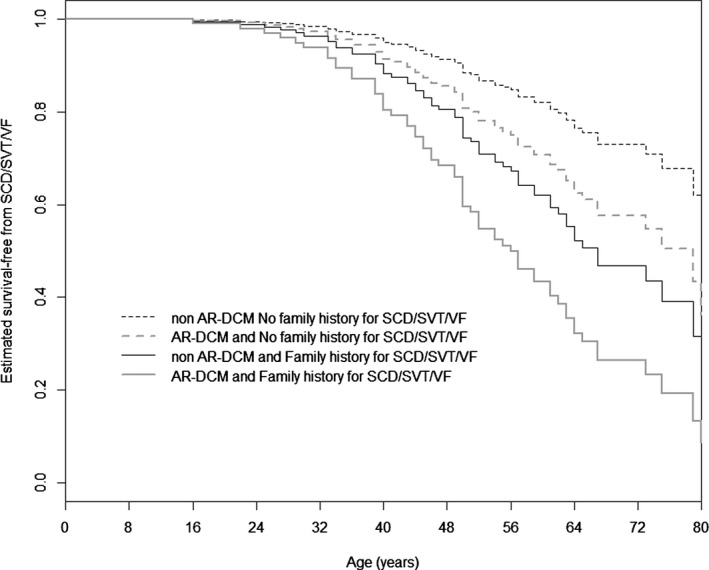
Cox‐estimated SCD/ventricular arrhythmias–free survival stratified by 2 risk factors. Cox‐estimated SCD/SVT/VF‐free survival stratified by 2 risk factors, family history of SCD or ventricular arrhythmias and AR‐DCM diagnosis, in the overall DCM population (285 patients). The AR‐DCM phenotype (hazard ratio 1.81, 95% CI 1.10–2.97, P=0.02) and family history of SCD or ventricular arrhythmias (hazard ratio 2.21, 95% CI 1.04–4.66, P=0.038) show an additive prognostic effect on mortality for arrhythmic events. AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation.
Baseline LVEF, LVEDD, male gender, family history of DCM or AR‐DCM, inverted T waves in leads V1 to V3, QRS >110 ms in leads V1 to V3, complete right bundle branch block, and complete left bundle branch block were not significantly related to the long‐term risk of SCD or ventricular arrhythmias.
Finally, we examined if each of the arrhythmic criteria used to define AR‐DCM (NSVT, >1000 PVCs/24 h, >50 ventricular couplets/24 h at Holter monitoring, history of unexplained syncope) had different prognostic significance in the AR‐DCM subgroup. The cumulative effect of having 1, 2, 3, or 4 criteria was not significant (Figure 4).
Figure 4.
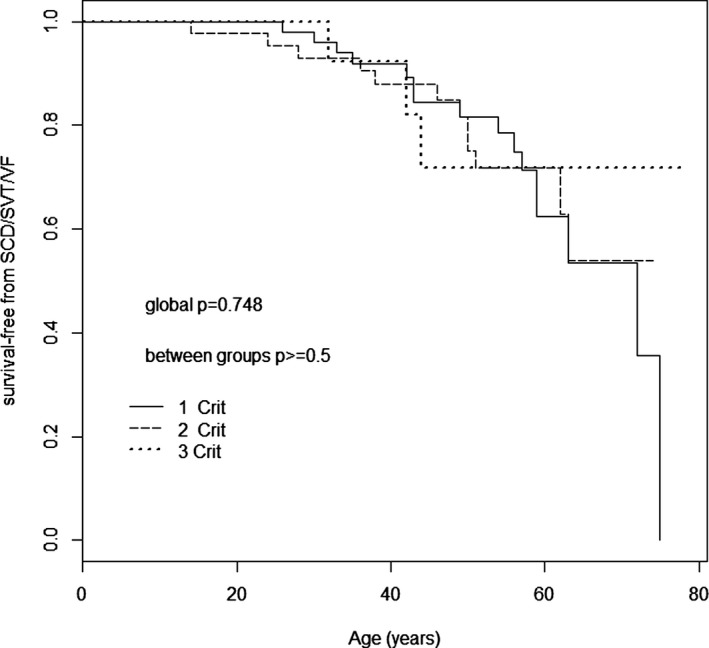
Kaplan–Meier SCD/ventricular arrhythmias–free survival. Kaplan–Meier SCD ventricular arrhythmias–free survival stratified by AR‐DCM criteria in the AR‐DCM population (109 patients). The cumulative effect of having 1, 2, 3, or 4 AR‐DCM criteria (NSVT ≥5 beats and ≥150 bpm; PVCs ≥1000/24 h; ventricular couplets ≥50/24 hours; syncope) is not significant. AR‐DCM indicates arrhythmogenic dilated cardiomyopathy; NSVT, nonsustained ventricular tachycardia; PVCs, premature ventricular contractions; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation.
Discussion
Phenotype and Prevalence of AR‐DCM
This was a study of a subset of DCM patients with a prominent arrhythmic phenotype (arrhythmogenic DCM or AR‐DCM), in the early stages of the disease, characterized by frequent ventricular arrhythmias and unexplained syncope in the absence of overt heart failure. In our large DCM study cohort, extensively investigated and followed for >20 years, the prevalence of the AR‐DCM phenotype was approximately one‐third of the overall DCM population. Therefore, an arrhythmogenic phenotype occurs in a sizable proportion of DCM patients. The long‐term natural history of AR‐DCM was characterized by a significantly higher rate of major arrhythmic events (SCD, SVT, VF), compared with the other DCM patients. The “AR‐DCM phenotype” and the family history of SCD or SVT/VF were found to be prognostic factors useful in the risk stratification of DCM patients, with an additive prognostic effect on the arrhythmic risk. Indeed, as illustrated in Figure 2, the risk of SCD/SVT/VF in the AR‐DCM group increased over time.
Interestingly, the prognosis of AR‐DCM was not related to the severity of LV dysfunction and LV dilatation. When we stratified the AR‐DCM cohort by LVEF ≤35% and LVEF >35%, there were no significant differences in the risk of SCD/SVT/VF. Neither LVEF nor LVEDD was a predictor of SCD or SVT/VF in the multivariable analysis. In addition, in the subset of our patients with LVEF between 35% and 45%, AR‐DCM patients had a higher incidence of major arrhythmic events (SCD/SVT/VF) compared with non–AR‐DCM subjects, thus reinforcing the concept that AR‐DCM may be associated with SCD even in those patients with only mild systolic dysfunction. AR‐DCM was not associated with poorer long‐term prognosis due to nonarrhythmic events, including death due to heart failure and heart transplantation. These findings suggest that subjects with AR‐DCM phenotype represent a distinct subset of DCM patients, rather than a different stage of the disease. In fact, the AR‐DCM phenotype shows similarities and overlap with other arrhythmogenic cardiomyopathies, such as left dominant arrhythmogenic cardiomyopathy and arrhythmogenic right ventricular (RV) cardiomyopathy.1, 25
Prognostic Role of Family History
Another predictor of long‐term life‐threatening arrhythmic events identified in our study was a family history of SCD or ventricular arrhythmias. This finding emphasizes the importance of the systematic multigenerational analysis of family history in DCM.8, 10 Although the role of family history is well established in other genetically determined arrhythmogenic cardiomyopathies and represents an important element for risk stratification, management, and decision‐making, this finding is novel in DCM and may represent an important risk factor for the primary prevention of SCD.1, 2, 25
Diagnostic Criteria of AR‐DCM
The inclusion criteria of the AR‐DCM cohort used in our study (Table 1) were stringent, based on other investigations in comparable study populations. The duration of NSVT is an established risk factor of major arrhythmic events and SCD in DCM. Grimm et al found that NSVT with a duration of ≥5 beats was significantly associated with increased risk of major arrhythmic events including SCD.17 Furthermore, the heart rate in NSVT has been arbitrary,26, 27, 28 ranging from ≥100 to 150 bpm, and not associated with increased risk of life‐threatening arrhythmias and SCD in DCM.17, 18, 29 In our study, we arbitrarily defined NSVT as ≥5 consecutive ventricular premature beats with a rapid heart rate (≥150 bpm), corresponding to the median ventricular rate previously reported in our DCM population.29 The definition of PVCs >1000/24 h and ventricular couplets were based on the experience with left‐dominant arrhythmogenic cardiomyopathy.1 In our inclusion criteria for AR‐DCM, as in previous studies on laminopathies,14 we included a history of syncope, an established risk factor for major arrhythmic events in ARVC30 and hypertrophic cardiomyopathy.31, 32
Clinical Implications
Our results suggest that in DCM, an accurate family history is important not only for diagnostic purposes8, 9 but also for improved prognostic assessment. In DCM, a family history of SCD/SVT/VF is associated with an increased risk of life‐threatening arrhythmias, a concept well established in other forms of arrhythmogenic cardiomyopathies such as ARVC33, 34 and hypertrophic cardiomyopathy.32, 35 Even though none of our AR‐DCM patients fulfilled new task force criteria of ARVC24 with LV involvement, the potential overlap with desmosomal diseases (ARVC, arrhythmogenic cardiomyopathy, left‐dominant arrhythmogenic cardiomyopathy), considered to be distinct disorders,10 warrants further investigations.36
Our findings support the use of systematic Holter monitoring in DCM for a more accurate phenotypic characterization to improve the prognostic stratification.
DCM patients with both an arrhythmogenic phenotype and a family history of SCD or ventricular arrhythmias are the highest risk group and should be considered candidates for careful follow‐up and possibly for ICD implantation for primary prevention, regardless of their LVEF or LVEDD.15 In our study, the risk of arrhythmic events was associated with these risk factors and significantly increased when they were combined (see Figure 3).
Study Limitations and Future Perspectives
The prevalence of AR‐DCM in our study may be overestimated because we enrolled patients with familial or suspected familial DCM, and we analyzed only patients who had baseline Holter monitoring. However, the prevalence of AR‐DCM in the Italian subcohort, where Holter monitoring was systematically performed at baseline and during follow‐up, was not significantly different (96/244 patients, 39%).
Patients excluded from the study because of the lack of baseline Holter monitoring (n=183) had slightly less severe DCM in terms of LV dysfunction, dilatation, and dyssynchrony (Table 5). However, these differences seem clinically marginal.13
Table 5.
Comparison of Clinical and Instrumental Characteristics at Enrollment of Our Study Population and Patients Excluded From the Study Because of the Lack of Baseline Holter Monitoring Data
| Baseline Characteristics | Our Study Population (n=285) | Patients Excluded From the Study (n=183) | P Valuea |
|---|---|---|---|
| Male sex, n (%) | 202 (70.9) | 103 (56.3) | 0.001 |
| Age at diagnosis, y | 41±14 | 43±16 | 0.177 |
| LVEDD, mm | 66±10 | 61±11 | <0.001 |
| LVEF, % | 33±13 | 36±14 | 0.007 |
| Complete LBBB, n (%) | 69 (24.2) | 22 (12) | 0.004 |
| Complete RBBB, n (%) | 11 (3.9) | 5 (3) | 0.794 |
| QRS >110 ms in V1 to V3, n (%) | 102 (36.3) | 61 (36.7) | 0.924 |
| Family history of SCD/SVT/VF, n (%) | 25 (8.8) | 23 (12.6) | 0.187 |
| β‐Blocker therapy, n (%) | 252 (88.4) | 131 (71.6) | 0.103 |
| ACEI therapy, n (%) | 226 (80.1) | 131 (71.6) | 0.395 |
LVEF indicates left ventricular ejection fraction; LVEDD, left ventricular end‐diastolic diameter; LBBB, left bundle branch block; RBBB, right bundle branch block; SCD/SVT/VF, sudden cardiac death, sustained ventricular tachycardia, and ventricular fibrillation; ACEI, angiotensin‐converting enzyme inhibitor;
Between groups.
Patients with an arrhythmogenic phenotype may be more likely to receive an ICD and ICD therapies are known to exceed the number of observed sudden deaths. Antitachycardia pacing as a surrogate of arrhythmic death is controversial. In our study population, of 25 ICD therapies, only 4 were antitachycardia pacing and preceded an arrhythmic storm in 1 case. In the remaining 3 cases, antitachycardia pacing treatment was for rapid and prolonged episodes of sustained VT (>185 bpm), consistent with potentially life‐threatening arrhythmias.
Special investigations, such with cardiac magnetic resonance, were not systematically available due to the extended enrollment period (1991–2012). The origin of ventricular arrhythmias, from either the left ventricle or right ventricle, could not be fully evaluated due to incomplete availability of data on their morphology. The definition of family history of SCD or rapid sustained ventricular arrhythmias (which included up to third‐degree relatives younger than age 60) could have introduced a bias due to nonfamilial/genetic causes, such as coronary heart disease.
The limited availability of genotyping data prevented analysis of the prognostic implications of specific gene mutations in the natural history of AR‐DCM. Large‐scale genotype–phenotype association studies are ongoing to confirm our observations. Endomyocardial biopsy was not systematically performed in this DCM cohort. Further studies should address the potential risk stratification differences in fibrosis, inflammation, and fat infiltration between AR‐DCM and non–AR‐DCM patients. Finally, the study population was enrolled in tertiary referral centers for cardiomyopathies and heart failure, potentially introducing a selection bias that limits the generalizability of extending our data to the general DCM population.
Conclusions
We have studied a specific subgroup of DCM patients with a predominant arrhythmogenic phenotype, AR‐DCM. Our results indicate that AR‐DCM is frequent and is characterized by a high risk of life‐threatening arrhythmias, while the long‐term incidence of heart failure and heart transplantation appears comparable to that of other DCM patients. Based on these data, we suggest that a comprehensive evaluation of DCM patients by Holter monitoring and family history, with particular attention to SCD, will improve the prognostic stratification and the global management of DCM patients. The subgroup of overall DCM patients who have an AR‐DCM phenotype should be considered at increased risk of future events compared with the remaining DCM group. The possible genetic overlap with other arrhythmogenic cardiomyopathies, such as ARVC, left‐dominant arrhythmogenic cardiomyopathy, and channelopathies, needs to be further explored.
Sources of Funding
This study was supported by National Institutes of Health grants 1R01HL116906 to Drs Towbin, Mestroni, Saffitz, Zareba, and Marcus; UL1 RR025780 and R01 HL69071 to Dr Mestroni; and K23 JL067915 and 1R01HL109209‐01A1 to Dr Taylor. This work was supported in part by a Trans‐Atlantic Network of Excellence grant from the Leducq Foundation (14 CVD 03). CRTrieste and Generali Assicurazioni Foundations support to Dr Sinagra are gratefully acknowledged.
Disclosures
None.
Acknowledgments
The authors thank the family members for their participation in these studies.
(J Am Heart Assoc. 2015;4:e002149 doi: 10.1161/JAHA.115.002149)
The manuscript's contents have been presented as an abstract at the 2013 American Society of Human Genetics in Boston, MA, October 22 to 26, 2013; 2013 Scientific Sessions of the American Heart Association in Dallas, TX, November 16 to 20, 2013; and American College of Cardiology (ACC) Scientific Sessions, March 29 to 31, 2014, in Washington, DC.
References
- 1. Sen‐Chowdhry S, Syrris P, Prasad SK, Hughes SE, Merrifield R, Ward D, Pennell DJ, McKenna WJ. Left‐dominant arrhythmogenic cardiomyopathy: an under‐recognized clinical entity. J Am Coll Cardiol. 2008;52:2175–2187. [DOI] [PubMed] [Google Scholar]
- 2. Sen‐Chowdhry S, Syrris P, Pantazis A, Quarta G, McKenna WJ, Chambers JC. Mutational heterogeneity, modifier genes, and environmental influences contribute to phenotypic diversity of arrhythmogenic cardiomyopathy. Circ Cardiovasc Genet. 2010;3:323–330. [DOI] [PubMed] [Google Scholar]
- 3. Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ. Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000;36:2212–2218. [DOI] [PubMed] [Google Scholar]
- 4. Ritter M, Oechslin E, Sutsch G, Attenhofer C, Schneider J, Jenni R. Isolated noncompaction of the myocardium in adults. Mayo Clin Proc. 1997;72:26–31. [DOI] [PubMed] [Google Scholar]
- 5. Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation. 1990;82:507–513. [DOI] [PubMed] [Google Scholar]
- 6. Oechslin EN, Attenhofer Jost CH, Rojas JR, Kaufmann PA, Jenni R. Long‐term follow‐up of 34 adults with isolated left ventricular noncompaction: a distinct cardiomyopathy with poor prognosis. J Am Coll Cardiol. 2000;36:493–500. [DOI] [PubMed] [Google Scholar]
- 7. Rigopoulos A, Rizos IK, Aggeli C, Kloufetos P, Papacharalampous X, Stefanadis C, Toutouzas P. Isolated left ventricular noncompaction: an unclassified cardiomyopathy with severe prognosis in adults. Cardiology. 2002;98:25–32. [DOI] [PubMed] [Google Scholar]
- 8. Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J. 1999;20:93–102. [DOI] [PubMed] [Google Scholar]
- 9. Hershberger RE, Cowan J, Morales A, Siegfried JD. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail. 2009;2:253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA; Heart Failure Society of America . Genetic evaluation of cardiomyopathy–a heart failure society of America practice guideline. J Card Fail. 2009;15:83–97. [DOI] [PubMed] [Google Scholar]
- 11. Henry WL, Gardin JM, Ware JH. Echocardiographic measurements in normal subjects from infancy to old age. Circulation. 1980;62:1054–1061. [DOI] [PubMed] [Google Scholar]
- 12. Mancia G, Fagard R, Narkiewicz K, Redon J, Zanchetti A, Bohm M, Christiaens T, Cifkova R, De Backer G, Dominiczak A, Galderisi M, Grobbee DE, Jaarsma T, Kirchhof P, Kjeldsen SE, Laurent S, Manolis AJ, Nilsson PM, Ruilope LM, Schmieder RE, Sirnes PA, Sleight P, Viigimaa M, Waeber B, Zannad F, Redon J, Dominiczak A, Narkiewicz K, Nilsson PM, Burnier M, Viigimaa M, Ambrosioni E, Caufield M, Coca A, Olsen MH, Schmieder RE, Tsioufis C, van de Borne P, Zamorano JL, Achenbach S, Baumgartner H, Bax JJ, Bueno H, Dean V, Deaton C, Erol C, Fagard R, Ferrari R, Hasdai D, Hoes AW, Kirchhof P, Knuuti J, Kolh P, Lancellotti P, Linhart A, Nihoyannopoulos P, Piepoli MF, Ponikowski P, Sirnes PA, Tamargo JL, Tendera M, Torbicki A, Wijns W, Windecker S, Clement DL, Coca A, Gillebert TC, Tendera M, Rosei EA, Ambrosioni E, Anker SD, Bauersachs J, Hitij JB, Caulfield M, De Buyzere M, De Geest S, Derumeaux GA, Erdine S, Farsang C, Funck‐Brentano C, Gerc V, Germano G, Gielen S, Haller H, Hoes AW, Jordan J, Kahan T, Komajda M, Lovic D, Mahrholdt H, Olsen MH, Ostergren J, Parati G, Perk J, Polonia J, Popescu BA, Reiner Z, Ryden L, Sirenko Y, Stanton A, Struijker‐Boudier H, Tsioufis C, van de Borne P, Vlachopoulos C, Volpe M, Wood DA. 2013 ESH/ESC guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Hypertension (ESH) and of the European Society of Cardiology (ESC). Eur Heart J. 2013;34:2159–2219. [DOI] [PubMed] [Google Scholar]
- 13. Merlo M, Pivetta A, Pinamonti B, Stolfo D, Zecchin M, Barbati G, Di Lenarda A, Sinagra G. Long‐term prognostic impact of therapeutic strategies in patients with idiopathic dilated cardiomyopathy: changing mortality over the last 30 years. Eur J Heart Fail. 2014;16:317–324. [DOI] [PubMed] [Google Scholar]
- 14. van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans‐van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, Jenkins S, Rowland C, Aslam U, Wilde AA, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Pinto YM. Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59:493–500. [DOI] [PubMed] [Google Scholar]
- 15. Kusumoto FM, Calkins H, Boehmer J, Buxton AE, Chung MK, Gold MR, Hohnloser SH, Indik J, Lee R, Mehra MR, Menon V, Page RL, Shen WK, Slotwiner DJ, Stevenson LW, Varosy PD, Welikovitch L. HRS/ACC/AHA expert consensus statement on the use of implantable cardioverter‐defibrillator therapy in patients who are not included or not well represented in clinical trials. Circulation. 2014;130:94–125. [DOI] [PubMed] [Google Scholar]
- 16. Phang RS, Kang D, Tighiouart H, Estes NA III, Link MS. High risk of ventricular arrhythmias in patients with nonischemic dilated cardiomyopathy presenting with syncope. Am J Cardiol. 2006;97:416–420. [DOI] [PubMed] [Google Scholar]
- 17. Grimm W, Christ M, Maisch B. Long runs of non‐sustained ventricular tachycardia on 24‐hour ambulatory electrocardiogram predict major arrhythmic events in patients with idiopathic dilated cardiomyopathy. Pacing Clin Electrophysiol. 2005;28(suppl 1):S207–S210. [DOI] [PubMed] [Google Scholar]
- 18. Watanabe J, Shinozaki T, Shiba N, Fukahori K, Koseki Y, Karibe A, Sakuma M, Miura M, Kagaya Y, Shirato K. Accumulation of risk markers predicts the incidence of sudden death in patients with chronic heart failure. Eur J Heart Fail. 2006;8:237–242. [DOI] [PubMed] [Google Scholar]
- 19. Paul T, Marchal C, Garson A Jr. Ventricular couplets in the young: prognosis related to underlying substrate. Am Heart J. 1990;119:577–582. [DOI] [PubMed] [Google Scholar]
- 20. Protonotarios N, Tsatsopoulou A, Anastasakis A, Sevdalis E, McKoy G, Stratos K, Gatzoulis K, Tentolouris K, Spiliopoulou C, Panagiotakos D, McKenna W, Toutouzas P. Genotype‐phenotype assessment in autosomal recessive arrhythmogenic right ventricular cardiomyopathy (Naxos disease) caused by a deletion in plakoglobin. J Am Coll Cardiol. 2001;38:1477–1484. [DOI] [PubMed] [Google Scholar]
- 21. Taylor MR, Sun AY, Davis G, Fiuzat M, Liggett SB, Bristow MR. Race, common genetic variation, and therapeutic response disparities in heart failure. JACC Heart Fail. 2014;2:561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lakdawala NK, Dellefave L, Redwood CS, Sparks E, Cirino AL, Depalma S, Colan SD, Funke B, Zimmerman RS, Robinson P, Watkins H, Seidman CE, Seidman JG, McNally EM, Ho CY. Familial dilated cardiomyopathy caused by an alpha‐tropomyosin mutation: the distinctive natural history of sarcomeric dilated cardiomyopathy. J Am Coll Cardiol. 2010;55:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grambsch PM, Therneau TM. Proportional hazards tests and diagnostics based on weighted residuals. Biometrika. 1994;81:515–526. [Google Scholar]
- 24. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sen‐Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61:233–253. [DOI] [PubMed] [Google Scholar]
- 26. Katritsis DG, Zareba W, Camm AJ. Nonsustained ventricular tachycardia. J Am Coll Cardiol. 2012;60:1993–2004. [DOI] [PubMed] [Google Scholar]
- 27. Pedersen CT, Kay GN, Kalman J, Borggrefe M, Della‐Bella P, Dickfeld T, Dorian P, Huikuri H, Kim YH, Knight B, Marchlinski F, Ross D, Sacher F, Sapp J, Shivkumar K, Soejima K, Tada H, Alexander ME, Triedman JK, Yamada T, Kirchhof P, Lip GY, Kuck KH, Mont L, Haines D, Indik J, Dimarco J, Exner D, Iesaka Y, Savelieva I, Ep‐Europace UK. EHRA/HRS/APHRS expert consensus on ventricular arrhythmias. Heart Rhythm. 2014;11:e166–e196. [DOI] [PubMed] [Google Scholar]
- 28. Authors/Task Force members , Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–2779. [DOI] [PubMed] [Google Scholar]
- 29. Zecchin M, Di Lenarda A, Gregori D, Merlo M, Pivetta A, Vitrella G, Sabbadini G, Mestroni L, Sinagra G. Are nonsustained ventricular tachycardias predictive of major arrhythmias in patients with dilated cardiomyopathy on optimal medical treatment? Pacing Clin Electrophysiol. 2008;31:290–299. [DOI] [PubMed] [Google Scholar]
- 30. Corrado D, Calkins H, Link MS, Leoni L, Favale S, Bevilacqua M, Basso C, Ward D, Boriani G, Ricci R, Piccini JP, Dalal D, Santini M, Buja G, Iliceto S, Estes NA III, Wichter T, McKenna WJ, Thiene G, Marcus FI. Prophylactic implantable defibrillator in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia and no prior ventricular fibrillation or sustained ventricular tachycardia. Circulation. 2010;122:1144–1152. [DOI] [PubMed] [Google Scholar]
- 31. Spirito P, Autore C, Rapezzi C, Bernabo P, Badagliacca R, Maron MS, Bongioanni S, Coccolo F, Estes NA, Barilla CS, Biagini E, Quarta G, Conte MR, Bruzzi P, Maron BJ. Syncope and risk of sudden death in hypertrophic cardiomyopathy. Circulation. 2009;119:1703–1710. [DOI] [PubMed] [Google Scholar]
- 32. O'Mahony C, Jichi F, Pavlou M, Monserrat L, Anastasakis A, Rapezzi C, Biagini E, Gimeno JR, Limongelli G, McKenna WJ, Omar RZ, Elliott PM; Hypertrophic Cardiomyopathy Outcomes I . A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk‐SCD). Eur Heart J. 2014;35:2010–2020. [DOI] [PubMed] [Google Scholar]
- 33. Silvano M, Corrado D, Kobe J, Monnig G, Basso C, Thiene G, Eckardt L. Risk stratification in arrhythmogenic right ventricular cardiomyopathy. Herzschrittmacherther Elektrophysiol. 2013;24:202–208. [DOI] [PubMed] [Google Scholar]
- 34. Corrado D, Leoni L, Link MS, Della Bella P, Gaita F, Curnis A, Salerno JU, Igidbashian D, Raviele A, Disertori M, Zanotto G, Verlato R, Vergara G, Delise P, Turrini P, Basso C, Naccarella F, Maddalena F, Estes NA III, Buja G, Thiene G. Implantable cardioverter‐defibrillator therapy for prevention of sudden death in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circulation. 2003;108:3084–3091. [DOI] [PubMed] [Google Scholar]
- 35. Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, Society of Thoracic Surgeons . 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2011;124:2761–2796. [DOI] [PubMed] [Google Scholar]
- 36. Haas J, Frese KS, Peil B, Kloos W, Keller A, Nietsch R, Feng Z, Muller S, Kayvanpour E, Vogel B, Sedaghat‐Hamedani F, Lim WK, Zhao X, Fradkin D, Kohler D, Fischer S, Franke J, Marquart S, Barb I, Li DT, Amr A, Ehlermann P, Mereles D, Weis T, Hassel S, Kremer A, King V, Wirsz E, Isnard R, Komajda M, Serio A, Grasso M, Syrris P, Wicks E, Plagnol V, Lopes L, Gadgaard T, Eiskjaer H, Jorgensen M, Garcia‐Giustiniani D, Ortiz‐Genga M, Crespo‐Leiro MG, Deprez RH, Christiaans I, van Rijsingen IA, Wilde AA, Waldenstrom A, Bolognesi M, Bellazzi R, Morner S, Bermejo JL, Monserrat L, Villard E, Mogensen J, Pinto YM, Charron P, Elliott P, Arbustini E, Katus HA, Meder B. Atlas of the clinical genetics of human dilated cardiomyopathy. Eur Heart J. 2015;36:1123–35a. [DOI] [PubMed] [Google Scholar]