

Abstract
Background
We have demonstrated previously that enzymatically degraded low‐density lipoprotein (eLDL) is an essential causative component for the initiation of atherosclerosis. Here, we investigated the different stages of human aortic valve sclerosis for the presence of eLDL and effectors of the innate immune system, as well as the interaction of eLDL with isolated valvular interstitial cells/myofibroblasts to discover possible pathways leading to aortic valve sclerosis.
Methods and Results
Human aortic valvular tissue was obtained from 68 patients undergoing valve replacement surgery. Patients were classified into 3 groups (mild, moderate, or severe aortic valve sclerosis), and clinical data for statistical analysis were gathered from all patients. Immunohistochemical staining demonstrated extensive extracellular deposits of eLDL throughout all grades of aortic valve sclerosis. Complementary analysis of lipid composition revealed higher concentrations of the decisive components of eLDL (ie, unesterified cholesterol and linoleic acid) compared with internal control tissues. Further, the complement component C3d and terminal complement complexes colocalized with eLDL compatible with the proposal that subendothelially deposited eLDL is enzymatically transformed into a complement activator at early stages of valvular cusp lesion development. Gene expression profiles of proteases and complement components corroborated by immunohistochemistry demonstrated an upregulation of the protease cathepsin D (a possible candidate for LDL degradation to eLDL) and the complement inhibitor CD55. Surprisingly, substantial C‐reactive protein expression was not observed before grade 2 aortic valve sclerosis as investigated with microarray analysis, reverse transcription–polymerase chain reaction analysis, and immunohistochemistry. Finally, we demonstrated cellular uptake of eLDL by valvular interstitial cells/myofibroblasts.
Conclusions
The present study is a startup of a hypothesis on the pathogenesis of aortic valve sclerosis declaring extracellular lipoprotein modification, subsequent complement activation, and cellular uptake by valvular interstitial cells/myofibroblasts as integral players.
Keywords: aortic valve stenosis, C‐reactive protein, low‐density lipoprotein
Although nonrheumatic aortic valve sclerosis is the most common form of valve disease in the elderly, its pathogenesis is not yet fully understood. Risk factors for the development of aortic valve sclerosis include older age, male sex, hypertension, smoking, diabetes mellitus, and hypercholesterolemia.1 This listing already suggests a comparable pathogenesis of atherosclerosis and aortic valve sclerosis.2 Actually, morphological and functional analyses of both aortic valve sclerosis and atherosclerosis bear close resemblance: Comparable to the latter, aortic valve sclerosis displays the characteristics of an active inflammatory process.3 On the other hand, studies elucidating the effect of statins on aortic valve sclerosis and atherosclerosis showed contradictory outcomes. This could be based on differences in pathogenesis but also on functional conditions. During statin therapy, increasing amounts of collagen and calcification stabilize atherosclerotic lesions, while the same effect leads to enhanced rigidity and, therefore, functional losses in aortic valve sclerosis.4
Consequently, the question arises of whether the pathogenesis of aortic valve sclerosis is comparable to the pathogenesis of atherosclerosis. The latter is regarded as a chronic inflammatory disease that develops in response to low‐density lipoprotein (LDL) entrapment.5 Among several other candidates, 2 concepts of lipoprotein modification are propagated: the widespread oxidation hypothesis6 and the less common Mainz hypothesis, which proposes that modification of LDL occurs through the action of ubiquitous hydrolytic enzymes (enzymatically modified LDL [eLDL]).7, 8 In fact, these different lipoprotein modifications do not really compete but rather complement each other.9
The Mainz hypothesis states that native LDL insudated into the intima is enzymatically modified by ubiquitous hydrolytic enzymes (ie, eLDL).10 In fact, extracellular deposits of eLDL were demonstrated by the use of monoclonal antibodies.11 Under normal circumstances (eg, normocholesterolemia), native LDL entrapped within the arterial intima is enzymatically modified (ie, eLDL), leading to a sequence of events that serve to clear the vessel wall of cholesterol. Binding of C‐reactive protein (CRP) to eLDL is the first trigger for complement activation, but in this early stage the terminal sequence is spared. Subsequent to cellular uptake of eLDL and foam cell formation,10 the physiological sequence of events is concluded by reverse cholesterol transport.7 If the capacity of the system is overburdened (eg, hypercholesterolemia), accumulation of eLDL is the consequence with subsequent generation of potentially harmful C5b‐9 complexes by both the classic and the alternative pathway.12, 13 Further, free fatty acids contained in the eLDL particle play multifaceted roles through their dual capacity to exert stimulatory and cytotoxic effects on neighboring cells.14, 15 These findings suggest a crucial role for inflammation in the initiation of atherosclerosis, with emphasis on eLDL as one important causative component.
In the present study, we investigated the different stages of human aortic valve sclerosis for the presence of eLDL and effectors of the innate immune, system as well as the interaction of eLDL with isolated valvular interstitial cells (VICs)/myofibroblasts, to discover possible pathways leading to aortic valve sclerosis.
Methods
Human Aortic Valvular Tissue
Formalin‐fixed nonrheumatic stenotic aortic valves with varying degrees of macroscopic disease were obtained from 68 patients undergoing valve replacement surgery for symptomatic aortic valve sclerosis. Patients were classified into 3 groups (mild, moderate, or severe aortic valve stenosis) according to the American College of Cardiology/American Heart Association Practice Guidelines 2006.16 Table 1 shows the detailed characteristic of the patients. Data are expressed as median (minimum difference; maximum difference; interquartile range) or (percentage) number of subjects. Sections for histological and immunohistochemical analyses were taken vertically through grade 1 to 4 lesions according to Warren and Young17 (see later).
Table 1.
Relations of Clinical Variables
| Variable | Mild Aortic Stenosis (n=4) | Moderate Aortic Stenosis (n=10) | Severe Aortic Stenosis (n=54) | P Value |
|---|---|---|---|---|
| Continuous variablesa | ||||
| Age, y | 77.0 (13.5) | 73.5 (14.25) | 76.0 (10.0) | P=0.660 |
| BMI, kg/m2 | 28.5 (3.1) | 25.9 (5.95) | 27.3 (4.8) | P=0.565 |
| Cholesterol, mg/dL | 157.0 (46.0) | 175.0 (57.0) | 191.0 (44.0) | P=0.298 |
| Creatinine, μmol/L | 110.5 (108.35) | 66.3 (70.8) | 79.6 (17.7) | P=0.269 |
| CRP, mg/dL | 0.6 (1.1) | 0.3 (1.1) | 0.2 (0.3) | P=0.283 |
| HbA1c, %Hb | 6.6 (1.7) | 5.5 (1.3) | 5.8 (0.8) | P=0.104 |
| LDL, mg/dL | 76.5 (25.0) | 101.0 (130.0) | 112.0 (54.0) | P=0.353 |
| N‐terminal of the prohormone brain natriuretic peptide, pg/mL | 573.0 (0.0) | 788.0 (3220.7) | 513.0 (1301.0) | P=0.810 |
| Categorical variablesb | ||||
| Smoking | ||||
| Never | 3 (100%) | 5 (62.5%) | 33 (73.3%) | P=1.0 |
| Past | 0 (0%) | 1 (12.5%) | 9 (20.0%) | |
| Present | 0 (0%) | 2 (25.0%) | 3 (6.7%) | |
| Hypertension | ||||
| No | 0 (0%) | 1 (10%) | 4 (7.6%) | P=0.778 |
| Yes | 4 (100%) | 9 (90%) | 49 (92.4%) | |
| Diabetes | ||||
| No | 0 (0%) | 9 (90%) | 40 (74.1%) | P=0.057 |
| Yes | 4 (100%) | 1 (10%) | 14 (25.9%) | |
Continuous variables are presented as median and interquartile range (in parentheses). BMI indicates body mass index; CRP, C‐reactive protein; LDL, low‐density lipoprotein.
Continous variables were tested with Kruskal–Wallis test.
Categorical variables were tested with Cochran–Armitage–Trend test.
Early valvular cusp lesions (grade 1) of 4 of the 68 aortic valves were kept in RNAlater (Thermo Fisher Scientific) until RNA isolation (see later). Likewise, normal and grade 1 valvular cusp areas of 11 of the 68 aortic valves were snap‐frozen in liquid nitrogen and stored at −80°C until lipid analysis (see later). The use of human tissue was approved by the Ethics Committee of the Medical Faculty of the University of Tuebingen, Germany (661/2011 BO2). All patients gave written informed consent.
Histological, Immunohistochemical, and Immunofluorescent Analyses
Paraffin‐embedded specimens were stained with Elastica van Gieson (Merck Millipore) to illustrate the layered architectural pattern and with alizarin red S to determine calcific deposition of the chosen cusp areas. Immunohistochemistry was performed by using the Dako REAL En Vision Detection System, rabbit/mouse kit (K5007; Dako Corporation). Serial sections of 3‐μm‐thick sections of paraffin‐embedded aortic valve leaflets were deparaffinized and treated with 0.3% Peroxidase Block (Dako) to block endogenous peroxidase activity, and then blocked with host serum of secondary antibody. After blocking, slides were incubated with primary antibodies listed in Table 2 for 30 minutes. In particular, the characterization of the murine monoclonal antibody AIL‐3 recognizing cryptic epitopes in human apolipoprotein B that become exposed after enzymatic degradation of LDL has been reported.11 Antigen retrieval of CD68 was achieved by heating the sections in target retrieval solution (Dako) in a steamer for 20 minutes. Application of the primary antibody was followed either directly by the secondary antibody or a bridge antibody (rabbit anti‐goat; VWR International) and the secondary antibody for 30 minutes. The reaction products were revealed by immersing the slides in diaminobenzidine tetrachloride to give a brown reaction product. Finally, the slides were counterstained with hemalaun (Merck Millipore) and mounted. An established scoring system was adopted for visual interpretation of the serial slices to allow semiquantitative analysis of the data.13 The proportion of the areas stained for C3d, C5b‐9, and CD55 relative to the overlapping area stained for eLDL (designated as 100%) was estimated and assigned to 1 of 5 scores: 0, <5%; 1, 6% to 25%; 2, 26% to 50%; 3, 51% to 75%; or 4, 76% to 100%.
Table 2.
Primary Antibodies Used for Immunohistochemical Studies
| Antigen | Clone | Source | Host | Dilution |
|---|---|---|---|---|
| α‐Actin | HHF35 | Dako | Mouse | 1:200 |
| α‐Actin | HHF35 (IgG1/κ) | Cell Marque | Mouse | 1:50 |
| Apolipoprotein B | Polyclonal | Acris | Goat | 1:1000 |
| Cathepsin D | 4G2 (IgG2b) | Acris | Mouse | 1:200 |
| C3d | Polyclonal | Dako | Rabbit | 1:500 |
| C5b‐9 | 3B1 (IgG1) | S. Bhakdi | Mouse | 1:200 |
| CD31 | JC70 (IgG1/κ) | Cell Marque | Mouse | 1:50 |
| CD55 | MEM‐118 (IgM) | Acris | Mouse | 1:100 |
| CD68 | PG‐M1 (IgG3) | Dako | Mouse | 1:100 |
| C‐reactive protein | CRP‐8 (IgG1) | Sigma | Mouse | 1:100 |
| Enzymatically modified low‐density lipoprotein | AIL‐3 (IgG1) | S. Bhakdi17 | Mouse | 1:1000 |
| Vimentin | V9 (IgG1/κ) | Cell Marque | Mouse | 1:200 |
For simultaneous localization of eLDL and C5b‐9, a double‐staining immunofluorescence was used. The primary antibodies were detected by using Alexa Fluor 488–conjugated AffiniPure F(ab′)2 Fragment Goat Anti‐Mouse IgG (H+L) and DyLightTM549‐conjugated AffiniPure Fragment Goat Anti‐Mouse IgG (H+L) (Jackson ImmunoResearch Laboratories, Inc). The sections were mounted by using Fluoroshield Mounting Medium With DAPI (Abcam).
RNA Extraction, Microarray Analysis, and Quantitative Real‐Time Polymerase Chain Reaction
Total RNA from aortic valves of 4 male patients was extracted by using a glass‐fiber filter–based protocol (mirVana miRNA Isolation Kit; Applied Biosystems/Ambion) according to the manufacturer's instructions. The RNA was eluted and resuspended in 100 μL of elution buffer. The quantity and quality of RNA extracted from aortic valves were measured both photometrically (NanoDrop ND 1000; Promega) and with the Agilent RNA 6000 Nano LabChip on a Bioanalyzer 2100 (Agilent Technologies). RNA integrity numbers (RIN 1=degraded, RIN 10=intact) were derived by using the system software. RIN scores for all 4 selected aortic valves were >6 (7.4–9.8), and both quantity and quality of the RNA were hence considered as sufficient for microarray analysis.
Expression profiles from aortic valve RNA specimens were obtained by using GeneChip Human Gene 1.0 ST Arrays (Affymetrix) according to the manufacturer's protocol. Briefly, 150 ng of total RNA was amplified and converted to sense‐strand cDNA by using the Ambion WT Expression Kit (Applied Biosystems). Fragmentation and labeling of amplified sense‐strand cDNA were carried out by using the GeneChip WT Terminal Labeling Kit (Affymetrix) according to the manufacturer`s instructions. GeneChips were hybridized, washed, and stained with use of the GeneChipHybridization, Wash and Stain Kit (Affymetrix). Hybridization for 16 hours at 60 rpm and 45°C was performed in a hybridization oven (GeneChip Hybridization Oven 640). GeneChip Arrays were subsequently washed and stained on the Affymetrix Gene Chip Fluidics Station 450DX and scanned on a GeneChip Scanner 3000 7G (Affymetrix).
As controls, we used 2 independent control sets―the data for 5 normal aortic valves as provided by Bossé et al18 and the data for 3 normal aortic valves performed by Derbali et al19 with the use of Human U133 plus 2.0 Affymetrix GeneChip microarrays. Due to low RNA integrity, one control sample (ID VAHN69) was removed by Bossé et al. Consequently, 4 remaining controls were available for our analysis. Raw and preprocessed data of the arrays can be downloaded from the National Center for Biotechnology Information website (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE12644). Expression Console (Version 1.20, Affymetrix) was applied to preprocess raw data separately for each group (cases and controls) by using robust multichip average.20 Resulting data were on log2 scale. Only the best matching probe sets between the 2 different microarray types (HuGene‐1_0‐st and HG‐U133_Plus_2) as defined by Affymetrix (http://www.affymetrix.com/Auth/support/downloads/comparisons/U133PlusVsHuGene_BestMatch.zip) were considered for further analysis. Because there was no 1‐to‐1 mapping between the probe sets, HuGene‐1_0‐st probe sets were multiplied according to the number of their corresponding best matches on HG‐U133_Plus_2, yielding 29118 probe set pairs. Finally, median normalization was applied to adjust expression levels of both groups. Patients and controls were specifically selected as white nonsmokers without type 2 diabetes (Table 3).
Table 3.
Characteristics of Patients Providing Aortic Valves for the Microarray Experiments
| ID | Age, y | BMI | Sex | Valve Area (qcm) | Mean Gradient (mm Hg) |
|---|---|---|---|---|---|
| Controls | |||||
| GSM377370 | 58 | 29.3 | Male | >2 | |
| GSM377371 | 67 | 31.1 | Male | >2 | |
| GSM377368 | 32 | 23.5 | Male | >2 | |
| GSM377369 | 51 | 21.5 | Male | >2 | |
| Average | 52.0±14.85 | 26.35±4.58 | >2 | ||
| Cases | |||||
| A‐33 | 63 | 28.7 | Male | 0.8 | 57 |
| A‐36 | 59 | 28.7 | Male | NS | 63 |
| A‐66 | 78 | 28.4 | Male | 0.8 | 46 |
| A‐67 | 76 | 23.7 | Male | 0.9 | 46 |
| Average | 69.0±9.42 | 27.38±2.45 | 0.83±0.05 | 53±8.45 | |
BMI indicates body mass index; NS, not specified.
Expressions of CRP, CD55, and cathepsin D were measured by using quantitative real‐time polymerase chain reaction (qRT‐PCR) and normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) according to the 2−ddCT method. Reverse transcription of total RNA from aortic valves was done with use of the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qRT‐PCR was performed by using the 7900HT Fast Real‐Time PCR system with TaqMan Universal Master Mix II (Applied Biosystems). The following TaqMan Gene Expression Assays (Applied Biosystems) were used for PCR amplification: CRP (Hs04183452_g1), CTSD (Hs00157205_m1), CD55 (Hs00892618_m1), and GAPDH (Hs99999905_m1).
Determination of Linoleic Acid and Free Cholesterol With Mass Spectrometry
Reference standards for linoleic acid were obtained from Nu‐Chek Prep Inc (Elysian). [2H4]Linoleic acid was purchased from Cayman Chemical (Biomol GmbH). Cholesterol was purchased from Sigma‐Aldrich, and [2H5]cholesterol was obtained through chemical synthesis.
Twelve tissue samples (10–40 mg) were homogenized with a mixture of 400 μL methanol and 125 μL water in a FastPrep 24 homogenizer (MP Biomedicals GmbH) at speed 6.0 using by lysing matrix D. Aliquots of the homogenate were immediately frozen at −80°C until analysis. Protein content was determined according to Smith et al.21
To 50 μL of tissue homogenate, 10 μL of internal standard mixture (50 pmol/μL [2H4]linoleic acid and 500 ng/μL [2H5]cholesterol in 2‐propanol), 35 μL of water and 110 μL of methanol were added, and the samples were sonicated for 20 seconds. After the addition of 160 μL of chloroform and 190 μL of water, the samples were mixed for 5 minutes, kept on ice for 10 minutes, and centrifuged for 10 minutes at 16 360g, and the organic phase was dried in a stream of nitrogen. Residues were redissolved in 250 μL of 2‐propanol.
The concentration of linoleic acid was determined by liquid chromatography (LC)‐mass spectrometry (MS)‐MS analysis in a volume of 25 μL of 2‐propanol extract. After the addition of 25 μL of mobile phase A (acetonitrile:water 6:4 v/v with 10 mmol/L ammonium formate), samples were centrifuged and the clear supernatant was transferred to autosampler vials. For LC‐MS‐MS analysis, an Agilent 6460 triple quadrupole mass spectrometer coupled to an Agilent 1200 HPLC system consisting of degasser G1379B, binary pump G1312B, well‐plate sampler G1367D, and column thermostat G1316B was used. Ionization mode was electrospray, polarity negative. The mass spectrometer was operated in Dynamic MRM mode.
For determination of free cholesterol, a volume of 50 μL of 2‐propanol extract was evaporated and derivatized to the trimethylsilyl derivatives with 20 μL of BSTFA [N,O‐bis(trimethylsilyl)trifluoroacetamide] at room temperature for 30 minutes. Gas chromatography–MS analysis was performed on a 5975C inert XL MSD, coupled to a 7890A GC (Agilent) in the EI mode. The trimethylsilyl derivatives of cholesterol and the internal standard [2H5]cholesterol were detected in SIM mode at m/z 458 and 463, respectively.
Calibration samples were extracted as the samples and analyzed together with the unknown samples. Calibration curves based on internal standard calibration were obtained by weighted (1/x) linear regression for the peak area ratio of the analyte to the respective internal standard against the amount of the analyte. The concentration in unknown samples was obtained from the regression line. Assay accuracy and precision were determined by analyzing quality controls that were prepared like the calibration samples.
VIC/Myofibroblast Culture
Five stenotic aortic valves were rinsed 3 times in PBS, placed in Dulbecco′s modified Eagle′s medium (DMEM; Biochrom) supplemented with 5% heat‐inactivated fetal bovine serum, 1% nonessential amino acids, 1% penicillin, 1% streptomycin, and 1% fungizone, and cut into small pieces. The tissue was then rinsed in serum‐free medium and incubated in 2.5 mg/mL type I collagenase (Sigma) diluted in serum‐free medium for 30 minutes at 37°C. Endothelial cells were then removed from the valve surface by slightly scraping both surfaces of the leaflets. The valves were then incubated in type I collagenase (0.8 mg/dL diluted in serum‐free medium) for an additional 30 minutes at 37°C. The tissue was then rinsed in DMEM, cut into 1‐ to 2‐mm2 pieces, and cultured in the aforementioned medium. After outgrowing VICs/myofibroblasts were removed through the use of trypsin‐EDTA (Sigma) and cultured until passages 5 or 6, the myofibroblast phenotype of the interstitial cells was confirmed by immunocytochemistry by using antibodies against smooth muscle cells and vimentin (Table 2).
Cellular Uptake of DiI‐labeled eLDL
Labeling of eLDL with 1,1′‐Dioctadecyl‐3,3,3′,3′‐Tetramethylindocarbocyanine Perchlorate (‘DiI’; DiIC18(3)) stain (Sigma) was performed as described previously.22 VICs/myofibroblasts were incubated with DiI‐labeled eLDL (20, 40, and 80 μg/mL protein eLDL) for 48 hours at 37°C. Cells were then washed 3 times with PBS and lysed with 0.2 mol/L NaOH and 0.1% SDS. The concentration of DiI‐eLDL was fluorometrically measured by using the EnSpire Multimode Reader (PerkinElmer) and calculated according to the standard curve of DiI‐eLDL (excitation and emission wavelengths 520 and 578 nm, respectively). For confocal microscopy, cells were washed 3 times with PBS, fixed in 4% paraformaldehyde, and counterstained with 4′,6‐diamidino‐2‐phenylindole mounting medium (Vector Laboratories). Confocal microscopy images were acquired by using Leica TCS SP8 laser scanning microscope (Leica Microsystems) with a ×63 oil‐immersion objective. Excitation and emission wavelengths for DiI are 540 to 550 and 575 to 640 nm, respectively.
Statistical Analyses
Kruskal–Wallis test was used for analysis of continuous clinical variables and Cochran‐Armitage for analysis of categorical variables. The goal was to evaluate whether differences regarding the immunohistochemical semiquantitative scoring system, the lipid measurements, and the microarray study could be confounded by clinical variables. Main research questions were whether eLDL by itself is present in aortic valve sclerosis, whether there are differences regarding the immunohistochemical semiquantitative scoring system between the different stages of aortic valve sclerosis, and whether there are differences regarding lipid content and gene expression between grade 1 lesions and nondiseased regions. Statistical analyses of the immunohistochemical semiquantitative scoring system and of lipoprotein measurement were performed by using the Wilcoxon matched‐pairs signed rank test. Differences in microarray expression levels between cases and controls were investigated with the package limma (Version 3.14.4) of statistical software R (Version 2.15.2; www.r-project.org). Resulting P values were adjusted for multiple testing by using the Bonferroni–Holm procedure. Statistical tests were 2‐sided and P<0.05 were considered statistically significant. Sample sizes were n=68 for histological, immunohistochemical, and immunofluorescent analyses; n=12/11 for determination of free cholesterol and linoleic acid by MS; n=4 for RNA extraction, microarray analysis, and quantitative real‐time PCR; and n=5 for VIC/myofibroblast culture.
Results
Patients
Table 1 shows statistical analyses for factors associated with aortic stenosis, classified as mild, moderate, or severe.16 Kruskal–Wallis test was used for analysis of continuous clinical variables and Cochran–Armitage test for analysis of categorical variables. None of the variables of age, body mass index, cholesterol, creatinine, CRP, hemoglobin A1c, LDL or N‐terminal of the prohormone brain natriuretic peptide level, smoking habits, hypertension, or diabetes reached statistical significance when comparing these 3 groups. Altogether, only a few patients had any comorbidities. For example, only 27.9% had diabetes, 27.3% were former or current smokers and 14.7% were renal insufficient. The same holds true for the other parameters except that 92.5% of the patients had high blood pressure.
General Morphological Findings—Grading of Aortic Valve Calcification and Structural Damage
Figure 1 shows representative examples of the 4 grades of aortic valve calcification and structural damage according to Warren and Yong.17 Early valvular cusp lesions (grade 1) are characterized by focal yellowish areas caused by incipient lipid deposition and fibrosis (Figure 1, first panel and inset) without calcification. Histologically, the overall structural integrity is retained. Grade 2 shows thickening of the respective valvular cup area with early development of calcified nodules (Figure 1, second panel). Grade 3 is characterized by large calcified nodules (Figure 1, third panel). At this stage, there may also be cholesterol crystal deposits (see later). Finally, the respective valvular cusp lesions of grade 4 are grossly distorted, and the structural integrity has been destroyed. There is much fibrosis and calcification (Figure 1, fourth panel).
Figure 1.
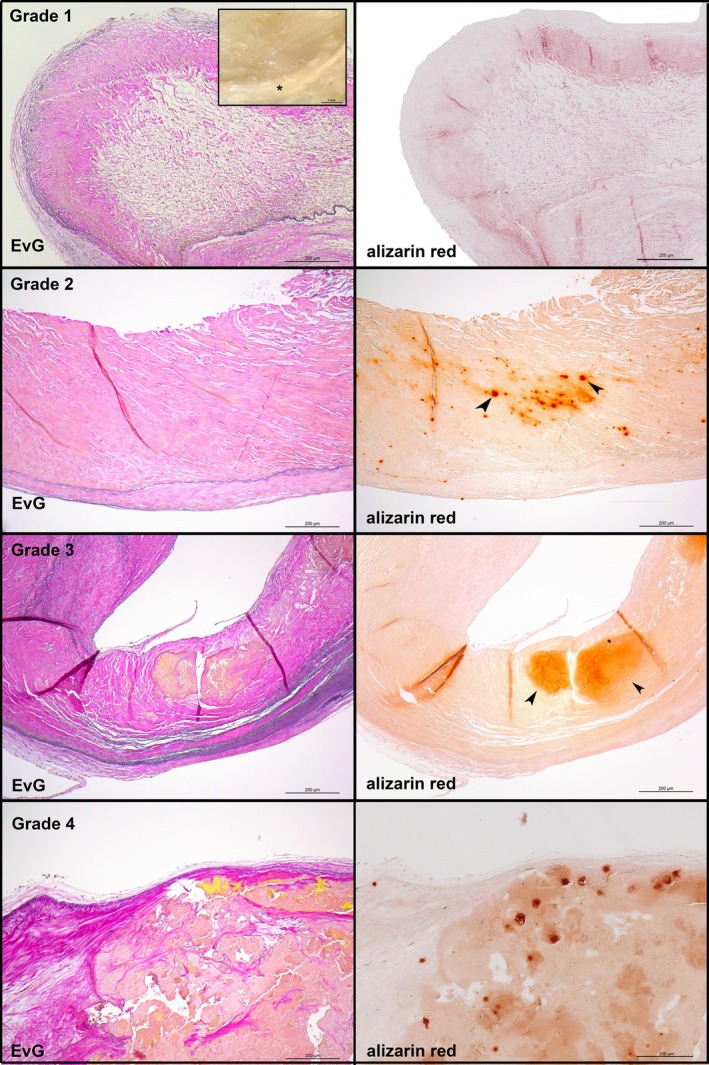
General morphological findings by grading aortic valve calcification and structural damage according to Warren and Yong.17 Left panels, van Gieson's staining demonstrating the 3 anatomic layers: fibrosa, spongiosa, and ventricularis. The fibrosa with the aortic side of the valve is to the top. Right panels, adjacent sections stained with alizarin red S to depict calcified areas. First panel, grade 1 (inset: macroscopic appearance of a representative early valvular cusp lesion. The valvular cusp was carefully inspected under a dissection microscope, and areas characterized by focal yellowish patches or streaks [asterisks] were defined as grade 1 lesions). Second panel, grade 2. Third panel, grade 3. Fourth panel, grade 4. EvG indicates Elastica van Gieson.
eLDL Is Present in All of the 4 Grades of Aortic Valve Calcification
Sixty‐eight specimens of aortic lesions fulfilling the criteria of grades 1 to 4 as defined here earlier were examined, and similar findings were made in all cases. With the use of a specific monoclonal antibody, eLDL was detectable in every lesion examined. Grade 1 and grade 2 typically showed a predominant focal deposition of eLDL in a pale insudative zone of the fibrosa below the layer of macrophage foam cells (Figure 2A, first and second panels). Nondiseased regions did not show any staining (not shown). eLDL was localized mainly extracellularly. This is exemplified in the first panel of Figure 2A, which shows staining of parallel sections with monoclonal antibody AIL‐3 and van Gieson's stain (to demonstrate connective tissue, inset). The extracellular matrix was suffused with lipid that caused distention of the tissue fibers, and positive staining for eLDL was always seen within these areas. Grade 3 and grade 4 also showed a predominant extracellular localization of eLDL, mainly around cholesterol crystal deposits (Figure 2A, third panel) and/or calcified areas (Figure 2A, fourth panel).
Figure 2.
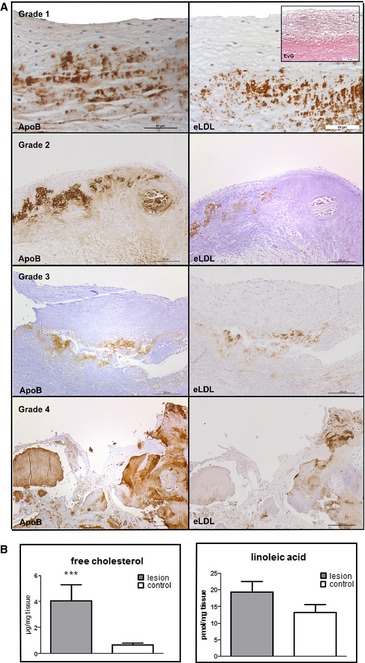
eLDL is present in all of the 4 grades of aortic valve calcification and structural damage. A, Left panels, apolipoprotein B staining demonstrating presence of LDL in general. The fibrosa with the aortic side of the valve is to the top. Right panels, adjacent sections stained with monoclonal antibody AIL‐3 for eLDL demonstrating extensive enzymatic modification of LDL. First panel, grade 1 (adjacent sections stained for eLDL with monoclonal antibody AIL‐3 and connective tissue by van Gieson's staining (inset), respectively. Note the extracellular matrix of the fibrosa suffused with aggregates of the modified lipoprotein). Second panel, grade 2. Third panel, grade 3. Fourth panel, grade 4. B, Analysis of lipid composition in early valvular cusp lesions (grade 1, dark gray) and internal control tissue (nondiseased areas, white) by GC‐MS for free cholesterol (left, n=12) and LC‐MS‐MS for linoleic acid (right, n=11). Note the higher concentrations of both constituents compared with the internal control tissue. ***P < 0.001. eLDL indicates enzymatically modified low‐density lipoprotein; EvG, Elastica van Gieson; GC, gas chromatography; MS, mass spectrometry; LC, liquid chromatography.
As evidenced from the left panels of Figure 2A, eLDL staining largely overlapped with apolipoprotein B staining demonstrating extensive enzymatic modification of LDL. Internal control tissue showed neither eLDL nor apolipoprotein B staining (Figure S1).
Free Cholesterol and Linoleic Acid Are Present in Early Valvular Cusp Lesions
Both unesterified cholesterol and linoleic acid are main and decisive constituents of eLDL.8 Therefore, lipids were extracted from 11 or 12 early valvular cusp lesions (grade 1) and from 11 or 12 visually nondiseased areas of the same valvular cusp as internal control. Lipid composition was analyzed by using GC‐MS for free cholesterol and LC‐MS‐MS for linoleic acid; statistical analysis was performed by using Wilcoxon matched‐pairs signed rank test. In early valvular cusp lesions, a significant higher concentration of free cholesterol was found compared with the internal control tissues (P=0.001; median difference 2.86; minimum difference 0.07, maximum difference 14.5; Figure 2B, left panel). The concentration of linoleic acid was higher in early valvular cusp lesions, too. However, due to high variations, these data did not reach statistical significance (P=0.137; median difference 3.095; minimum difference 2.26, maximum difference 17.69, Figure 2B, right panel).
Colocalization of eLDL With C3d and C5b‐9
Complement activation is a leading effect of eLDL in atherosclerotic lesions,10, 12, 13 and C3d and C5b‐9 deposits were also present in every grade 1 to 4 valvular cusp lesion examined. Grade 1 and grade 2 typically showed a predominant focal deposition of C3d in the pale insudative zone of the fibrosa below the layer of macrophage foam cells and in the deeper part of the fibrosa adjacent to the spongiosa. The same holds true for C5b‐9 (Figure 3A, first and second panels). In grade 1, the proportion of the area stained for C3d relative to the area stained for eLDL (eLDL/C3d), however, was significantly larger than for eLDL/C5b‐9 (median difference 2, minimum difference 1, maximum difference 4, P=0.0052, Wilcoxon matched‐pairs signed rank test), indicating that the complement cascade partially halts before the terminal sequence. From grade 2 up, however, there are no longer significant differences, indicating a prevalence of complete complement activation in intermediate and advanced stages of aortic valve calcification. Internal control tissue showed neither C3d nor C5b‐9 staining (Figure S1).
Figure 3.
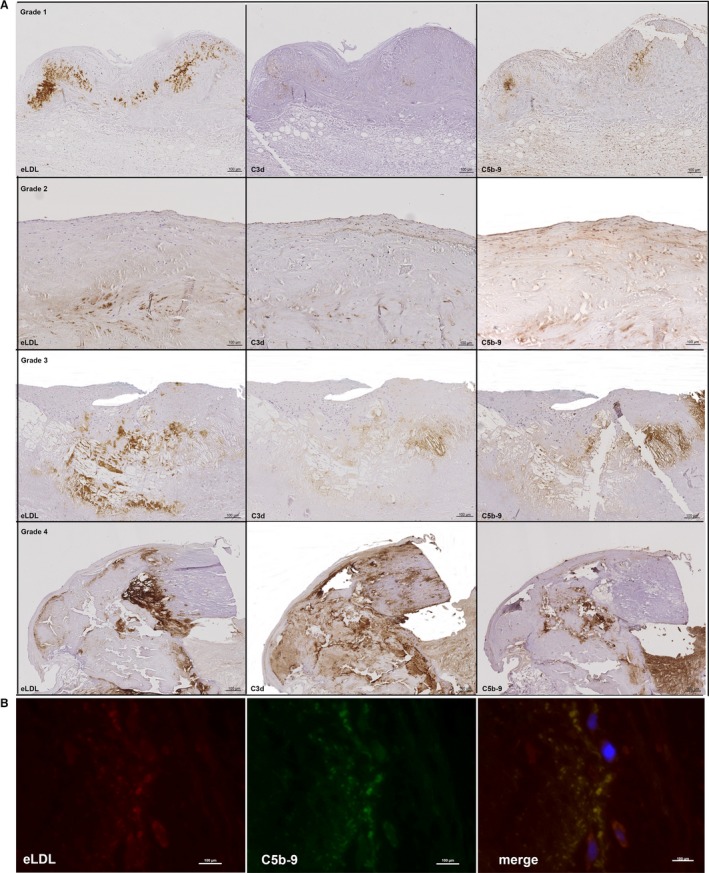
Colocalization of eLDL with C3d and C5b‐9. A, Sequential sections of the 4 grades of aortic valve calcification and structural damage stained for eLDL (left panels), C3d (middle panels), and C5b‐9 (right panels). Note the close intermingling and overlap of the different antigens in all of the 4 grades. In all panels, the fibrosa with the aortic side of the valve is to the top. B, Representative double immunofluorescence staining of another early valvular cusp lesion demonstrating perfect overlap of eLDL and C5b‐9 (merge, yellow indicates colocalization). The aortic side of the valve is to the right. eLDL indicates enzymatically modified low‐density lipoprotein.
The close spatial relation between the different antigens was further illustrated by double immunofluorescence staining for eLDL and C5b‐9 (Figure 3B) demonstrating almost perfect concordance. Nondiseased regions did not show any staining. The controls processed with the irrelevant isotype‐matched monoclonal mouse antibody instead of the specific antibody were completely negative (not shown).
Differential Regulation of Proteases and Complement Components in Early Valvular Cusp Lesions Compared With Control Probes
Enzymatic modification of LDL requires proteases,10, 23 and both binding to CRP and complement activation12, 13 are important capacities of eLDL in atherogenesis. We therefore performed expression studies by using Affymetrix HuGene 1‐0 st v1 arrays to determine genes differentially regulated in early valvular cusp lesions (grade 1) compared with healthy probe sets. Figure 4 shows gene expression profiles of proteases and complement components depicted as a heat map with upregulation and downregulation in red and blue, respectively, and expression between indicated in white (Figure 4A), as well as fold‐ change and 95% CI (Figure 4B). Notably, there was an upregulation of the protease cathepsin D (CTSD; fold‐change 5.53, adjusted P value 9.68×10−6) and the complement inhibitor CD55 (decay accelerating factor; fold‐change 8.97, adjusted P value 5.47×10−7). In contrast, there was just a low expression of CRP (fold‐change 0.51, adjusted P value 1.62×10−3) as log2 expression levels were <6 in both lesions and controls (for more details, see Table S1). To further support this finding, we compared our cases with 3 additional controls19 who were 58, 62, and 73 years old. As before, CD55 and CTSD were significantly higher expressed in our cases compared with the 3 controls in unmatched (unadjusted Welch's t test P≤0.0025) as well as in age‐matched analysis (paired t test P≤0.042; pairing expression of 58 and 59, 62 and 63, and 73 and mean of 76 and 78 years for controls and cases), although fold‐changes were less prominent compared with the 4 controls (CD55: fold‐change [case/control]=2.5–2.7, CTSD 7.1). CRP expression did not differ significantly between cases and the 3 controls, although expression in controls was slightly elevated compared with cases (fold‐change: unmatched 0.85–0.96, matched 0.78–0.89) and controls showed very similar CRP expression (data not shown). These results were confirmed both by qRT‐PCR of the same and 4 additional independent early valvular cusp lesions (grade 1) not used for the microarray study before (Figure 5A) as well as immunohistochemistry (see later and Figure 5B).
Figure 4.
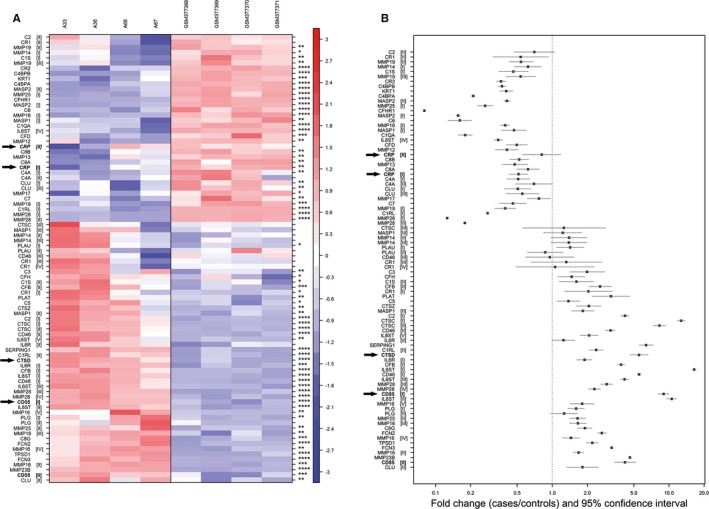
CTSD and CD55 are present in early valvular cusp lesions (grade 1). A, Gene expression profiles of proteases and complement components. The rows (probe sets) in the heat map were hierarchically clustered (complete linkage) based on Pearson's correlation. Rows were centered to mean 0 and scaled to SD 1. Upregulation and downregulation are shown in red and blue, respectively; expression between is indicated in white. Columns on the left side represent samples of early valvular cusp lesions (A‐33, A‐36, A‐66, A‐67); those shown on the right side are controls (GSM377368, GSM377369, GSM377370, GSM37737118). Each row represents a different probe set tagging a specific protease or complement component indicated on the left. Different probe sets of one specific gene are marked with roman numerals in parenthesis. (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001). CRP, CTSD and CD55 are highlighted by arrows. For more details, see Table S1. B, Fold‐changes (cases vs controls) and 95% CIs of microarray expression profiles. Genes represented by probe sets that were not significantly differentially expressed between cases and controls and/or low expressed (defined as mean log2 signal intensities <5 in both, cases and controls) are not displayed. Probe sets (y‐axis) are sorted according to (A). Fold‐changes and 95% CIs (x‐axis) are indicated by black squares and vertical lines, respectively. The dashed horizontal line represents a fold‐change of 1 (ie, no difference between cases and controls); fold‐change above (below) indicate an upregulation (downregulation) in cases compared with controls. CRP indicates C‐reactive protein; CTSD, cathepsin D.
Figure 5.
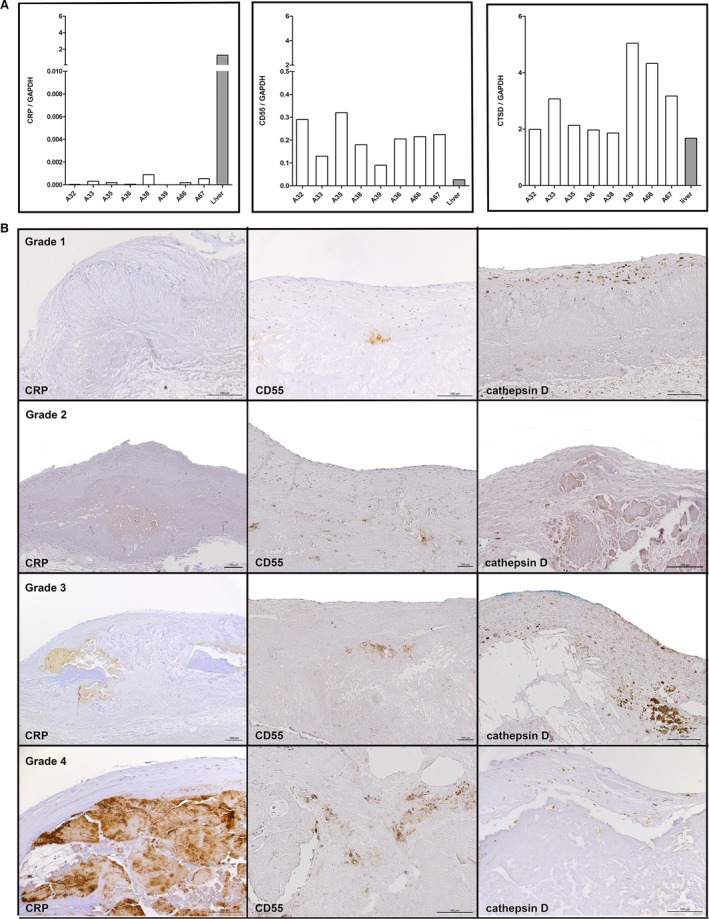
A, Confirmation of gene expression profiles of CRP, CD55, and CTSD by qRT‐PCR. The mRNA‐levels were assessed in the same tissue used for the microarrays (A33, A36, A66, A67) and in 4 additional independent early valvular cusp lesions (grade 1) not used for the microarray study before (A32, A35, A38, A39) and normalized to GAPDH as reference gene. Liver tissue was used as positive control. B, Representative (not sequential) sections of the 4 grades of aortic valve calcification and structural damage stained for CRP (left panels), CD55 (middle panels), and CTSD (right panels). Note the lack of extracellular CRP staining in early valvular cusp lesions (grade 1) and increasing extracellular staining around calcified areas of grades 2 to 4 lesions, focal extracellular deposition of CD55, and the marked intracellular staining of foam cells with CTSD. The fibrosa with the aortic side of the valve is to the top. CRP indicates C‐reactive protein; CTSD, cathepsin D; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; qRT‐PCR, quantitative real‐time polymerase chain reaction.
Lack of CRP in Early Valvular Cusp Lesions
One of the hallmarks of atherosclerosis is the close spatial and functional association between eLDL, complement components and CRP within the vessel wall.11, 12, 13 It was thus self‐evident to investigate whether the same holds true for the 4 grades of aortic valve calcification and structural damage. As already indicated here, array analysis did show only low CRP expression within early valvular cusp lesions (grade 1). This result was corroborated by RT‐PCR analysis (Figure 5A). It is well known, however, that most of the CRP within atherosclerotic lesions derives from plasma CRP insudation.24 We therefore performed extensive immunohistochemical studies using a well‐established mouse monoclonal anti‐CRP antibody.12, 13, 24, 25 Surprisingly, besides increasing extracellular staining around calcified areas of grade 2 to 4 lesions (rather impressive in grade 4 lesions) as well as rare intracellular staining, we were not able to detect any extracellular staining in early lesions (Figure 5B, left panels).
Cathepsin D and CD55 Are Present in All of the 4 Grades of Aortic Valve Calcification
Next, we sought to corroborate the results obtained with microarray analysis (Figure 4) and RT‐PCR (Figure 5A) to both confirm and specify cathepsin D and CD55 expression in early valvular cusp lesions (grade 1). To this end, tissue sections were stained for cathepsin D or CD55 (Table 2) and compared with eLDL staining (not shown). As shown in Figure 5B (upper right panel), the predominant manifestation of cathepsin D was a marked intracellular staining of foam cells. Scattered cathepsin D–positive cells were also detectable in the pale insudative zone of the fibrosa below the layer of macrophage foam cells suffused with eLDL. Within this part of the fibrosa, focal deposits of CD55 were present (Figure 5B, upper middle panel). The proportion of the area stained for C5b‐9 relative to the area stained for eLDL (eLDL/C5b‐9), however, was significantly larger than that for eLDL/CD55 (median difference 2, minimum difference 1, maximum difference 3, P=0.0126), indicating that CD55 is not able to completely inhibit formation of the terminal complement complex. From grade 2 to grade 4, the same staining pattern of both cathepsin D and CD55 as in grade 1 lesions was observed, although with increasing staining areas and intensities for CD55 (Figure 5B, second to fourth panels, middle and right). Internal control tissue showed neither cathepsin D nor CD55 staining (Figure S1).
Uptake of eLDL by Cultured VICs/Myofibroblasts
The next experiments were undertaken to determine whether eLDL would be taken up by VICs/myofibroblasts. As a measure for cholesterol uptake, we analyzed DiI‐labeled eLDL internalization by using both confocal microscopy and fluorometric quantification measurements. The phenotype of VICs/myofibroblasts was confirmed by positive staining for both vimentin and α‐actin (Figure 6A). After incubation with DiI‐labeled eLDL for 48 hours at 37°C, VICs/myofibroblasts demonstrated a strong perinuclear signal from DiI‐labeled eLDL compared with control cells incubated with medium alone. Likewise, fluorometric analyses of 3 independent experiments showed that VICs/myofibroblasts accumulate up to 40 μg of DiI‐eLDL/mg protein (Figure 6B).
Figure 6.
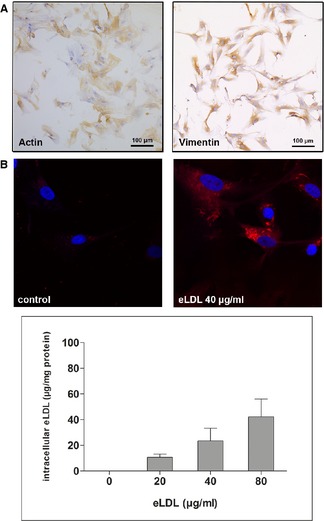
Uptake of eLDL by cultured valvular VICs/myofibroblasts. A, immunocytochemistry with monoclonal antibodies against vimentin and α‐actin (see Table 2) confirming the phenotype of VICs/myofibroblasts. B, Upper panel, confocal microscopy demonstrating a strong perinuclear signal from DiI‐labeled eLDL. Lower panel, fluorometric analyses of 3 independent experiments showing that VICs/myofibroblasts incubated with increasing eLDL concentrations accumulate up to 40 μg DiI‐eLDL/mg protein. eLDL indicates enzymatically modified low‐density lipoprotein; VICs, valvular interstitial cells.
Discussion
Until now, the pathogenesis of aortic valve sclerosis has not been yet fully understood. Because of comparable risk factors and functional processes of aortic valve sclerosis and atherosclerosis, a related pathogenesis of both entities is very likely. Certainly, modified lipoproteins are intimately involved.3 eLDL is thought to be one of the initiators of atherosclerosis.7, 8, 10, 11 The novel findings of the present study are the evidence for the presence of this modified lipoprotein in all stages of aortic valve sclerosis, its role as a possible complement activator, and its cellular uptake by VICs/myofibroblasts.
For the appropriate interpretation of these results, one has to consider 2 different classifications of aortic valve sclerosis―on the one hand, the clinical classification related to a variety of hemodynamic and natural history data,16 and on the other hand, the histological grading related to the aortic valve calcification and structural damage.17 Regarding the latter, it is important to keep in mind that a single excised aortic valve usually shows ≥2 of the 4 histological grades and even unaffected cusp areas.
By means of histological and immunohistological staining, we could demonstrate predominant extracellular localization of eLDL as early as in grade 1 lesions. As initiating pathophysiological effects of modified lipoproteins depends on their extracellular localization, the question of localization is of paramount importance. Macrophages produce a plethora of enzymes, so detection of modified LDL in cells may not attain the significance for initiating pathogenic events attributed to extracellular modified LDL. Acquisition of direct evidence that eLDL is indeed present extracellularly became a pressing issue, and immunohistochemical localization appeared to us the method of choice. If we may assume that enzymatic degradation and fusion of lipid particles are essentially synonymous,10 our findings are in perfect accordance with the recently published electron micrographs of isolated extracellular lipid particles from aortic valves.26 Compared with the histological structure of adaptive intimal thickening of human arteries prone to atherosclerotic lesion development,27 the composition of the human aortic valve cusp with the fibrosa suffused with insudated lipoproteins is quite different. This dense matrix might favor entrapment of insudated lipoproteins counteracting sufficient statin effects.
Both unesterified cholesterol and linoleic acid are the decisive components of eLDL.8 The analysis of the lipid composition of early valvular cusp lesions (grade 1) compared with internal control tissues by mass spectrometry detecting both unesterified cholesterol and linoleic acid corroborated our immunohistochemical findings. While it is known that free cholesterol has an intrinsic complement‐activating capacity (see later),28 free fatty acids contained in the eLDL particle not only bind and activate C129 but also play multifaceted roles through their dual capacity to exert stimulatory and cytotoxic effects on neighboring cells.14, 15
The concept that we have been pursuing proposes extracellular enzymatic degradation of LDL to represent a key step leading to generation of an atherogenic lipoprotein.7, 10, 11, 30 eLDL generated by all tested proteases is endowed with the same atherogenic properties as the originally described trypsin‐generated eLDL.23, 31 Evidence for the presence of proteases like matrix metalloproteinases or cathepsins in aortic valve sclerosis is abundant.4 Cathepsin D, which we have now identified as an important intracellular constituent of aortic valve sclerosis, is a hydrolytic enzyme, known to be released from macrophages in atherosclerotic lesions that may induce hydrolytic modification of LDL.32 The present demonstration of cathepsin D and eLDL at close range in all stages of aortic valve sclerosis is of distinct interest. It is conceivable that proteases like cathespin D serve a dual role: (1) they convert the trapped lipoprotein into a molecule with proinflammatory properties and (2) play an important role in matrix remodeling as a prerequisite for valve calcification.4 Of course, we cannot speculate which of the known proteases assumes the decisive role of LDL modification in vivo. Proteases are highly redundant, and proteolytic activity on a given substrate is not dependent on the presence of a single enzyme.23
In parallel to findings in early atherosclerotic lesions, we could demonstrate immunohistochemical staining for C3d and C5b‐9,11, 13, 33 the terminal membrane attack complex, in all stages of aortic valve sclerosis. Complement activation in aortic stenosis has already been detected before,34 but in the present study we were able to show close scrutiny of the staining patterns of complement components with eLDL for the first time, and our semiquantitative immunohistochemical analysis of eLDL and C5b‐9 suggests that C5b‐9 deposits reflect the presence of extensively modified LDL, whereby other possible complement activating mechanisms are not excluded. The significance of local complement activation itself can hardly be overemphasized. Complement plays a role in promoting the progression of cardiovascular disease, for example by acting as a drive to inflammation by increasing secretion of monocyte chemotactic protein‐1 and/or interleukin‐8.34, 35 Possible targets for sublytic complement attack in aortic valve sclerosis are VICs/myofibroblasts, the main cell type of the aortic valve (Figure 6A).36
Besides its extracellular effects, the observed cellular uptake of eLDL by VICs/myofibroblasts may also initiate a plethora of harmful proinflammatory, profibrotic, and calcifying events leading to aortic valve sclerosis. As similar atherogenic effects of eLDL are well documented37 and other LDL modifications like oxidation have been reported to exert such effects on uptake by VICs/myofibroblasts,38 these data call for further detailed investigation.
We have previously shown that eLDL triggers complement activation via 2 pathways. The first is CRP dependent, occurs at low eLDL concentrations, and halts before the terminal sequence. The terminal sequence is thereby spared, rendering CRP a putative protective function that is overrun at higher eLDL concentrations. Accordingly, the second is CRP independent, occurs at higher eLDL concentrations, and drives complement activation to completion.12, 13 Unlike in early atherosclerotic lesions, in the present study we were not able to detect mentionable amounts of CRP in early valvular cusp lesions (grade 1), using both immunohistochemical staining and gene expression analysis or RT‐PCR. Increasing extracellular staining was observed not before grade 2 aortic valve sclerosis and was rather impressive around calcified areas of 4 lesions consistent with increased CRP levels in sera of patients with aortic stenosis.39 Regardless, the lack of extracellular staining of CRP in early valvular cusp lesions (grade 1) in contrast to early atherosclerotic lesions is remarkable and might contribute to the disparities in the pathogenesis of both entities. Specifically, tempering of terminal complement activation by CRP bound to eLDL obviously does not apply to early valvular cusp lesions, thus clearing the way for detrimental terminal complement activation of eLDL not even inhibited by late‐onset CRP accumulation.
Another mediator involved in the regulation of the complement pathway is decay accelerating factor (CD55). In the present study, we were able to detect both upregulation of CD55 expression compared with control cusps in early valvular cusp lesions (grade 1) and focal deposition of CD55 in all stages of aortic valve sclerosis. However, our semiquantitative immunohistochemical analysis of CD55 and C5b‐9 suggests that the protective effect of CD55 is rather limited and overrun by eLDL‐driven complement activation.
Coming back to the ambivalent relationship between atherosclerosis and aortic valve sclerosis, the present findings that provide evidence of extracellularly degraded LDL, complement activation and cellular uptake of eLDL point to similarities, on the one hand, while the lack of significant CRP expression in early valvular cusp lesions points to disparities, on the other hand. In any event, the present study is a startup of a hypothesis on the pathogenesis of aortic valve sclerosis declaring extracellular lipoprotein modification, subsequent complement activation, and cellular uptake by VICs/myofibroblasts as integral players. Future research demands investigation of the interaction between eLDL and VICs/myofibroblasts with special emphasis on proinflammatory and/or anti‐inflammatory cytokine patterns evoked by cellular uptake of eLDL. It is tempting to evaluate possible strategies for the prevention of the well‐known proinflammatory effects of eLDL.
Sources of Funding
This work was supported by the innovation fund of the Robert‐Bosch‐Hospital, Stuttgart, Germany, and the Robert Bosch Foundation, Germany. Drs Schwab and Schaeffeler were supported in part by the Deutsche Forschungsgemeinschaft KFO 274 (grant SCHW858/1‐1).
Disclosures
None.
Supporting information
Figure S1. Internal control tissues for both histochemistry and immunohistochemistry comprising unaffected areas of 8 different aortic valves. In (A) note the aortic valve leaflet composed of 3 anatomic layers: the fibrosa, a collagen‐rich layer on the aortic side of the valve (top layer); the spongiosa, a layer of variable thickness lying between the fibrosa and ventricularis, with a less dense matrix and often containing adipose cells and the ventricularis, a layer with prominent elastin fibers located on the ventricular side of the valve (bottom layer). In (B through F), note the lack of calcified areas (B) as well as any of the antigens detected from grade 1 upward (C through F, compare with Tables 1, 2, 3 and 5).
Table S1. Results of Comparison Between Cases (n=4) and Controls (n=4) for Selected Genes by Microarray Analysis
Acknowledgments
We owe many thanks to Sucharit Bhakdi, MD (University of Mainz, Germany), for providing the monoclonal antibodies against eLDL and the neoantigens of C5b‐9. We thank Kerstin Winter, Monika Seiler, and Andrea Jarmuth for expert technical assistance.
(J Am Heart Assoc. 2015;4:e002156 doi: 10.1161/JAHA.115.002156)
Accompanying Figure S1 and Table S1 are available at http://jaha.ahajournals.org/content/4/10/e002156/suppl/DC1
References
- 1. Rajamannan NM, Evans FJ, Aikawa E, Grande‐Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: a review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: calcific aortic valve disease‐2011 update. Circulation. 2011;124:1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Milin AC, Vorobiof G, Aksoy O, Ardehali R. Insights into aortic sclerosis and its relationship with coronary artery disease. J Am Heart Assoc. 2014;3:e001111 doi: 10.1161/JAHA.114.001111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carabello BA. Introduction to aortic stenosis. Circ Res. 2013;113:179–185. [DOI] [PubMed] [Google Scholar]
- 4. Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: methods, models, and mechanisms. Circ Res. 2011;108:1392–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Williams KJ, Tabas I. The response‐to‐retention hypothesis of atherogenesis reinforced. Curr Opin Lipidol. 1998;9:471–474. [DOI] [PubMed] [Google Scholar]
- 6. Steinberg D. Low density lipoprotein oxidation and its pathobiological significance. J Biol Chem. 1997;272:20963–20966. [DOI] [PubMed] [Google Scholar]
- 7. Bhakdi S, Lackner KJ, Han SR, Torzewski M, Husmann M. Beyond cholesterol: the enigma of atherosclerosis revisited. Thromb Haemost. 2004;91:639–645. [DOI] [PubMed] [Google Scholar]
- 8. Torzewski M, Bhakdi S. Complement and atherosclerosis‐united to the point of no return? Clin Biochem. 2013;46:20–25. [DOI] [PubMed] [Google Scholar]
- 9. Torzewski M, Lackner KJ. Initiation and progression of atherosclerosis–enzymatic or oxidative modification of low‐density lipoprotein? Clin Chem Lab Med. 2006;44:1389–1394. [DOI] [PubMed] [Google Scholar]
- 10. Bhakdi S, Dorweiler B, Kirchmann R, Torzewski J, Weise E, Tranum‐Jensen J, Walev I, Wieland E. On the pathogenesis of atherosclerosis: enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J Exp Med. 1995;182:1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Torzewski M, Klouche M, Hock J, Messner M, Dorweiler B, Torzewski J, Gabbert HE, Bhakdi S. Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler Thromb Vasc Biol. 1998;18:369–378. [DOI] [PubMed] [Google Scholar]
- 12. Bhakdi S, Torzewski M, Klouche M, Hemmes M. Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler Thromb Vasc Biol. 1999;19:2348–2354. [DOI] [PubMed] [Google Scholar]
- 13. Bhakdi S, Torzewski M, Paprotka K, Schmitt S, Barsoom H, Suriyaphol P, Han SR, Lackner KJ, Husmann M. Possible protective role for C‐reactive protein in atherogenesis: complement activation by modified lipoproteins halts before detrimental terminal sequence. Circulation. 2004;109:1870–1876. [DOI] [PubMed] [Google Scholar]
- 14. Reiss K, Cornelsen I, Husmann M, Gimpl G, Bhakdi S. Unsaturated fatty acids drive disintegrin and metalloproteinase (ADAM)‐dependent cell adhesion, proliferation, and migration by modulating membrane fluidity. J Biol Chem. 2011;286:26931–26942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suriyaphol P, Fenske D, Zahringer U, Han SR, Bhakdi S, Husmann M. Enzymatically modified nonoxidized low‐density lipoprotein induces interleukin‐8 in human endothelial cells: role of free fatty acids. Circulation. 2002;106:2581–2587. [DOI] [PubMed] [Google Scholar]
- 16. Bonow RO, Carabello BA, Kanu C, de Leon AC Jr, Faxon DP, Freed MD, Gaasch WH, Lytle BW, Nishimura RA, O'Gara PT, O'Rourke RA, Otto CM, Shah PM, Shanewise JS, Smith SC Jr, Jacobs AK, Adams CD, Anderson JL, Antman EM, Faxon DP, Fuster V, Halperin JL, Hiratzka LF, Hunt SA, Lytle BW, Nishimura R, Page RL, Riegel B. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to revise the 1998 guidelines for the management of patients with valvular heart disease): developed in collaboration with the Society of Cardiovascular Anesthesiologists: endorsed by the Society for Cardiovascular Angiography and Interventions and the Society of Thoracic Surgeons. Circulation. 2006;114:e84–e231. [DOI] [PubMed] [Google Scholar]
- 17. Warren BA, Yong JL. Calcification of the aortic valve: its progression and grading. Pathology. 1997;29:360–368. [DOI] [PubMed] [Google Scholar]
- 18. Bosse Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2:489–498. [DOI] [PubMed] [Google Scholar]
- 19. Derbali H, Bosse Y, Cote N, Pibarot P, Audet A, Pepin A, Arsenault B, Couture C, Despres JP, Mathieu P. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via toll‐like receptor 2. Am J Pathol. 2010;176:2638–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of affymetrix genechip probe level data. Nucleic Acids Res. 2003;31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. [DOI] [PubMed] [Google Scholar]
- 22. Kapinsky M, Torzewski M, Buchler C, Duong CQ, Rothe G, Schmitz G. Enzymatically degraded LDL preferentially binds to CD14(high) CD16(+) monocytes and induces foam cell formation mediated only in part by the class B scavenger‐receptor CD36. Arterioscler Thromb Vasc Biol. 2001;21:1004–1010. [DOI] [PubMed] [Google Scholar]
- 23. Torzewski M, Suriyaphol P, Paprotka K, Spath L, Ochsenhirt V, Schmitt A, Han SR, Husmann M, Gerl VB, Bhakdi S, Lackner KJ. Enzymatic modification of low‐density lipoprotein in the arterial wall: a new role for plasmin and matrix metalloproteinases in atherogenesis. Arterioscler Thromb Vasc Biol. 2004;24:2130–2136. [DOI] [PubMed] [Google Scholar]
- 24. Torzewski M, Rist C, Mortensen RF, Zwaka TP, Bienek M, Waltenberger J, Koenig W, Schmitz G, Hombach V, Torzewski J. C‐reactive protein in the arterial intima: role of C‐reactive protein receptor‐dependent monocyte recruitment in atherogenesis. Arterioscler Thromb Vasc Biol. 2000;20:2094–2099. [DOI] [PubMed] [Google Scholar]
- 25. Torzewski J, Torzewski M, Bowyer DE, Frohlich M, Koenig W, Waltenberger J, Fitzsimmons C, Hombach V. C‐reactive protein frequently colocalizes with the terminal complement complex in the intima of early atherosclerotic lesions of human coronary arteries. Arterioscler Thromb Vasc Biol. 1998;18:1386–1392. [DOI] [PubMed] [Google Scholar]
- 26. Lehti S, Kakela R, Horkko S, Kummu O, Helske‐Suihko S, Kupari M, Werkkala K, Kovanen PT, Oorni K. Modified lipoprotein‐derived lipid particles accumulate in human stenotic aortic valves. PLoS One. 2013;8:e65810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stary HC, Blankenhorn DH, Chandler AB, Glagov S, Insull W Jr, Richardson M, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of the intima of human arteries and of its atherosclerosis‐prone regions. A report from the committee on vascular lesions of the council on arteriosclerosis, American Heart Association. Circulation. 1992;85:391–405. [DOI] [PubMed] [Google Scholar]
- 28. Alving CR, Richards RL, Guirguis AA. Cholesterol‐dependent human complement activation resulting in damage to liposomal model membranes. J Immunol. 1977;118:342–347. [PubMed] [Google Scholar]
- 29. Biro A, Ling WL, Arlaud GJ. Complement protein C1q recognizes enzymatically modified low‐density lipoprotein through unesterified fatty acids generated by cholesterol esterase. Biochemistry. 2010;49:2167–2176. [DOI] [PubMed] [Google Scholar]
- 30. Seifert PS, Hugo F, Tranum‐Jensen J, Zahringer U, Muhly M, Bhakdi S. Isolation and characterization of a complement‐activating lipid extracted from human atherosclerotic lesions. J Exp Med. 1990;172:547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Han SR, Momeni A, Strach K, Suriyaphol P, Fenske D, Paprotka K, Hashimoto SI, Torzewski M, Bhakdi S, Husmann M. Enzymatically modified LDL induces cathepsin H in human monocytes: potential relevance in early atherogenesis. Arterioscler Thromb Vasc Biol. 2003;23:661–667. [DOI] [PubMed] [Google Scholar]
- 32. Hakala JK, Oksjoki R, Laine P, Du H, Grabowski GA, Kovanen PT, Pentikainen MO. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2003;23:1430–1436. [DOI] [PubMed] [Google Scholar]
- 33. Torzewski M, Torzewski J, Bowyer DE, Waltenberger J, Fitzsimmons C, Hombach V, Gabbert HE. Immunohistochemical colocalization of the terminal complex of human complement and smooth muscle cell alpha‐actin in early atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 1997;17:2448–2452. [DOI] [PubMed] [Google Scholar]
- 34. Helske S, Oksjoki R, Lindstedt KA, Lommi J, Turto H, Werkkala K, Kupari M, Kovanen PT. Complement system is activated in stenotic aortic valves. Atherosclerosis. 2008;196:190–200. [DOI] [PubMed] [Google Scholar]
- 35. Torzewski J, Oldroyd R, Lachmann P, Fitzsimmons C, Proudfoot D, Bowyer D. Complement‐induced release of monocyte chemotactic protein‐1 from human smooth muscle cells. A possible initiating event in atherosclerotic lesion formation. Arterioscler Thromb Vasc Biol. 1996;16:673–677. [DOI] [PubMed] [Google Scholar]
- 36. Mulholland DL, Gotlieb AI. Cell biology of valvular interstitial cells. Can J Cardiol. 1996;12:231–236. [PubMed] [Google Scholar]
- 37. Klouche M, Gottschling S, Gerl V, Hell W, Husmann M, Dorweiler B, Messner M, Bhakdi S. Atherogenic properties of enzymatically degraded LDL: selective induction of MCP‐1 and cytotoxic effects on human macrophages. Arterioscler Thromb Vasc Biol. 1998;18:1376–1385. [DOI] [PubMed] [Google Scholar]
- 38. Syvaranta S, Alanne‐Kinnunen M, Oorni K, Oksjoki R, Kupari M, Kovanen PT, Helske‐Suihko S. Potential pathological roles for oxidized low‐density lipoprotein and scavenger receptors SR‐AI, CD36, and LOX‐1 in aortic valve stenosis. Atherosclerosis. 2014;235:398–407. [DOI] [PubMed] [Google Scholar]
- 39. Galante A, Pietroiusti A, Vellini M, Piccolo P, Possati G, De Bonis M, Grillo RL, Fontana C, Favalli C. C‐reactive protein is increased in patients with degenerative aortic valvular stenosis. J Am Coll Cardiol. 2001;38:1078–1082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Internal control tissues for both histochemistry and immunohistochemistry comprising unaffected areas of 8 different aortic valves. In (A) note the aortic valve leaflet composed of 3 anatomic layers: the fibrosa, a collagen‐rich layer on the aortic side of the valve (top layer); the spongiosa, a layer of variable thickness lying between the fibrosa and ventricularis, with a less dense matrix and often containing adipose cells and the ventricularis, a layer with prominent elastin fibers located on the ventricular side of the valve (bottom layer). In (B through F), note the lack of calcified areas (B) as well as any of the antigens detected from grade 1 upward (C through F, compare with Tables 1, 2, 3 and 5).
Table S1. Results of Comparison Between Cases (n=4) and Controls (n=4) for Selected Genes by Microarray Analysis