

Abstract
Background
Transcription of the myristoylated alanine‐rich C kinase substrate (MARCKS) is upregulated in animal models of intimal hyperplasia. MARCKS knockdown inhibits vascular smooth muscle cell (VSMC) migration in vitro; however, the mechanism is as yet unknown. We sought to elucidate the mechanism of MARCKS‐mediated motility and determine whether MARCKS knockdown reduces intimal hyperplasia formation in vivo.
Methods and Results
MARCKS knockdown blocked platelet‐derived growth factor (PDGF)‐induced translocation of cortactin to the cell cortex, impaired both lamellipodia and filopodia formation, and attenuated motility of human coronary artery smooth muscle cells (CASMCs). Activation of the small GTPases, Rac1 and Cdc42, was prevented by MARCKS knockdown. Phosphorylation of MARCKS resulted in a transient shift of MARCKS from the plasma membrane to the cytosol. MARCKS knockdown significantly decreased membrane‐associated phosphatidylinositol 4,5‐bisphosphate (PIP 2) levels. Cotransfection with an intact, unphosphorylated MARCKS, which has a high binding affinity for PIP 2, restored membrane‐associated PIP 2 levels and was indispensable for activation of Rac1 and Cdc42 and, ultimately, VSMC migration. Overexpression of MARCKS in differentiated VSMCs increased membrane PIP 2 abundance, Rac1 and Cdc42 activity, and cell motility. MARCKS protein was upregulated early in the development of intimal hyperplasia in the murine carotid ligation model. Decreased MARKCS expression, but not total knockdown, attenuated intimal hyperplasia formation.
Conclusions
MARCKS upregulation increases VSMC motility by activation of Rac1 and Cdc42. These effects are mediated by MARCKS sequestering PIP 2 at the plasma membrane. This study delineates a novel mechanism for MARCKS‐mediated VSMC migration and supports the rational for MARCKS knockdown to prevent intimal hyperplasia.
Keywords: membrane lipids, migration, Rac1, restenosis, vascular smooth muscle
Introduction
Each year, there are over 7 million cardiovascular bypass and angioplasty procedures performed in the United States.1 More than one third of these procedures will have limited durability owing to the formation of intimal hyperplasia, the pathological response of a blood vessel to injury. Although there is no therapy to prevent intimal hyperplasia in bypass grafts, there are some strategies to prevent intimal hyperplasia after angioplasty and stenting.2 The current antiproliferative agents used to prevent intimal hyperplasia formation inhibit both smooth muscle cell and endothelial cell proliferation. Because these agents prevent the reestablishment of an intact endothelium, patients need to remain on dual antiplatelet therapy indefinitely. An improved strategy to prevent intimal hyperplasia in both surgical bypass and angioplasty and stenting is an unmet clinical need.
Pathologically, intimal hyperplasia is a cell‐migration–dependent, intimal outgrowth, in which vascular smooth muscle cells (VSMCs) dedifferentiate to a migratory, proliferative, and secretory phenotype. These dedifferentiated VSMCs migrate through the medial layer and invade the intima, where they proliferate and form the hyperplastic neointima.3 The mechanism by which VSMCs gain motility during intimal hyperplasia development is not fully understood. However, gene expression profiling in large mammal models of intimal hyperplasia suggest that regulation of actin cytoskeleton is a predominant process in the early phase of intimal hyperplasia.4, 5 Increased cytoskeletal actin assembly dynamics are known to promote cell migration. Targeting molecules associated with actin and cytoskeleton signaling and that restrain VSMC translation at the initial phase of restenosis could inhibit VSMC migration6, 7 and therefore is an appealing alternative therapeutic strategy to prevent intimal hyperplasia development.
The myristoylated alanine‐rich C‐kinase substrate (MARCKS) is a filamentous actin (F‐actin) binding protein that shuttles from cell membrane to cytoplasm upon signal transduction and regulates actin assembly dynamics during migration in multiple types of cells. MARCKS contains a unique F‐actin binding motif, the effector domain (ED domain).8 The ED domain and the flanking N‐terminal sequence are highly conserved among species. Twenty‐five amino acids long and rich in basic amino acids, the ED domain is positively charged and has high electrostatic affinity to the cell membrane and the membrane lipid messenger, phosphatidylinositol 4,5‐bisphosphate (PIP2).9 MARCKS is known to participate in a variety of signaling pathways, including the protein kinase C (PKC) pathway, that lead to actin cytoskeleton reorganization and increased branched actin polymerization at the cell membrane. Serines 152 and 156 in the ED domain are targets of both PKC and p42/44 mitogen‐activated protein kinase and are known to be involved in regulating MARCKS interaction with the actin cytoskeleton and cell membrane.10, 11 Phosphorylation of the ED domain causes dissociation of MARCKS from the cell membrane, disrupts its interaction with F‐actin,12 and prevents association with PIP2.13, 14 Under physiological conditions, expression of MARCKS is cell‐type specific, with high abundance in both embryonic and adult neural tissues.15
Previous studies in large animal models of intimal hyperplasia have demonstrated a 2‐ to 4‐fold increase in MARCKS mRNA transcripts at early time points postsurgery.16, 17 Knocking down MARCKS inhibits human VSMC migration in vitro and prevents intimal hyperplasia in the human saphenous vein ex vivo.18 MARCKS is a well‐established regulator of migration in multiple cell types. Dysregulation of MARCKS is closely associated with metastasis in a wide range of cancers and is an indicator of poor prognosis.19, 20 MARCKS increases cell motility through the protein kinase B pathway in fibroblasts21 and glial cells.22 Motility is also potentiated in macrophages,23 neutrophils,24 skeletal myoblasts,25 and endothelial cells26 through direct binding of MARCKS to actin. However, the mechanism through which MARCKS regulates VSMC motility is as yet unknown.
In the present study, we examined the expression and function of MARCKS in both dedifferentiated VSMCs, human coronary artery vascular smooth muscle cells (CASMCs)27 and rat aortic vascular smooth muscle cells, A10 cells,28 and differentiated VSMCs, rat aortic vascular smooth muscle cells, and A7r5 cells.29 Surprisingly, MARCKS does not modulate cell motility through its direct interactions with actin, but rather through regulation of the small GTPases, Rac1 and Cdc42, by controlling availability of PIP2 at the plasma membrane. Finally, we hypothesize that even partial MARCKS knockdown reduces intimal hyperplasia formation in vivo in the murine carotid ligation model.
Materials and Methods
Cell Culture
Primary human CASMCs (Lonza, Walkersville, MD) were cultured in smooth muscle cell medium (SmGM‐2; Lonza), following the manufacturer's instructions. Experiments were performed on CASMCs between passages 4 and 7. A7r5 and A10, VSMC lines derived from the embryonic rat thoracic aorta were purchased from the American Type Culture Collection (Manassas, VA) and cultured following the supplier's instructions. Antibiotics and DMEM were purchased from Lonza. FBS was purchased from Atlanta Biologicals Inc (Norcross, GA).
Reagents and Antibodies
GFP‐ and RFP‐tagged pleckstrin homology (PH) domain of PLC‐δ (GFP‐PH and RFP‐PH) plasmids were kindly provided by Dr Tamas Balla (National Institutes of Health, Bethesda, MD). Alexa Fluor 568– or Alexa Fluor 488–conjugated phalloidin were obtained from Invitrogen (Thermo Fisher Scientific, Grand Island, NY). Antibodies used in this study were from the following sources: anti‐human MARCKS (Catalog No.: 5607S; Cell Signaling Technology, Danvers, MA); polyclonal anti‐murine MARCKS (Catalog No.: AB9298; EMD Millipore, Billerica, MA); phospho‐MARCKS (Catalog No.: 07‐1238; EMD Millipore); GAPDH (Catalog No.: 2118L; Cell Signaling Technology); monoclonal anti‐Rac1 (Catalog No.: 05389, EMD Millipore); polyclonal anti‐Cdc42 (Catalog No.: 2462; Cell Signaling Technology); polyclonal anti‐PAK (Catalog No.: 4570; Cell Signaling Technology); polyclonal anti‐phospho‐PAK (Catalog No.: 2601; Cell Signaling Technology); monoclonal anti‐cortactin (Catalog No.: 05‐180; EMD Millipore); monoclonal anti‐beta‐actin (Catalog No.:A5316; Sigma‐Aldrich, St. Louis, MO); monoclonal anti–alpha‐smooth muscle actin (αSMA; Catalog No.: A5228; Sigma‐Aldrich); anti–platelet endothelial cell adhesion molecule‐1 (PECAM‐1/CD31; Catalog No.: 550 274; BD, Franklin Lakes, NJ); anti‐platelet‐derived growth factor (PDGF) receptor alpha (Catalog No.: mab322; R&D Systems, Minneapolis, MN); anti‐PDGF receptor beta (Catalog No.: 05‐1135; EMD Millipore); polyclonal anti‐calponin (Catalog No.: TA300720; OriGene Technologies, Inc., Rockville, MD); monoclonal anti‐smoothelin (Catalog No.: MAB3242, EMD Millipore); and anti‐SM 22‐α (Catalog No.: ab14106; Abcam, Cambridge, MA). Secondary antibodies, including horseradish peroxidase–conjugated antibodies against goat, mouse, and rabbit immunoglobulin IgG and FITC‐conjugated, rhodamine‐conjugated, and Alexa Fluor 633–conjugated secondary antibodies were from Invitrogen (Thermo Fisher Scientific).
Small Interfering RNA and Plasmid DNA Transfection
Human MARCKS small interfering RNA (siRNA; 5′‐GGU GCC CAG UUC UCC AAG AUU‐3′), and nontargeting control siRNA (5′‐CGC ACC AGA ACA AAC ACA UU ‐3′) were used as previously described.18 Rat‐specific MARCKS siRNA was designed and purchased from GE Healthcare (Catalog No.: M‐11507500; Waukesha, WI). Cells were transfected with 100 nmol/L of siRNA with or without plasmids in Opti‐MEM (Invitrogen, Carlsbad, CA) for 24 hours using the DharmaFECT‐Duo transfection reagent, following the manufacturer's instructions (GE Healthcare).
Wound‐Healing Assay
The wound‐healing assay was performed as described previously.30, 31 Cells were seeded onto 6‐cm2 plates and cultured in normal growth media until full confluence was reached. A wound was created by scrapping the cell layer with a sterile rubber policeman. Cells were then gently washed and grown in culture. Migration was assessed over a period of 16 hours for CASMC and A7r5 cells and 10 hours for A10 cells. The healing process was digitally recorded, and the reference marks were used to align during the subsequent image acquisition. Migration of cells was determined by reduction of the width of the wound area (3 sites were randomly measured and the average was counted for each image) or by cell number within the wound. Additionally, migration was quantified by cell count in the wound and was presented as the change in the number of cells in the wound from the creation of the wound to the predetermined time point. Data are presented as the mean and SE of 3 independent experiments. Significance of association was determined by the Wilcoxon rank‐sum test.
Cell Morphological Analysis
VSMCs were seeded onto fibronectin‐coated coverslips and starved for 48 or 72 hours with DMEM containing 0.2% FBS. Starved cells were then stimulated with 20 ng/mL of PDGF for 10 minutes. Stimulated cells were then fixed with 4% paraformaldehyde and stained with antibodies as specified. Stained cells were inspected under a Zeiss 510 Duo laser confocal microscope (Carl Zeiss GmbH, Oberkochen, Germany) with a 60× oil objective lens. To quantify membrane ruffling and lamellipodia formation, the area representing ruffling and lamellipodia on captured images were digitally outlined and measured with Zeiss Zen software. Data are presented as the mean and SEs. Statistical analysis was based on 3 independent experiments with at least 40 cells counted in each experiment. Significance of association was determined by the Wilcoxon rank‐sum test.
Small GTPase Rac1 and Cdc42 Activity Pull‐Down Assay
Rac1 and Cdc42 activity was measured by GTP pull‐down assay as described previously.31 Briefly, VSMCs were cultured to subconfluence and lysed with buffer containing 50 mmol/L of Tris‐HCl (pH 7.40, 1 mmol/L of EDTA, 150 mmol/L of NaCl, 1 mmol/L of phenylmethylsulphonyl fluoride, 1% Triton X‐100, 1 mmol/L of sodium fluoride, and proteinase inhibitor cocktail tablet (Roche, San Francisco, CA). Lysates were incubated with glutathione S‐transferase/p21‐activated protein kinase/Cdc42/Rac interactive binding domain (GST‐PAK‐CRIB) and glutathione Sepharose‐4B beads (Thermo Fisher Scientific). Beads were then briefly washed. Bound proteins were eluted by boiling in 2× SDS sample buffer and then subjected to 12% SDS‐PAGE followed by immunoblotting with either the Rac1 or Cdc42 antibody. Blots were analyzed with densitometry. Experiments were performed in triplicate. Data are presented as means and SEs. Significance of association was determined by 2‐way ANOVA.
Membrane‐Associated PIP2 Quantification
VSMCs were transfected with the green fluorescent protein (GFP)‐ or red fluorescent protein (RFP)‐tagged PH domain of PLC‐δ (GFP‐PH or RFP‐PH), as specified, using the Superfect reagent (Qiagen, Germantown, MD). After transfection, cells were starved for 48 hours and then treated with 20 ng/mL of PDGF for 10 minutes. Live cell images of transfected cells before and after PDGF treatment were collected with a Zeiss 510 Duo laser confocal microscope and analyzed with Zeiss Zen imaging software. Live cell quantification of membrane‐associated PIP2 level using GFP‐PH has been described in detail previously.32, 33 Pixel quantification along a single line across the cell was used to capture the specific florescence emission for both the region immediately adjacent to the membrane, the fluorescence in plasma membrane (Fpm), and the cytosol, the fluorescence in cytosol (Fc). To generate the proportion of fluorescence that was associated with the plasma membrane, we calculated the Fpm/Fc ratio. For each experimental condition, 8 cells were analyzed and data are presented as the mean and SEs. Significance of association was determined by 2‐way ANOVA.
Murine Carotid Ligation Model
CL57/B6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). MARCKS heterozygous mice (MARCKS+/−) were previously generated by Dr Perry J. Blackshear and Dr Deborah J. Stumpo. Mice were maintained following the guidelines and protocols of the animal care and use committee of the University of Maryland School of Medicine (Baltimore, MD). Mice were anesthetized and a 1‐cm midline anterior cervical incision was made. To induce intimal hyperplasia, the carotid artery was ligated at the bifurcation as described in the literature.34 Animals were allowed to recover and were subsequently euthanized at predetermined time points. At the time of euthanasia, mice were perfused with PBS solution and bilateral common carotid arteries were retrieved and snap‐frozen in optimal cutting temperature (OCT) compound (Sakura, Torrance, CA). The uninjured, contralateral carotid artery served as an internal control.
Immunohistochemistry
Tissues from the carotid ligation experiments were processed for immunohistochemistry. Frozen tissues were sectioned at 5‐μm intervals beginning 500 μm proximal to the ligature at the carotid bifurcation. Tissues were fixed with 10% formalin in PBS for 5 minutes and then treated with Verhoeff‐Van Gieson (VVG) stain or fixed with 4% paraformaldehyde for 5 minutes, then treated with antibodies against PECAM‐1/CD31, αSMA, and MARCKS in conjunction with appropriate secondary antibodies. Confocal microscopic images of stained sections were acquired using a Zeiss 510 Duo laser confocal microscope (Carl Zeiss GmbH).
Statistical Analysis
All experiments were performed in triplicate, unless otherwise noted. Data are presented as means±SEs. The Wilcoxon rank‐sum test was used to determine significance of association between conditions with categorical predictor variables and continuous, nonparametric outcome variables. For conditions with multiple predictor variables, a 1‐ or 2‐way ANOVA was used to determine significance of association as appropriate. The Shapiro–Wilk test was used to determine whether the data met criteria for normal distribution. Data with a Shapiro–Wilk test P value greater than 0.05 was considered to fit a normal distribution.
Results
MARCKS Knockdown Inhibits Dedifferentiated VSMC Migration
To examine the effects of MARCKS knockdown on cell motility of dedifferentiated VSMCs, we transfected MARCKS siRNA into cultured human CASMCs. At 72 hours post‐siRNA treatment, protein expression of MARCKS was decreased by 93.3%±2.9% compared to cells treated with control (nontargeting) siRNA (Figure 1A). Using the wound‐healing assay, we examined the effects of MARCKS on CASMC motility under normal cell‐culture conditions (Figure 1B). MARCKS knockdown severely impaired CASMC migration. In cells treated with control siRNA, the width of the wound decreased 1780±117 μm. However, after MARCKS knockdown, the wound width decreased only 417±102 μm (Figure 1C; P<0.05). MARCKS knockdown also lowered the increase in number of cells within the wound after 16 hours. After treatment with control siRNA, there were 135±4 cells within the wound. After MARCKS knockdown, there were only 38±3 cells within the wound (Figure 1D; P<0.05). The contribution of cell proliferation to the quantity of cells in the wound was negligible at the time points studied (data not shown).
Figure 1.
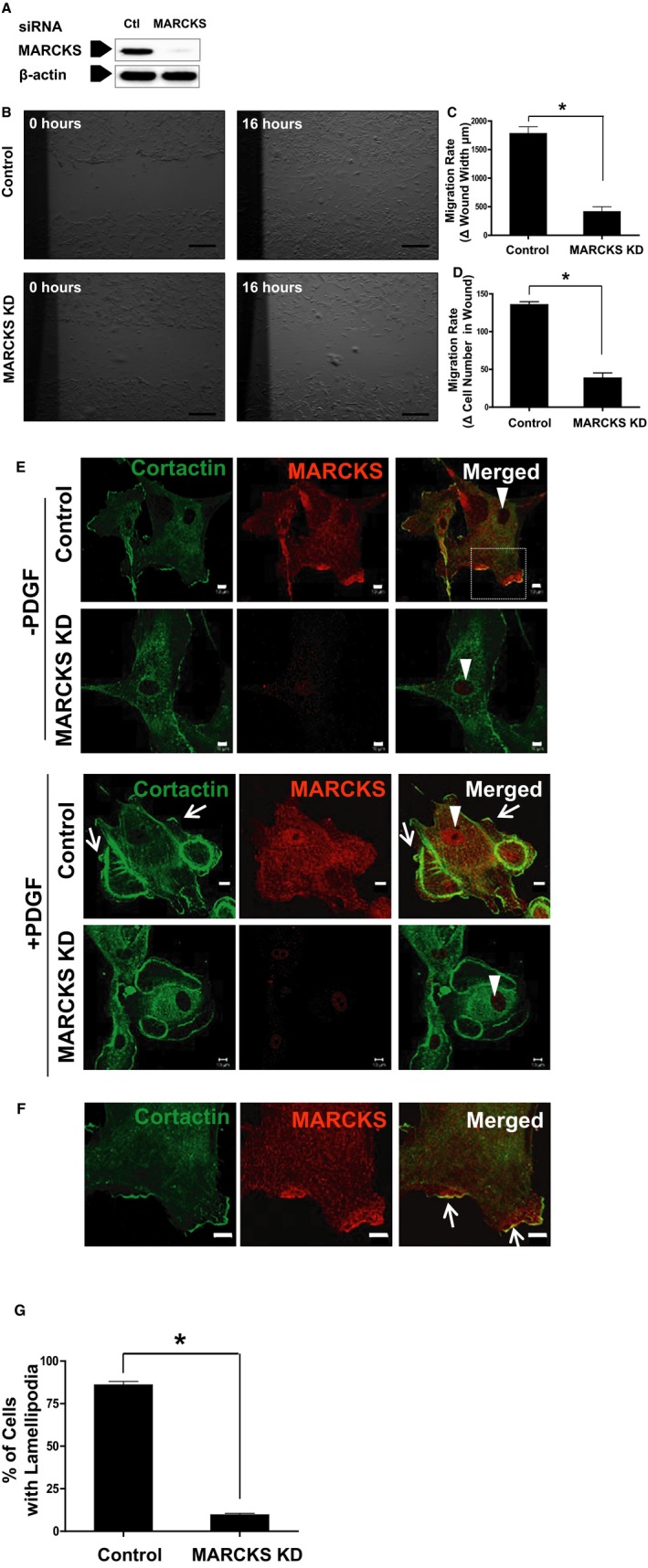
MARCKS KD impairs dedifferentiated VSMC migration. Human CASMCs were transfected with 100 nM of MARCKS siRNA (KD) or nontargeting, control siRNA (Control). A, At 72 hours post‐siRNA treatment, protein expression of MARCKS was decreased by 93%±3% compared to cells treated with control (nontargeting) siRNA. B, MARCKS KD significantly attenuated VSMC migration in the wound‐healing scratch assay. C, Cell migration was determined by both reduction of the width of the wound. D, The density of cells within the wound. *P<0.05. Scale bars=500 μm. E, Starved CASMCs were stimulated with PDGF (20 ng/mL for 10 minutes), fixed, and stained with anti‐cortactin (green) and anti‐MARCKS (red) antibodies. White triangles denote the nucleus which stains for neither cortactin nor MARCKS. Lamellipodia, white arrows, formed around the periphery of the cell in the control group, which were seldom detected after MARCKS KD. White arrow heads denote cell nucleus. Scale bars=10 μm; shown are representative images of 3 independent experiments. F, These morphological changes are best appreciated at higher magnification. MARCKS and cortactin colocalized in lamellipodia (white arrows). Scale bar=10 μm. G, Significantly more cells formed lamellipodia in repsonse to PDGF stimulation in the control group compared to the MARCKS KD group. At least 40 cells were counted in each group per experiment. Data are presented as the mean and SE of 3 independent experiments. CASMCs indicates coronary artery smooth muscle cells; KD, knockdown; MARCKS, myristoylated alanine‐rich protein kinase substrate; PDGF, platelet‐derived growth factor; siRNA, small interfering RNA; VSMC, vascular smooth muscle cell. *P<0.001.
Lamellipodia and Filopodia Formation Is Severely Impaired by MARCKS Knockdown in Dedifferentiated VSMCs
The cellular location of MARCKS and cortactin was determined in both the presence and absence of PDGF. Nuclei were identified by the absence of staining (Figure 1E, white arrow heads). MARCKS was located adjacent to the cell membrane in quiescent CASMCs and translocated to the cytoplasm after PDGF stimulation. After exposure to PDGF (20 ng/mL for 10 minutes), lamellipodia and filopodia were easily detected at the periphery of CASMCs treated with control siRNA (Figure 1E, white arrows). In contrast, after MARCKS knockdown, CASMCs were round and demonstrated minimal polarization. There were few lamellipodia and filopodia detected by staining with cortactin (Figure 1E; scale bar=10 μm). Colocalization of MARCKS and cortactin in lamellipodia is evident when viewed with a high‐powered objective (Figure 1F, white arrows; scale bars=10 μm). After MARCKS knockdown, cortactin failed to be recruited to the cortical area, a process dependent on Rac1 activation.35 PDGF stimulation (20 ng/mL for 10 minutes) predictably induced lamellipodia formation in 86%±2% of cells in control siRNA‐treated CASMCs, but only 10%±1% in the setting of MARCKS knockdown (Figure 1G; P<0.05).
MARCKS Is Indispensable for the Activation of Small GTPase Rac1 and Cdc42 in Response to PDGF in Dedifferentiated VSMC
Rac1 and Cdc42 are small GTPases that are essential in regulating lamellipodia and filopodia formation during cell migration. MARCKS knockdown did not affect the total protein expression of Rac1 or Cdc42 or the small GTPase effector, PAK1, in CASMCs (Figure 2A and 2B). We then examined the activity of Rac1 and Cdc42 in response to PDGF. The PAK‐CRIB pull‐down assay was used to quantify activated Rac1 (Rac1‐GTP) and activated Cdc42 (Cdc42‐GTP) (Figure 2C). In cells treated with control siRNA, treatment with 20 ng/mL of PDGF for 10 minutes resulted in an expected 318% increase in Rac1‐GTP (2.13±0.05 with stimulation vs 0.51±0.05 in quiescent conditions; P<0.05) and a 108.3% increase in Cdc42‐GTP (1.74±0.02 with stimulation vs 0.83±0.12 in quiescent conditions; P<0.05). However, this response was not observed in CASMCs after MARCKS knockdown. Rac1‐GTP and Cdc42‐GTP levels did not increase in response to PDGF. Rac1‐GTP was 0.70±0.05 with stimulation versus 0.68±0.05 in the quiescent condition, and Cdc42‐GTP was 0.74±0.04 with stimulation versus 0.71±0.09 in the quiescent condition (Figure 2D and 2E). Furthermore, MARCKS knockdown decreased phospholrylation of PAK1, the common downstream effector of Rac1 and Cdc42 signaling. CASMCs treated with control siRNA had a significant increase in phospho‐PAK1 after PDGF stimulation (Figure 2F). Stimulation with PDGF increased the fraction of phosphorylated PAK1 from 0.37±0.01 to 2.02±0.07, a 446% increase. However, CASMCs depleted of MARCKS did not exhibit a similar response and the fraction of phosphorylated PAK1 only increased from 0.6±0.09 to 0.89±0.16, a nonsignificant 48% increase (Figure 2G). Significance of association was determined by 2‐way ANOVA. These findings suggest that MARCKS knockdown impairs Rac1 and Cdc42 activation and downstream signaling in dedifferentiated human CASMCs.
Figure 2.
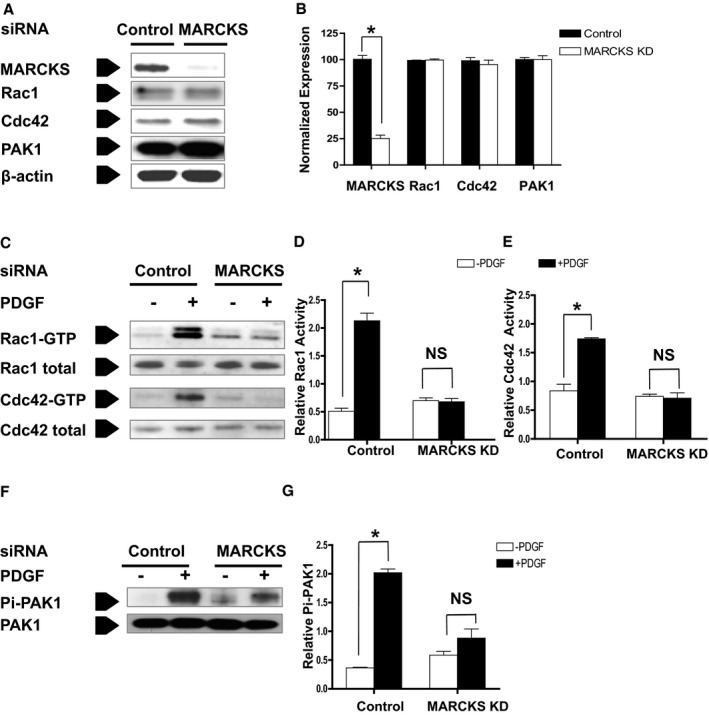
MARCKS KD inhibits small GTPase Rac1 and Cdc42 activation and blocks downstream signaling transduction in dedifferentiated VSMCs. A, Human CASMCs were treated with control (Control) siRNA or MARCKS siRNA (MARCKS KD). B, MARCKS siRNA reduced MARCKS protein expression by 94.7%±3.1%. *P<0.0001. MARCKS KD did not affect the basal expression of small GTPase Rac1 or Cdc42, or the GTPase effector, PAK1. C, Rac1 and Cdc42 activation was detected in the presence (+PDGF) or absence (−PDGF) of PDGF (20 ng/mL for 10 minutes) with the PAK‐CRIB pull‐down assay, as described in the Materials and Methods section. D, PDGF stimulation increased the activation of Rac1 in control cells, but had no effect after MARCKS KD. E, Similarly, PDGF stimulation increased the activation of Cdc42 in control cells, but also had no effect after MARCKS KD. F and G, MARCKS KD also prevented the activation of the Rac1 and Cdc42 effector, PAK1. Data are presented as the mean and SE of 3 independent experiments. *P<0.05. CASMCs indicates coronary artery smooth muscle cells; CRIB, Cdc42/Rac interactive binding domain; KD, knockdown; MARCKS, myristoylated alanine‐rich protein kinase substrate; NS, not significant; PAK, p21‐activated kinase; PDGF, platelet‐derived growth factor; siRNA, small interfering RNA; VSMC, vascular smooth muscle cell.
Human CASMCs and A10 Cells Have Lower Expression of Markers of Differentiation and Higher Constitutive Expression of MARCKS Than A7r5 Cells
In the present investigation, we used CASMCs and A10 cells for loss‐of‐function experiments. We chose these cells because they have lower expression of commonly cited markers of smooth muscle cell differentiation and thus more closely resemble medial VSMCs in intimal hyperplasia (Figure 3A). More important, these 2 cell lines had higher constitutive expression of MARCKS than A7r5 cells, increasing the power of the knockdown experiments. A7r5 cells had increased expression of the markers of smooth muscle cell differentiation and lower constitutive MARCKS expression. Thus, these cells were used in the gain‐of‐function experiments (Figure 3B).
Figure 3.
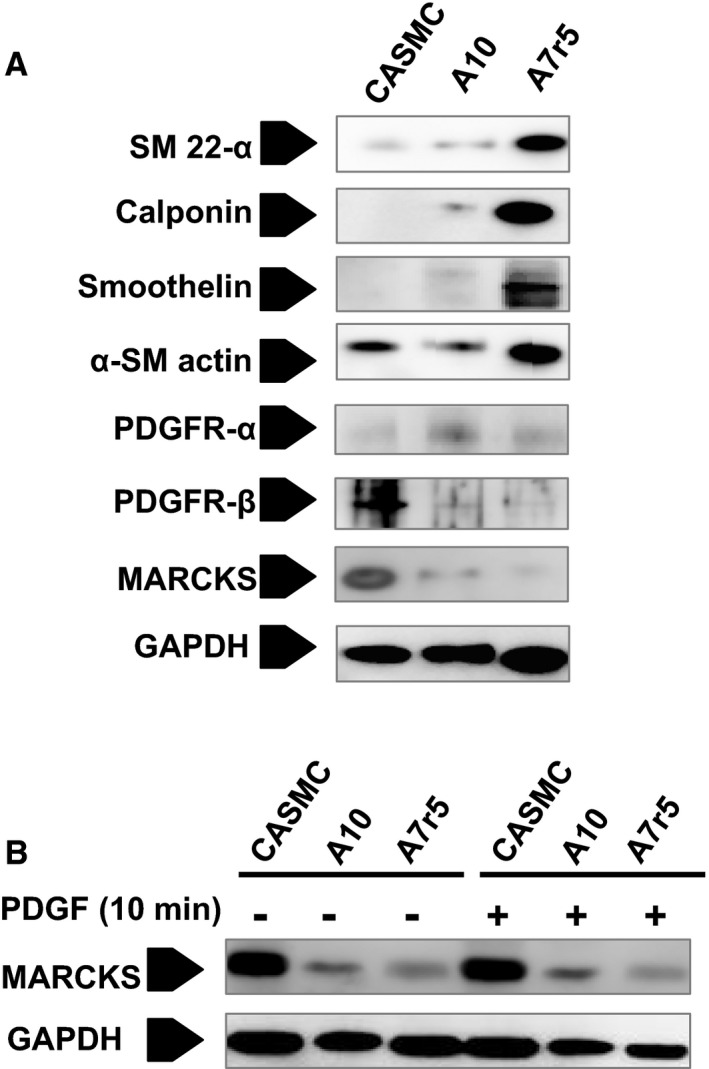
Markers of differentiation and MARCKS expression in human CASMCs, rat A10 cells, and rat A7r5 cells. In the present investigation, we used CASMCs and A10 cells for loss‐of‐function experiments. A, We chose these cells because they have lower expression of commonly cited markers of smooth muscle cell differentiation and thus more closely resemble the medial VSMCs in intimal hyperplasia. B, These 2 cell lines had higher constitutive expression of MARCKS than A7r5 cells, increasing the power of the KD experiments. A7r5 cells had increased expression of the markers of differentiation and lower constitutive MARCKS expression. Thus, these cells were used in the gain‐of‐function experiments. CASMCs indicates coronary artery smooth muscle cells; MARCKS, myristoylated alanine‐rich protein kinase substrate; PDGF, platelet‐derived growth factor; PDGFR, platelet‐derived growth factor receptor; SM, smooth muscle; VSMC, vascular smooth muscle cell.
Overexpression of Wild‐Type MARCKS, but Not the PseudoPhosphorylated ED Domain Mutant (S4D) MARCKS, Rescues Migration and Rac1 and Cdc42 Activation Defects Caused by MARCKS Knockdown in Dedifferentiated VSMCs
To explore the mechanism of how MARCKS regulates VSMC migration, we transiently expressed plasmids in pEGFP‐N1 vector carrying GFP‐fused, bovine wild‐type (WT), or functional domain mutant MARCKS in dedifferentiated A10 VSMCs with endogenous MARCKS knockdown. A schematic representation of WT and MARCKS mutants is shown in Figure 4A. These plasmids and the function of MARCKS mutants have been well characterized.13, 36, 37, 38 The G2A mutant lacks the myristoylated tail, the S4G mutant expresses a phosphorylation‐deficient ED domain, and the S4D has a pseudophosphorylated ED domain. The rat aortic VSMC line, A10, was used in this experiment because, first, A10 cells are dedifferentiated VSMCs (Figure 3),28 representative of the VSMCs in intimal hyperplasia and, second, because of its higher transfection efficiency for plasmid DNA. Rat‐specific MARCKS siRNA efficiently knocked down endogenous MARCKS, but did not affect expression of bovine MARCKS. MARCKS knockdown decreased A10 cell migration by 52.6% (47.4%±2.1% vs 100%±2.3%; P<0.05). Expression of WT bovine MARCKS rescued migration defects in A10 cells depleted of endogenous MARCKS. G2A and S4G mutants also rescued migration. However, the pseudophosphorylated ED domain mutant, S4D, failed to rescue the cell migration defects caused by endogenous MARCKS knockdown (49%±13.7% vs control, 100%±2.3%; P<0.05; Figure 4B).
Figure 4.
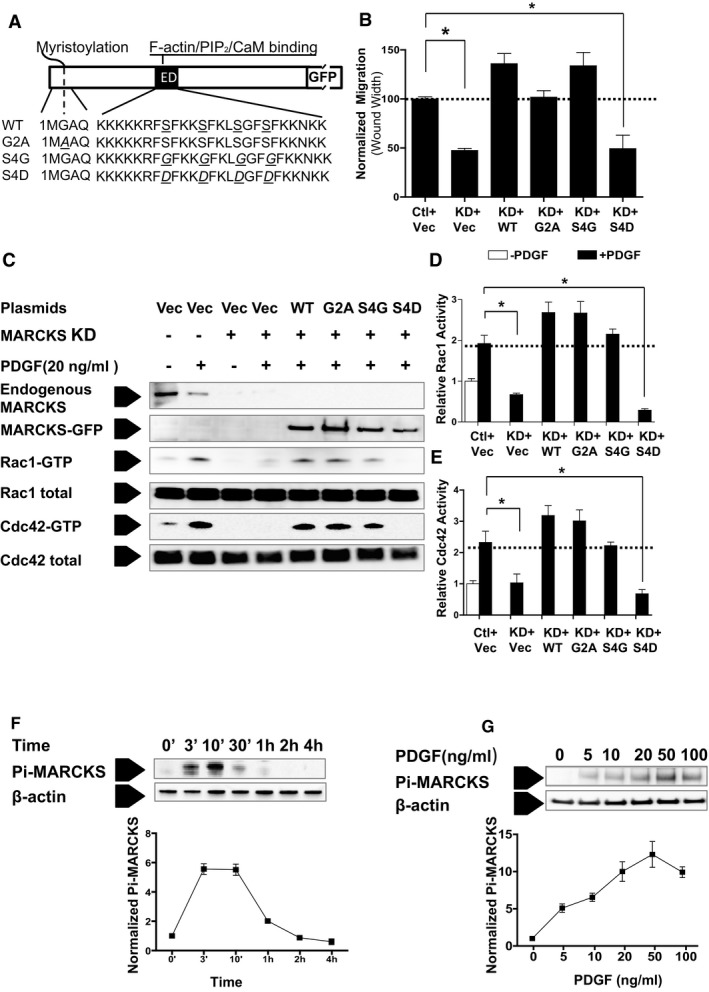
Ectopic expression of wild‐type (WT) MARCKS, but not the pseudophosphorylation mutant rescues the migration defects observed in MARCKS KD in dedifferentiated VSMCs. A, Dedifferentiated rat A10 VSMCs were treated with control siRNA (Ctl) or MARCKS siRNA (KD) and cotransfected with plasmids for either pEGFP‐N1 vector alone (Vec), WT bovine MARCKS (MARCKS WT), the myristoylation‐deficient MARCKS mutant (G2A), the phosphorylation‐deficient ED domain MARCKS mutant (S4G), or the pseudophosphorylated ED domain MARCKS mutant (S4D), respectively. B, Cell migration was determined by the wound‐healing assay in normal growth media as described in the Materials and Methods section. Cotransfection with WT MARCKS, myristolation‐deficient MARCKS, and phosphorylation‐deficient MARCKS all rescued migration. The pseudophosphorylation mutant did not rescue migration. Data are presented as the mean and SE of 3 independent experiments and normalized to the control treatment (Ctl+Vec). *P<0.05. C, Activation of the GTPases Rac1 and Cdc42 was determined in the presence of MARCKS KD and cotransfection with WT plasmids and each of the 3 mutant plasmids. D, The WT MARCKS, G2A, and S4G, but not the pseudophosphorylated S4D mutant, rescued both Rac1 and (E) cdc42 activation. Data are presented as the mean and SE of 3 independent experiments normalized to the quiescent condition (−PDGF) in control cells (Ctl+Vec). *P<0.05. F, PDGF stimulates MARCKS phosphorylation (Pi‐MARCKS) in CASMCs in a time‐dependent manner. Maximal MARCKS protein expression was observed after 10 minutes of stimulation. G, MARCKS phosphorylation is also dependent on the dose of PDGF. Exposure of starved cells to 50 ng/mL yielded the greatest phosphorylation of MARCKS at 10 minutes (right panel). CaM indicates calmodulin; ED, effector domain; GFP, green fluorescent protein; KD, knockdown; MARCKS, myristoylated alanine‐rich protein kinase substrate; PDGF, platelet‐derived growth factor; pEGFP, enhanced green fluorescent protein plasmid; siRNA, small interfering RNA; VSMC, vascular smooth muscle cell; WT, wild type.
MARCKS knockdown prevented activation of both Rac1 and Cdc42. Cotransfection with WT MARCKS, G2A, or S4G MARCKS mutants increased Rac1 and Cdc42 activity. However, the S4D mutant which carries the pseudophosphorylated ED domain, did not rescue Rac1 or Cdc42 activation in response to PDGF (Figure 4C through 4E; P<0.05). This finding is consistent with the above observation that the S4D mutant failed to rescue VSMC migration after MARCKS knockdown.
PDGF Increases MARCKS Phosphorylation in VSMCs
In CASMCs, PDGF transiently induced phosphorylation of the MARCKS ED domain. Phospho‐MARCKS expression was maximal after 10 minutes and then gradually decreased. Phospho‐MARCKS was barely detected 30 minutes after PDGF stimulation (Figure 4F). Stimulation of CASMCs with PDGF ranging from 10 to 100 ng/mL for 10 minutes resulted in phosphorylation of MARCKS in a dose‐dependent manner. Exposure of starved cells to 50 ng/mL yielded the greatest phosphorylation of MARCKS at 10 minutes (Figure 4G).
MARCKS Knockdown Decreases Membrane PIP2 Abundance in VSMCs
The MARCKS ED domain binds and sequesters PIP2 on the cell membrane. Phosphorylation of the ED domain decreases MARCKS affinity to membrane‐bound PIP2. 13, 14 Therefore, we hypothesized that MARCKS knockdown affects membrane‐associated PIP2 abundance in VSMCs. Live cell imaging with fluorescent protein, GFP‐tagged pleckstrin homology domain of PLC‐δ (GFP‐PH), as a biosensor was used to quantify PIP2 levels at the plasma membrane (Figure 5A). Under quiescent conditions in control siRNA‐treated CASMCs, GFP‐PH was highly concentrated on the plasma membrane, and Fpm/Fc was 1.88±0.15. MARCKS knockdown significantly decreased membrane‐associated PIP2; Fpm/Fc was 1.17±0.04, or 37.9% less than control cells (Figure 5B, control vs knockdown; P<0.05). Stimulation of quiescent control cells with PDGF (20 ng/mL for 10 minutes) induced the expected redistribution of GFP‐PH into the cytoplasm, indicating the loss by hydrolysis of PIP2 on the cell membrane. Correspondingly, a 42.7% decrease of membrane‐bound PIP2 was observed in control cells after PDGF treatment. After MARCKS knockdown, PDGF treatment failed to induce any significant change and membrane‐bound PIP2 remained as the basal level (Fpm/Fc=1.08±0.02), which is similar to the PIP2 level in the control group after PDGF treatment (Fpm/Fc=1.08±0.04; Figure 5B). These data demonstrate that MARCKS is required to maintain a high level of PIP2 at the cell membrane in VSMCs.
Figure 5.
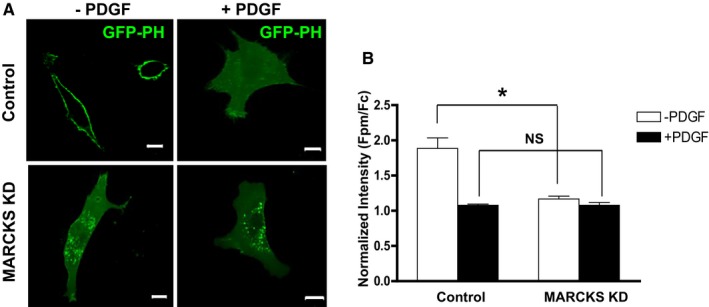
MARCKS KD decreases membrane bound PIP 2 in dedifferentiated VSMCs. A, GFP‐tagged pleckstrin homology domain of PLC‐δ (GFP‐PH) was used as a live cell biosensor to detect PIP 2 levels in human CASMCs. Representative images of cells in each treatment are presented. Scale bars=10 μm. B, Mean intensity values were determined by measuring the pixels along the plasma membrane (Fpm) and in the cytoplasm (Fc). Fluorescence of GFP‐PH in plasma membrane is normalized by the mean fluorescence value in the cytoplasm yielding the proportion of PIP 2 at the membrane (Fpm/Fc). Cells were treated with MARCKS siRNA (MARCKS KD) or control siRNA (Control). Starved quiescent cells were treated with (+PDGF) or without (−PDGF) PDGF (20 ng/mL for 10 minutes). Before exposure to PDGF, significantly more PIP 2 localized to the plasma membrane in control cells, compared to MARCKS KD cells (Fpm/Fc=1.88±0.42 and Fpm/Fc=1.17±0.11, respectively). *P<0.001; n=8. However, after stimulation with PDGF, there was no significant difference of proportion of PIP 2 localized to the plasma membrane (Fpm/Fc=1.08±0.05 and Fpm/Fc=1.08±0.11, respectively; n=8). CASMCs indicates coronary artery smooth muscle cells; Fc, fluorescence in cytosol; Fpm, fluorescence in plasma membrane; GFP, green fluorescent protein; KD, knockdown; MARCKS, myristoylated alanine‐rich protein kinase substrate; NS, not significant; PDGF, platelet‐derived growth factor; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5‐bisphosphate; PLC, phospholipase C; siRNA, small interfering RNA.
Ectopic Expression of MARCKS in Differentiated VSMCs Increases Membrane PIP2 Abundance, Rac1 and Cdc42 Activity, and Cell Motility
MARCKS‐GFP was overexpressed in A7r5 cells, the differentiated rat aortic smooth muscle cell line that displays a less‐motile phenotype (Figure 3A).29 A7r5 cells were used for overexpression experiments because MARCKS is expressed constituatively at lower levels than in CASMCs or A10 cells (Figure 3B). RFP‐tagged pleckstrin homology domain of PLC‐δ (RFP‐PH) was used as a biosensor to detect PIP2 (Figure 6A). MARCKS‐GFP overexpression significantly increased membrane‐associated PIP2 compared to GFP vector‐transfected cells n quiescent cells. (Figure 6B; P<0.001). MARCKS overexpression did not significantly change basal membrane‐associated PIP2 in A7r5 cells treated with PDGF. (Figure 6B). MARCKS overexpression also significantly increased A7r5 cell migration by 49.1% (149%±22.5% vs control, 100%±5.6%) as determined by the wound‐healing assay (Figure 6C; P<0.05). Lamellipodia and dorsal ruffles were more frequently detected in A7r5 cells overexpressing MARCKS‐GFP (Figure 6D, white arrows). After PDGF stimulation, 23.6%±4.9% of control cells displayed dorsal ruffles, whereas in A7r5 cells overexpressing MARCKS‐GFP, incidence of cells with dorsal ruffles was 42.4%±5.2% (Figure 6E; P<0.05). MARCKS overexpression increased the activation of small GTPases. Overexpression of MARCKS increased Rac1 activity; however, this increase did not reach statistical significance (Figure 6G). Cdc42 activity was significantly increased by MARCKS overexpression (P<0.05; Figure 6H). MARCKS overexpression in differentiated VSMCs increased membrane PIP2 abundance, Rac1/Cdc42 activity, induced formation of dorsal ruffles, and, ultimately, resulted in increased cell motility.
Figure 6.
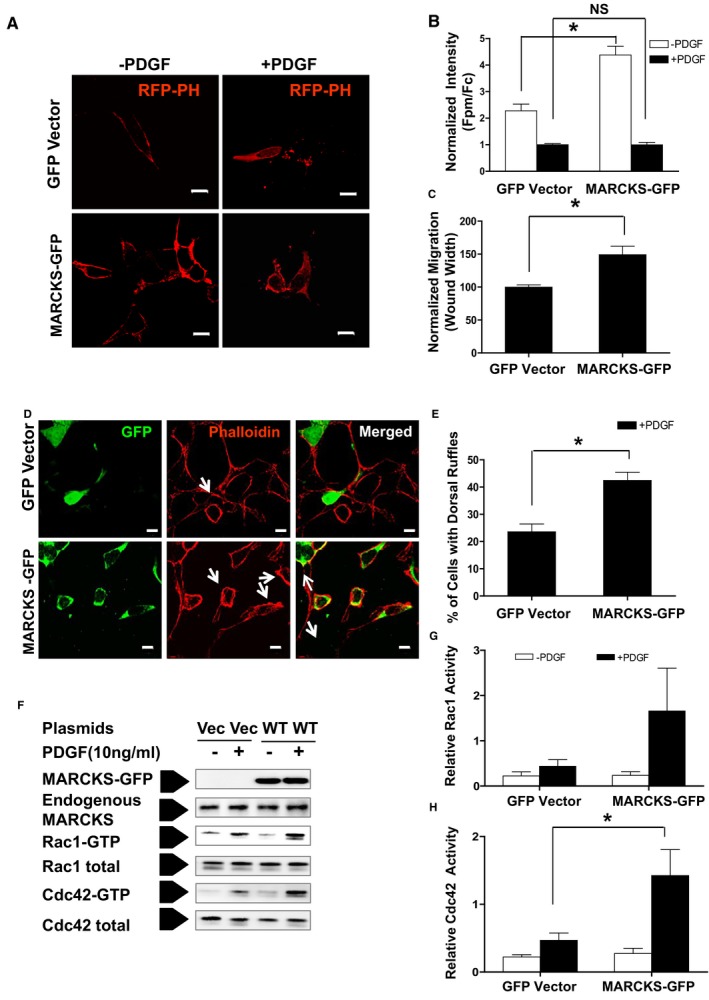
Overexpressing MARCKS increases differentiated vascular smooth muscle cell (VSMC) motility. Differentiated rat VSMC A7r5 cells that express low constituative levels of MARCKS protein were transfected with GFP‐vector (Vec) or WT bovine MARCKS‐GFP (+WT). A, Cells were cotransfected with RFP‐PH and with either GFP vector or MARCKS‐GFP plasmids. Membrane‐associated PIP 2 level (Fpm/Fc) was measured and quantified as described in the Materials and Methods section. B, Membrane‐associated PIP 2 level increased in A7r5 cells overexpressing MARCKS (MARCKS‐GFP) compared to control (Vector). *P<0.001, n=8. There was no difference between the 2 treatments in membrane‐associated PIP 2 after stimulation with PDGF; n=8. Representative images of subcellular location of the RFP‐PH biosensor in each treatment are shown on the right panel. Scale bars=10 μm. C, Cell migration increased in A7r5 cells overexpressing MARCKS as determined by the wound‐healing assay. *P<0.05; n=3. D, A7r5 Cells were starved for 48 hours before stimulation with PDGF (+PDGF, 10 ng/mL for 10 minutes). Cells were fixed and stained with phalloidin (red) to label the actin cytoskeleton. White arrows denote dorsal ruffles and lamellipodia. Shown are representative images of 3 experiments with independent cell preparations. E, Overexpression of MARCKS promotes dorsal ruffle and lamellipodia formation in differentiated VSMCs. Seventy cells were counted in each group per experiment. *P<0.01. Scale bars=10 μm. F, Activation of the small GTPases Rac1 and Cdc42 was assessed using the PAK‐CRIB pull‐down assay. G, Overexpression of MARCKS increased Rac1 activity, but not significantly. H, MARCKS overexpression significantly increased Cdc42 activity. Activity was normalized to unstimulated control cells (Vector, −PDGF). CRIB indicates Cdc42/Rac interactive binding domain; Fc indicates fluorescence in cytosol; Fpm, fluorescence in plasma membrane; GFP, green fluorescent protein; MARCKS, myristoylated alanine‐rich protein kinase substrate; NS, not significant; PAK, p21‐activated kinase; PDGF, platelet‐derived growth factor; PH, pleckstrin homology; PIP2, phosphatidylinositol 4,5‐bisphosphate; RFP, red fluorescent protein; VSMC, vascular smooth muscle cell; WT, wild type. *P<0.05.
MARCKS Protein Expression Increases in Migrating VSMCs During Intimal Hyperplasia Development in Mice
The mouse carotid ligation model of intimal hyperplasia was used to evaluate the in vivo function of MARCKS in vascular remodeling. Carotid arteries were sampled at days 0, 7, 14, and 28 postligation. Total lysates from 3 carotid arteries (6 μg of total protein) at each time point were analyzed with Western blot (Figure 7A). Ligation induced an increase of MARCKS protein at days 7 and 14, then decreased to baseline at day 28. An increase of β‐actin and decrease of αSMA were also observed at the early phase of intimal hyperplasia development. Interestingly, phospho‐MARCKS, which does not contribute to sequestering PIP2 at the membrane, significantly decreased after ligation. On day 7, the relative level of phospho‐MARCKS was only 21% of the control, unligated samples. Phospho‐MARCKS expression was decreased after carotid ligation at all time points examined after carotid ligation (Figure 7B). In unligated carotid artery sections, immunofluorescence staining detected weak expression MARCKS in VSMCs labeled with αSMA. However, after carotid ligation, MARCKS protein staining was much more prominent in VSMCs (Figure 7C). MARCKS was detected on the luminal surface of VSMCs just deep to the internal elastic lamina at the leading edge of VSMCs migrating toward the lumen (Figure 7B, white arrows). Increased expression of MARCKS was also detected in endothelial cells in the ligation samples. These in vivo data demonstrate that MARCKS expression increases during intimal hyperplasia development in the mouse carotid ligation model.
Figure 7.
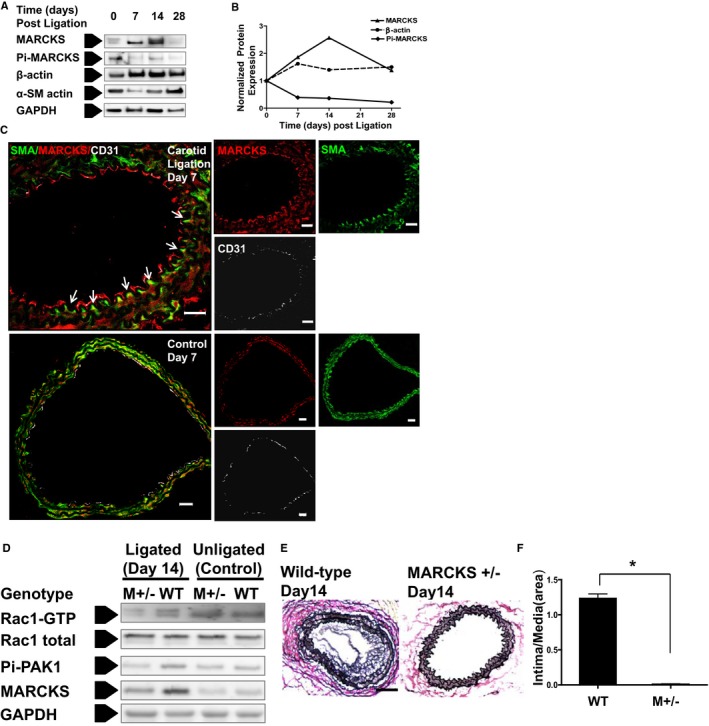
Decreased MARCKS expression inhibits intimal hyperplasia formation in vivo. A, MARCKS protein is up‐regulated during the formation of intimal hyperplasia in the murine carotid ligation model. Common carotid arteries in WT mice were ligated at the carotid bifurcation and harvested at days 0 (untreated), 7, 14, and 28. Total lysates from 3 arteries (6 μg of total protein) were used to assess phospho‐MARCKS (Pi‐MARCKS), MARCKS, α‐SMA, and β‐actin expression at each time point with Western blot analysis. B, Protein expression was normalized to GAPDH. MARCKS expression increases early in the formation of intimal hyperplasia. Maximal MARCKS expression occurs at 14 days. C, The caroitd artery was examined with immunostaining of frozen sections at an early phase of intimal hyperplasia formation (day 7 postligation). Confocal microscopy was used to identify MARCKS (red), α‐SMA (green), and the murine endothelial cell marker, CD31 (white). At day 7, the injured carotid arteries in WT mice had developed a significant hyperplastic lesion. The merged confocal images demonstrate that MARCKS was highly expressed in the injured arteries. MARCKS is localized in the polarized leading edge (white arrows) of invading VSMCs. Scale bars=20 μm. D, MARCKS expression increased in WT mice as a result of carotid ligation (day 14). MARCKS expression increased only slightly after carotid ligation in MARCKS+/− mice (M+/−). Rac1 and PAK1 activation were lower in the ligated carotid artery (day 14) of MARCKS+/− mice (M+/−) as compared to WT mice. E, Representative VVG‐stained sections of ligated carotid artery are shown at day 14 in WT (left) and MARCKS +/− (right) mice. There was a significantly greater hyperplastic response observed in WT mice compared to MARCKS+/− mice (M +/−). Scale bars=200 μm. F, Intimal hyperplasia was quantified with intima area/media area ratio, n=3. CD indicates cluster of differentiation; MARCKS, myristoylated alanine‐rich protein kinase substrate; PAK, p21‐activated kinase; SM, smoothelin; SMA, smooth muscle actin; VSMC, vascular smooth muscle cell; VVG, Verhoeff‐Van Gieson; WT, wild type. *P<0.0001.
Decreased MARCKS Expression Inhibits Intimal Hyperplasia Formation
We investigated the in vivo effects of MARCKS knockdown on intimal hyperplasia development. MARCKS knockout is lethal in mice; however, heterozygous mice (MARCKS+/−) are vital, but exhibit reduced MARCKS expression.15 MARCKS protein expression was lower in heterozygous carotid artery compared to WT under physiological conditions in nonligated samples. At day 14 postsurgery, ligation induced a significant increase of MARCKS protein in carotid arteries of WT animals, but not in MARCKS+/− mice (Figure 7D). Rac1 activation was lower in ligated MARCKS+/− samples as compared to WT samples. Accordingly, the common effector of Rac1 and Cdc42, PAK1, was less activated in ligated heterozygous samples as compared to WT mice (Figure 7D). WT mice developed robust intimal thickening at day 14 after carotid ligation; however, MARCKS+/− mice failed to develop significant intimal hyperplasia (Figure 7E). Although changes in proliferation can partially explain this finding, VSMCs need to first migrate to the neointima to create intimal hyperplasia. In contrast to the ample accumulation of VSMC in the neointima as observed in WT day 14 samples, there were few VSMCs in the intima on the luminal side of the internal elastic lamina in MARCKS+/− mice. The intima area/media area ratio at day 14 was significantly lower in MARCKS heterozygous mice as compared to WT mice (P<0.001; Figure 7E).
Discussion
In the present study, we show that MARCKS is an important regulator of VSMC migration both in vitro and in vivo. MARCKS knockdown decreases the fraction of PIP2 available at the plasma membrane. And, subsequently, MARCKS knockdown prevents activation of Rac1 and Cdc42, resulting in significantly attenuated VSMC motility. MARCKS protein expression is significantly increased at the early phase of intimal hyperplasia in the mouse carotid ligation model. This finding confirms the previously noted association between active MARCKS transcription and development of intimal hyperplasia after bypass surgery in large mammals.16, 17, 39 Genetically disrupting 1 allele of the MARCKS gene is sufficient to block protein upregulation of MARCKS in the carotid artery in response to ligation and prevents intimal hyperplasia formation.
This novel work is significant for 3 reasons. This is the first report of the mechanism by which MARCKS signaling regulates VSMC motility. Second, this is the first report wherein MARCKS knockdown prevents the formation of intimal hyperplasia in vivo. Finally, only a reduction in MARCKS, not a complete knockout, is sufficient to attenuate formation of intimal hyperplasia. MARCKS is an important intracellular messenger in a variety of cell types. Knockdown of MARCKS is more feasible than a complete knockout, making MARCKS an attractive target for therapy to prevent intimal hyperplasia formation.
The phenotype switch that increases motility of medial VSMCs is essential for the pathogenesis of intimal hyperplasia.40 Increased motility of medial VSMCs allows their migration from the media to neointima and is one of the early events in development of intimal hyperplasia. Our data demonstrate that overexpressing MARCKS in differentiated VSMCs (A7r5 cells) increases cell motility, whereas knocking down MARCKS in dedifferentiated VSMCs (A10 cells and human CASMCs) severely impairs cell migration. Consistent with previous observations, we confirmed that MARCKS is upregulated in the early phases of intimal hyperplasia. Taken together, these data support the assertion that MARCKS upregulation plays a causative, not a compensatory, or a coincidental role in the formation of intimal hyperplasia. The phospholipid messenger, PIP2, is an essential upstream signal in multiple cellular pathways. MARCKS stabilizes membrane‐bound PIP2, protects it from flank diffusion, and consumption by phospholipase C (PLC) or phosphoinositide 3‐kinase (PI3K) for second signal messenger synthesis.41 Regulation of the abundance of MARCKS and, correspondingly, the membrane composition of PIP2, is closely associated with cell motility.
Recent in vivo research has revealed that losing MARCKS expression is responsible for the paucity in PIP2 in hippocampal neurons, which accounts for the neurodegeneration and cognition deficiency with physiological brain aging.42 Increasing evidence has established a critical role for PIP2 metabolism in regulation of VSMC phenotype switch in the pathogenesis of intimal hyperplasia. Enhancing PI3K or PLC contributes to VSMC dedifferentiation and neointima formation.43, 44, 45 We found that there is a significant increase of MARCKS protein expression in the early phase of intimal hyperplasia formation when VSMCs revert to a more‐motile phenotype. Although total MARCKS protein is increased, phosphorylated MARCKS is dramatically decreased. However, this finding is not unexpected given that only unphosphorylated MARCKS has the ability to sequester PIP2 at the plasma membrane.12, 14 There is increased dynamic turnover of membrane PIP2 in vivo in migrating dedifferentiated VSMCs during early pathogenesis of intimal hyperplasia. Enrichment of PIP2 at the cell membrane is known to favor membrane polarization during cell migration.46 Early in the development of intimal hyperplasia (day 7), there is increased MARCKS expression predominantly on the apical surface of VSMCs along the polarized leading edge of the cell advancing toward the internal elastic lamina. MARCKS regulation of available PIP2 at the plasma membrane is a novel mechanism of regulating VSMC motility.
In the formation of intimal hyperplasia, VSMCs revert to a motile phenotype in which the actin cytoskeleton is reorganized and enables migration into the intima. The small GTPases, Rac1 and Cdc42, are key regulators of cortical actin polymerization and are essential for lamellipodia and filopodia formation during cell migration.47 Depleting MARCKS impairs lamellipodia and filopodia formation, which has been well documented in both in vivo48 and in vitro cell‐culture systems.36, 49 However, the effect of MARCKS knockdown on Rac1 and Cdc42 has not been reported on. We systematically quantified Rac1 and Cdc42 activity in VSMCs. MARCKS positively affects Rac1 and Cdc42 activation. First, depleting MARCKS inhibited Rac1 and Cdc42 activation and downstream signaling and was rescued by ectopically expressing WT MARCKS. Second, overexpressing MARCKS in differentiated VSMCs increased both Rac1 and Cdc42 activity, which was accompanied by increased cortical actin dynamics and cell motility. These data are the first direct evidence suggesting a regulatory role of MARCKS on small GTPase function in regulating cell migration.
To further support our hypothesis that MARCKS regulates Rac1 and Cdc42 activation by regulating PIP2 availability at the plasma membrane, we performed rescue experiments in dedifferentiated VSMC after MARCKS knockdown. We cotransfected WT or 1 of 3 MARCKS mutants with MARCKS siRNA. The mutants included a nonmyristolated mutant, an ED pseudophosphorylated mutant, and a phosphorylation‐deficient ED mutant. Predictably, WT MARCKS rescued both Rac1 and Cdc42 activity and motility in these cells. The myristoylation‐deficient mutant and the phosphorylation‐deficient mutant also rescued Rac1/Cdc42 activity and motility. However, the MARCKS mutant with pseudophosphorylated ED domain failed to rescue either Rac1 or Cdc42 activity or motility. This mutation prevents PIP2 binding and lends support to the hypothesis that it is MARCKS‐PIP2 binding that is responsible for the motile phenotype. These data support the assertion that MARCKS regulated activation of Rac1 and Cdc42 through controlling PIP2 availability and thus regulates formation of lamellipodia and filopodia.
In the in vivo experiments, MARCKS (+/−) mice failed to form intimal hyperplasia after carotid ligation. This result was unanticipated. We presumed that we would need complete knockout of MARCKS to observe a change in phenotype. In MARCKS (+/−) mice, few VSMCs were located on the luminal aspect of the internal elastic lamina. However, we did observe a modest thickening of media layer in MARCKS (+/−) samples, suggesting a local proliferation of VSMCs. The attenuated migration observed in MARCKS knockdown, as opposed to complete knockout, is actually consistent with previously reported proliferation data. We previously reported an attenuation of PDGF‐induced VSMC migration as determined by Transwell assay with a partial MARCKS knockdown mediated by only 25 nmol/L of MARCKS siRNA. This dose of siRNA resulted in an incomplete knockdown of MARCKS protein in vitro.18
The present investigation has several limitations. Three different cell lines were used in the in vitro studies. We selected these cell lines for specific reasons. MARCKS is upregulated in response to vascular injury. Injured VSMCs represent a dedifferentiated phenotype. To approximate this dedifferentiated, MARCKS‐overexpressing phenotype, we conducted knockdown experiments in human CASMCs and A10 cells. For gain‐of‐function experiments, we wanted to use a cell line that had low constitutive MARCKS expression. For these experiments, we selected the A7r5 cell line. Although these cells are derived from embryonic rat aorta, there is evidence in the literature that A10 and A7r5 cells represent dedifferentiated and differentiated VSMCs, respectively.28, 29 To further confirm that these cells represented different states of differentiation, we assayed expression of SM 22‐α, smoothelin, calponin, and αSMA in each of the 3 cell types. These data support the assertion that the A7r5 cell line is more differentiated than either A10 cells or human CASMC (Figure 3). Regardless, the pragmatic reason for selection of these cells is the different level of constitutive MARCKS expression. MARCKS has higher levels of expression in CASMCs and A10 cells, making them better suited for knockdown experiments, whereas MARCKS has lower levels of expression in A7r5 cells, making these cells better suited for gain‐of‐function experiments.
Another limitation of the present work is the use of the carotid ligation model for intimal hyperplasia. Although this is a well‐described, well‐accepted model of intimal proliferation, it does not cause an injury to the endothelium. It is a flow‐mediated model of vascular proliferative disease. In the clinical setting, vascular proliferative lesions are formed, in part, as a result of endothelial damage. Thus, extrapolation from findings in these experiments might have limited clinical relevance. A final limitation is that the MARCKS+/− mice are pan‐knockdown. MARCKS expression is not only decreased in vascular smooth muscle cells, but rather all the cells in the entire organism. In this experimental system, the reduction of intimal hyperplasia formation might be owing to other factors, for example, the immune response to injury in addition to a direct effect on VSMC motility.
In summary, we demonstrate that MARCKS is a critical regulator of VSMC migration both in vitro and in vivo. Consistent with previous studies in large animals, MARCKS is significantly upregulated in the first week after murine carotid artery ligation. At the time of surgery or angioplasty, local injury to the vessel, destruction of the endothelium, and the inflammatory milieu cause VSMCs to revert to a dedifferentiated, motile phenotype. This is the first report defining the mechanism through which MARCKS regulates cell motility in VSMCs. Although MARCKS is involved in many signaling pathways, we have demonstrated that its PIP2 binding is critical in regulating small GTPase Rac1 and Cdc42 activity and thus VSMC motility. MARCKS is so indispensible for increased motility that only a reduction of MARCKS, not complete abolishment, is needed to prevent the formation of intimal hyperplasia in the murine carotid ligation. For these reasons, inhibition of MARCKS at the time of surgery or angioplasty is a promising candidate for therapy to prevent intimal hyperplasia.
Sources of Funding
This work was funded, in part, by The Veterans Affairs Career Development Award 1IK2BX001553‐01 and through the Vascular Cures Foundation E.J. Wylie Scholarship to T.S.M. This work was also supported, in part, by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences.
Disclosures
None.
Acknowledgments
GFP‐tagged and RFP‐tagged pleckstrin homology domain of PLC‐δ (GFP‐PH and RFP‐PH) plasmids were kindly provided by Dr Tamas Balla of the National Institutes of Health.
(J Am Heart Assoc. 2015;4:e002255 doi: 10.1161/JAHA.115.002255)
References
- 1. Lloyd‐Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De SG, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel‐Smoller S, Wong ND, Wylie‐Rosett J. Executive summary: heart disease and stroke statistics–2010 update: a report from the American Heart Association. Circulation. 2010;121:948–954. [DOI] [PubMed] [Google Scholar]
- 2. Costa MA, Simon DI. Molecular basis of restenosis and drug‐eluting stents. Circulation. 2005;111:2257–2273. [DOI] [PubMed] [Google Scholar]
- 3. Chaabane C, Otsuka F, Virmani R, Bochaton‐Piallat ML. Biological responses in stented arteries. Cardiovasc Res. 2013;99:353–363. [DOI] [PubMed] [Google Scholar]
- 4. Bhasin M, Huang Z, Pradhan‐Nabzdyk L, Malek JY, LoGerfo PJ, Contreras M, Guthrie P, Csizmadia E, Andersen N, Kocher O, Ferran C, LoGerfo FW. Temporal network based analysis of cell specific vein graft transcriptome defines key pathways and hub genes in implantation injury. PLoS One. 2012;7:e39123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de la Cuesta F, Zubiri I, Maroto AS, Posada M, Padial LR, Vivanco F, Alvarez‐Llamas G, Barderas MG. Deregulation of smooth muscle cell cytoskeleton within the human atherosclerotic coronary media layer. J Proteomics. 2013;82:155–165. [DOI] [PubMed] [Google Scholar]
- 6. Wang D, Paria BC, Zhang Q, Karpurapu M, Li Q, Gerthoffer WT, Nakaoka Y, Rao GN. A role for Gab1/SHP2 in thrombin activation of PAK1: gene transfer of kinase‐dead PAK1 inhibits injury‐induced restenosis. Circ Res. 2009;104:1066–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu JH, Fanaroff AC, Sharma KC, Smith LS, Brian L, Eipper BA, Mains RE, Freedman NJ, Zhang L. Kalirin promotes neointimal hyperplasia by activating Rac in smooth muscle cells. Arterioscler Thromb Vasc Biol. 2013;33:702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartwig JH, Thelen M, Rosen A, Janmey PA, Nairn AC, Aderem A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium‐calmodulin. Nature. 1992;356:618–622. [DOI] [PubMed] [Google Scholar]
- 9. Wang J, Arbuzova A, Hangyas‐Mihalyne G, McLaughlin S. The effector domain of myristoylated alanine‐rich C kinase substrate binds strongly to phosphatidylinositol 4,5‐bisphosphate. J Biol Chem. 2001;276:5012–5019. [DOI] [PubMed] [Google Scholar]
- 10. Blackshear PJ. The MARCKS family of cellular protein kinase C substrates. J Biol Chem. 1993;268:1501–1504. [PubMed] [Google Scholar]
- 11. Hasegawa H, Nakai M, Tanimukai S, Taniguchi T, Terashima A, Kawamata T, Fukunaga K, Miyamoto E, Misaki K, Mukai H, Tanaka C. Microglial signaling by amyloid beta protein through mitogen‐activated protein kinase mediating phosphorylation of MARCKS. Neuroreport. 2001;12:2567–2571. [DOI] [PubMed] [Google Scholar]
- 12. Thelen M, Rosen A, Nairn AC, Aderem A. Regulation by phosphorylation of reversible association of a myristoylated protein kinase C substrate with the plasma membrane. Nature. 1991;351:320–322. [DOI] [PubMed] [Google Scholar]
- 13. Disatnik MH, Boutet SC, Pacio W, Chan AY, Ross LB, Lee CH, Rando TA. The bi‐directional translocation of MARCKS between membrane and cytosol regulates integrin‐mediated muscle cell spreading. J Cell Sci. 2004;117:4469–4479. [DOI] [PubMed] [Google Scholar]
- 14. Kim J, Blackshear PJ, Johnson JD, McLaughlin S. Phosphorylation reverses the membrane association of peptides that correspond to the basic domains of MARCKS and neuromodulin. Biophys J. 1994;67:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stumpo DJ, Bock CB, Tuttle JS, Blackshear PJ. MARCKS deficiency in mice leads to abnormal brain development and perinatal death. Proc Natl Acad Sci U S A. 1995;92:944–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalish JA, Willis DJ, Li C, Link JJ, Deutsch ER, Contreras MA, Quist WC, Logerfo FW. Temporal genomics of vein bypass grafting through oligonucleotide microarray analysis. J Vasc Surg. 2004;39:645–654. [DOI] [PubMed] [Google Scholar]
- 17. Willis DJ, Kalish JA, Li C, Deutsch ER, Contreras MA, LoGerfo FW, Quist WC. Temporal gene expression following prosthetic arterial grafting. J Surg Res. 2004;120:27–36. [DOI] [PubMed] [Google Scholar]
- 18. Monahan TS, Andersen ND, Martin MC, Malek JY, Shrikhande GV, Pradhan L, Ferran C, LoGerfo FW. MARCKS silencing differentially affects human vascular smooth muscle and endothelial cell phenotypes to inhibit neointimal hyperplasia in saphenous vein. FASEB J. 2009;23:557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Micallef J, Taccone M, Mukherjee J, Croul S, Busby J, Moran MF, Guha A. Epidermal growth factor receptor variant III‐induced glioma invasion is mediated through myristoylated alanine‐rich protein kinase C substrate overexpression. Cancer Res. 2009;69:7548–7556. [DOI] [PubMed] [Google Scholar]
- 20. Techasen A, Loilome W, Namwat N, Takahashi E, Sugihara E, Puapairoj A, Miwa M, Saya H, Yongvanit P. Myristoylated alanine‐rich C kinase substrate phosphorylation promotes cholangiocarcinoma cell migration and metastasis via the protein kinase C‐dependent pathway. Cancer Sci. 2010;101:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ott LE, Sung EJ, Melvin AT, Sheats MK, Haugh JM, Adler KB, Jones SL. Fibroblast migration is regulated by Myristoylated Alanine‐Rich C‐Kinase Substrate (MARCKS) protein. PLoS One. 2013;8:e66512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suk K. Unexpected role of lipocalin‐type prostaglandin D synthase in brain: regulation of glial cell migration and morphology. Cell Adh Migr. 2012;6:160–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Green TD, Park J, Yin Q, Fang S, Crews AL, Jones SL, Adler KB. Directed migration of mouse macrophages in vitro involves myristoylated alanine‐rich C‐kinase substrate (MARCKS) protein. J Leukoc Biol. 2012;92:633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Eckert RE, Neuder LE, Park J, Adler KB, Jones SL. Myristoylated alanine‐rich C‐kinase substrate (MARCKS) protein regulation of human neutrophil migration. Am J Respir Cell Mol Biol. 2010;42:586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iorio R, Bennato F, Mancini F, Colonna RC. ELF‐MF transiently increases skeletal myoblast migration: possible role of calpain system. Int J Radiat Biol. 2013;89:548–561. [DOI] [PubMed] [Google Scholar]
- 26. Kalwa H, Sartoretto JL, Sartoretto SM, Michel T. Angiotensin‐II and MARCKS: a hydrogen peroxide‐ and RAC1‐dependent signaling pathway in vascular endothelium. J Biol Chem. 2012;287:29147–29158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Satomi‐Kobayashi S, Kinugasa M, Kobayashi R, Hatakeyama K, Kurogane Y, Ishida T, Emoto N, Asada Y, Takai Y, Hirata K, Rikitake Y. Osteoblast‐like differentiation of cultured human coronary artery smooth muscle cells by bone morphogenetic protein endothelial cell precursor‐derived regulator (BMPER). J Biol Chem. 2012;287:30336–30345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rao RS, Miano JM, Olson EN, Seidel CL. The A10 cell line: a model for neonatal, neointimal, or differentiated vascular smooth muscle cells? Cardiovasc Res. 1997;36:118–126. [DOI] [PubMed] [Google Scholar]
- 29. Firulli AB, Han D, Kelly‐Roloff L, Koteliansky VE, Schwartz SM, Olson EN, Miano JM. A comparative molecular analysis of four rat smooth muscle cell lines. In Vitro Cell Dev Biol Anim. 1998;34:217–226. [DOI] [PubMed] [Google Scholar]
- 30. Rodriguez LG, Wu X, Guan JL. Wound‐healing assay. Methods Mol Biol. 2005;294:23–29. [DOI] [PubMed] [Google Scholar]
- 31. Yu D, Zhan XH, Niu S, Mikhailenko I, Strickland DK, Zhu J, Cao M, Zhan X. Murine missing in metastasis (MIM) mediates cell polarity and regulates the motility response to growth factors. PLoS One. 2011;6:e20845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Szentpetery Z, Balla A, Kim YJ, Lemmon MA, Balla T. Live cell imaging with protein domains capable of recognizing phosphatidylinositol 4,5‐bisphosphate; a comparative study. BMC Cell Biol. 2009;10:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Varnai P, Balla T. Live cell imaging of phosphoinositide dynamics with fluorescent protein domains. Biochim Biophys Acta. 2006;1761:957–967. [DOI] [PubMed] [Google Scholar]
- 34. Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. [DOI] [PubMed] [Google Scholar]
- 35. Weed SA, Du Y, Parsons JT. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J Cell Sci. 1998;111(Pt 16):2433–2443. [DOI] [PubMed] [Google Scholar]
- 36. Calabrese B, Halpain S. Essential role for the PKC target MARCKS in maintaining dendritic spine morphology. Neuron. 2005;48:77–90. [DOI] [PubMed] [Google Scholar]
- 37. Li H, Chen G, Zhou B, Duan S. Actin filament assembly by myristoylated alanine‐rich C kinase substrate‐phosphatidylinositol‐4,5‐diphosphate signaling is critical for dendrite branching. Mol Biol Cell. 2008;19:4804–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swierczynski SL, Siddhanti SR, Tuttle JS, Blackshear PJ. Nonmyristoylated MARCKS complements some but not all of the developmental defects associated with MARCKS deficiency in mice. Dev Biol. 1996;179:135–147. [DOI] [PubMed] [Google Scholar]
- 39. Taubman MB. Gene induction in vessel wall injury. Thromb Haemost. 1993;70:180–183. [PubMed] [Google Scholar]
- 40. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 41. McLaughlin S, Murray D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature. 2005;438:605–611. [DOI] [PubMed] [Google Scholar]
- 42. Trovo L, Ahmed T, Callaerts‐Vegh Z, Buzzi A, Bagni C, Chuah M, Vandendriessche T, D'Hooge R, Balschun D, Dotti CG. Low hippocampal PI(4,5)P(2) contributes to reduced cognition in old mice as a result of loss of MARCKS. Nat Neurosci. 2013;16:449–455. [DOI] [PubMed] [Google Scholar]
- 43. Caglayan E, Vantler M, Leppanen O, Gerhardt F, Mustafov L, Ten Freyhaus H, Kappert K, Odenthal M, Zimmermann WH, Tallquist MD, Rosenkranz S. Disruption of platelet‐derived growth factor‐dependent phosphatidylinositol 3‐kinase and phospholipase Cgamma 1 activity abolishes vascular smooth muscle cell proliferation and migration and attenuates neointima formation in vivo. J Am Coll Cardiol. 2011;57:2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holy EW, Jakob P, Eickner T, Camici GG, Beer JH, Akhmedov A, Sternberg K, Schmitz KP, Luscher TF, Tanner FC. PI3K/p110alpha inhibition selectively interferes with arterial thrombosis and neointima formation, but not re‐endothelialization: potential implications for drug‐eluting stent design. Eur Heart J. 2014;35:808–820. [DOI] [PubMed] [Google Scholar]
- 45. Furgeson SB, Simpson PA, Park I, Vanputten V, Horita H, Kontos CD, Nemenoff RA, Weiser‐Evans MC. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro‐inflammatory phenotype and enhances neointima formation. Cardiovasc Res. 2010;86:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sharma VP, DesMarais V, Sumners C, Shaw G, Narang A. Immunostaining evidence for PI(4,5)P2 localization at the leading edge of chemoattractant‐stimulated HL‐60 cells. J Leukoc Biol. 2008;84:440–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hall KV, Alstrup P. The prognostic factors of arterialized bypass veins in the lower extremities. Ann Chir Gynaecol. 1976;65:93–95. [PubMed] [Google Scholar]
- 48. Iioka H, Ueno N, Kinoshita N. Essential role of MARCKS in cortical actin dynamics during gastrulation movements. J Cell Biol. 2004;164:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yamaguchi H, Shiraishi M, Fukami K, Tanabe A, Ikeda‐Matsuo Y, Naito Y, Sasaki Y. MARCKS regulates lamellipodia formation induced by IGF‐I via association with PIP2 and beta‐actin at membrane microdomains. J Cell Physiol. 2009;220:748–755. [DOI] [PubMed] [Google Scholar]