

Abstract
The components of the Switch/Sucrose non-fermentable (SWI/SNF) complex are mutated in approximately 20% of human cancers. The A/T-rich interacting domain 1A (ARID1A) subunit has one of the highest mutation rates. Most notably, ARID1A is mutated in over 50% of ovarian clear cell carcinomas (OCCCs). We reported that inhibition of enhancer of zeste homology 2 (EZH2) is synthetically lethal in ARID1A-mutated OCCC.
Keywords: ARID1A, EZH2, ovarian cancer, SWI/SNF chromatin-remodeling complex, synthetic lethality
Genes encoding subunits of the ATP-dependent chromatin-remodeling complex are mutated in many cancer types.1 Most notably, the A/T-rich interacting domain 1A (ARID1A) gene, which encodes a subunit of the Switch/Sucrose non-fermentable (SWI/SNF) chromatin-remodeling complex, is mutated in up to 57% of ovarian clear cell carcinomas (OCCCs).2,3 Indeed, ARID1A is among the genes that show the highest mutation rates across multiple cancer types,1 including up to 27% of gastric carcinomas, 13% of hepatocellular carcinomas, 13% of bladder carcinomas, 15% of esophageal adenocarcinomas, and 17% of Burkitt lymphomas. In addition to mutation, loss of ARID1A expression has been reported in several cancer types, most frequently in breast and kidney cancers.4 However, despite the prevalence of ARID1A mutations in many cancer types, a rational therapeutic approach to target cancers with ARID1A mutations has not yet been explored.
Epithelial ovarian cancer remains the most lethal gynecologic malignancy in the developed world. OCCC ranks second as a cause of death from epithelial ovarian cancer and is associated with a poor prognosis compared to other histologic subtypes. OCCC typically has a low initial response rate to platinum-based standard care, and there is currently no effective therapy for the disease. More than 90% of the ARID1A mutations observed in OCCC are frame-shift or nonsense mutations that result in loss of ARID1A protein expression.3 Notably, loss of ARID1A expression in OCCC significantly correlates with a shorter progression-free survival and is associated with a worse response to chemotherapy compared with ARID1A-positive OCCC. Thus, there is an even greater need for targeted therapy that is selective for ARID1A-mutated OCCC. Similarly, ARID1A mutation and/or loss of expression have been reported to be a marker of poor prognosis in a number of other cancer types. Thus, new therapeutics based on ARID1A mutational status is of high clinical impact.
Cancer mutations that cause a loss of function are not directly druggable with conventional targeted approaches such as antibodies. Synthetic lethality is a phenomenon in which only the simultaneous perturbation of 2 factors results in cell death.5 Our recent study demonstrates that inhibition of enhancer of zeste homology 2 (EZH2) activity selectively suppresses the growth of ARID1A-mutated OCCC cells in a synthetic lethal manner.6 EZH2 is an epigenetic regulator that silences the expression of its target genes. Notably, EZH2 is often overexpressed in OCCC. Highly specific EZH2 inhibitors (such as GSK126) have been developed, and are now in clinical trials for hematopoietic malignancies.7 Significantly, the EZH2 inhibitor GSK126 caused the regression of established ARID1A-mutated OCCC and decreased the number of disseminated tumor nodules in xenograft models.6
Given the recent success in targeting chromatin regulators in cancer, our study has substantial translational potential for the management of ARID1A-mutated OCCC. In this context, ARID1A mutation status, determined by genome sequence in the upcoming era of precision medicine, or loss of ARID1A protein expression could serve as biomarkers to predict therapeutic efficacy to EZH2 inhibitors. Future studies are warranted to determine whether the observed synthetic lethality between EZH2 inhibition and ARID1A mutation extends beyond ARID1A-mutated OCCC. Moreover, genetic alterations in components of the SWI/SNF complex are a well-recognized feature of many cancer types. For example, in rhabdoid tumors, a rare childhood cancer, loss of expression of sucrose nonfermenting 5 (SNF5), a non-catalytic core subunit of SWI/SNF, directly upregulates EZH2 expression.8 Survival of SNF5-deficient cancer cells depends upon the upregulated EZH2.9 Therefore, it will be critical to determine whether the observed synthetic lethality also applies to mutations in other components of the SWI/SNF complex and to develop companion predictive markers for response to EZH2 inhibitors in these contexts.
To elucidate the mechanism underlying the observed synthetic lethality, we profiled changes in gene expression induced by restoration of wild type ARID1A or GSK126 treatment in ARID1A-mutated cells. This analysis revealed antagonistic roles of ARID1A and EZH2 in regulating a significant number of overlapping genes (Fig. 1). Notably, the antagonistic roles of SWI/SNF and polycomb proteins, which include ARID1A and EZH2 respectively, were initially suggested in genetic studies using Drosophila. The most interesting novel ARID1A/EZH2 target gene that we identified was phosphoinositide 3-kinase interacting protein 1 (PIK3IP1). Functionally, we demonstrated that PIK3IP1 contributes to the observed synthetic lethality in ARID1A-mutated cells treated with EZH2 inhibitor. At the chromatin level, our data indicate that ARID1A dominates in the expression of the ARID1A/EZH2 target genes when both ARID1A and EZH2 are present. In contrast, in the absence of ARID1A, the balance is tipped toward EZH2-dependent silencing of these genes. Consequently, inhibition of EZH2 activity in the absence of functional ARID1A leads to reactivation of target genes such as PIK3IP1 to trigger apoptosis. As such, EZH2 inhibitors selectively suppress the growth of ARID1A-mutated, but not wild type, cells.
Figure 1.
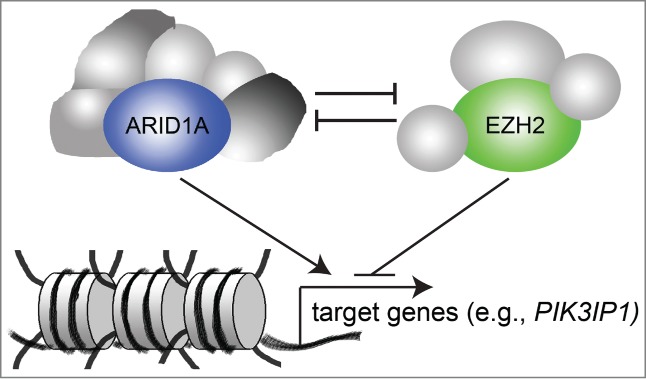
Antagonism between A/T-rich interacting domain 1A (ARID1A) and enhancer of zeste homology 2 (EZH2) underlies the observed synthetic lethality between EZH2 inhibition and ARID1A mutations. ARID1A and EZH2 are antagonistic in regulating the expression of the same set of target genes (such as phosphoinositide 3-kinase interacting protein 1 [PIK3IP1]) and ARID1A dominates over EZH2 in determining the expression pattern of these genes.
In addition to ARID1A mutation, the phosphoinositide 3-kinase (PI3K)/protein kinase B (best known as AKT), pathway is often activated in OCCC as a result of gain-of-function mutations in PIK3CA, the gene encoding the catalytic subunit of PI3K. Indeed, conditional Arid1a knockout together with activation of phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit α isoform (Pik3ca) leads to the development of OCCC in genetic mouse models.10 Interestingly, the validated ARID1A/EZH2 target gene PIK3IP1 is a negative regulator of PI3K. These findings suggest that ARID1A mutation cooperates with PI3K/AKT signaling to drive OCCC. Thus, a combination of the EZH2 inhibitor together with inhibition of the PI3K/AKT pathway may carry an even greater clinical benefit.
In summary, our recent studies demonstrate that targeting EZH2 activity using clinically applicable small molecule EZH2 inhibitors represents a novel synthetically lethal therapeutic strategy in ARID1A-mutated OCCC. Given that mutation and loss of expression of ARID1A and genetic alterations in other subunits of the ATP-dependent chromatin remodeling complex are observed at a high frequency in many cancer types, these findings will have far-reaching implications for the future development of epigenetic therapeutic strategies.
Funding
This work was supported by grants from the US National Institutes of Health/National Cancer Institute (R01CA160331 and R01CA163377 to R.Z. and Cancer Center Support grant CA010815 to the Wistar Institute), a US Department of Defense Ovarian Cancer Academy award (OC093420 to R.Z.), and an Ovarian Cancer Research Fund Program project (to R.Z.). R.Z. is an Ovarian Cancer Research Fund Liz Tilberis Scholar. B.G.B. is supported by an American Cancer Society postdoctoral fellowship (PF-13–058–01-TBE). K.M.A. is supported by a training grant from the US National Institutes of Health/National Cancer Institute (T32CA9171–35).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G., Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505(7484):495-501; PMID:24390350; http://dx.doi.org/ 10.1038/nature12912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Vogelstein B Jr. et al., Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010; 330(6001):228-31; PMID:20826764; http://dx.doi.org/ 10.1126/science.1196333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al., ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med 2010; 363(16):1532-43; PMID:20942669; http://dx.doi.org/ 10.1056/NEJMoa1008433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Nagl NG, Flowers S Jr., Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. Int J Cancer 2004; 112(4):636; PMID:15382044; http://dx.doi.org/ 10.1002/ijc.20450 [DOI] [PubMed] [Google Scholar]
- 5.Kaelin WG., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5(9):689-98; PMID:16110319; http://dx.doi.org/ 10.1038/nrc1691 [DOI] [PubMed] [Google Scholar]
- 6.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih IM, Conejo-Garcia JR, et al., Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med 2015; 21(3):231-8; PMID:25686104; http://dx.doi.org/23051747 10.1038/nm.3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A 3rd, Diaz E, et al., EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012; 492(7427):108-12; PMID:23051747; http://dx.doi.org/ 10.1038/nature11606 [DOI] [PubMed] [Google Scholar]
- 8.Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 2010; 18(4):316-28; PMID:20951942; http://dx.doi.org/ 10.1016/j.ccr.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, Porter Scott M, Chesworth R, Moyer MP, Copeland RA, et al., Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 2013; 110(19):7922-7; PMID:23620515; http://dx.doi.org/ 10.1073/pnas.1303800110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J, et al., Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun 2015; 6:6118; PMID:25625625; http://dx.doi.org/ 10.1038/ncomms7118 [DOI] [PMC free article] [PubMed] [Google Scholar]