

ABSTRACT
The role of altered lipid metabolism in pancreatic ductal adenocarcinoma (PDAC) is poorly appreciated. We recently identified the lipid signature of PDAC and revealed low-density lipoprotein receptor (Ldlr) as a metabolic driver of this disease. Here, we comment our findings that disruption of Ldlr leads to intratumoral cholesterol imbalance and improves chemotherapy efficiency.
KEYWORDS: Cholesterol, gemcitabine, Ldlr, metabolism, mechanisms of oncogenesis and tumor progression, novel therapeutic targets, pancreatic cancer, tumor metabolism
abbreviations
- Acly
ATP citrate lyase
- CE
cholesteryl ester
- FA
fatty acid
- Fasn
fatty acid synthase
- FC
free cholesterol
- GEMM
genetically engineered mouse model
- HBP
hexosamine biosynthetic pathway
- Hmgcr
3-Hydroxy-3-methylglutaryl coenzyme A reductase
- Ldlr
low-density lipoprotein receptor
- PDAC
pancreatic ductal adenocarcinoma
Pancreatic ductal adenocarcinoma (PDAC) is expected to surpass breast and colorectal cancer in terms of mortality and become the second leading cause of cancer death in 2020–2030. At the present time, only 15% of PDAC patients benefit from surgery, which is the only curative treatment, and 85% develop unresectable locally advanced or metastatic tumors. For patients who do not benefit from surgery the 5-year survival rate is less than 5%.1 Despite significant scientific advances in our understanding of PDAC pathobiology and extensive efforts to develop new therapeutic tools to tackle this cancer, the survival rate for PDAC patients has not improved significantly over past decades. Many obstacles specific to this tumor, such as late diagnosis, early metastatic spread, and resistance to chemotherapy and radiotherapy, have hampered clinical progress against PDAC. Histologically, pancreatic tumors are characterized by a prominent stroma made up of extracellular matrix components, activated cancer-associated fibroblasts, immune cells, nerve fibers, and few and abnormal blood vessels.1 This stromal architecture creates a physical barrier for drug delivery and for proper diffusion of oxygen and nutrients toward cancer cells. Despite nutrient deprivation and hypoxic stress the tumor cells are proliferating, attesting to their ability to rewire their metabolism in order to cope with adverse environmental conditions and form a tumor mass.2,3
In our studies, we used a genetically engineered mouse (GEM) model that harbors sustained activation of Kirsten rat sarcoma viral oncogene homolog (Kras) and a disruption of the Inhibitor of cyclin-dependent kinase type 4 (Ink4a)/Arf locus. In this GEMM, faithfully reproducing the evolution and malignancy of human disease (Pdx1-Cre; LSL-KrasG12D; Ink4a/Arffl/fl mice), we have shown that a fifth of the total tumor area is composed of hypoxic cancer cells.4 The presence of these hypoxic areas creates a glycolytic gradient within the tumor mass, with hypoxic tumor cells exhibiting a higher glycolytic rate than normoxic cells. The ensuing increase in lactate released by hypoxic cells serves as an energy source for the neighboring oxygenated cells.4 This illustrates that these 2 tumor cell populations function symbiotically. Under hypoxic stress, pancreatic cancer cells also show an intense appetite for glutamine, and the resulting enhanced glutaminolysis, coupled with increased glycolytic flux, promotes overactivation of the hexosamine biosynthetic pathway (HBP).4 In response to hypoxia, the HBP strongly contributes to tumor aggressiveness because it drives post-translational modifications, such as O-GlcNAcylation, that stabilize hypoxia-inducible factor 1, glucose transporter 1, and the glycolytic enzyme phosphofructokinase 1.5,6
Glucose and glutamine are commonly described as the main substrates for tumor growth. However, given the tremendous energetic and biomass demand of PDAC due to the high proliferative rate of the cancer cells, these 2 nutrients cannot be the only metabolic suppliers. Moreover, the breakdown of muscle proteins and burning of fat stores in PDAC patients suffering from cachectic syndrome reinforce the hypothesis that PDAC consumes various nutrients and consequently highlights its metabolic flexibility. Therefore, deciphering the complete picture of metabolic reprogramming underlying malignant progression of PDAC would highlight key and attractive therapeutic metabolic candidates to defeat this disease.
Using a global transcriptomic approach restricted to metabolism-related transcripts, we identified the metabolic routes preferentially used by pancreatic tumors to fulfill their energetic and biosynthetic needs.7 Our metabolic screening revealed an undeniable contribution of lipids to the impaired metabolic network encountered in PDAC, with a high enrichment of activated-pathways associated with cholesterol- and lipoprotein-related metabolisms, and with retinoic acid, glycosphingolipid, phosphatidylinositol, leukotriene, and steroid metabolism.7,8 In contrast, regulation of fatty acid (FA) metabolic pathways in PDAC appears to be more complex than that of lipoproteins. Long-chain FA synthesis, transport, and desaturation are downregulated in PDAC, suggesting that these FAs come preferentially from an exogenous source. On the other hand, saturated short- and medium-chain FA must come from de novo lipogenesis as key transcripts encoding fatty acid synthase (Fasn) and ATP citrate lyase (Acly) that are involved in their synthesis are found to be upregulated in PDAC (data not shown). Hence, our findings highlight marked differences in lipid metabolic reprogramming in PDAC compared to that in prostate tumors, which exhibit lower rates of glycolysis and increased dependence on energetic FA degradation through β-oxidation.9 Our results also reveal that the deregulation of lipid metabolism in PDAC extends beyond the commonly noted alteration of de novo lipogenesis and offers new targetable lipid metabolic pathways for PDAC treatment. Lipoprotein catabolism appears to be one such pathway.
We focused on the key player in lipoprotein catabolism—the low-density lipoprotein receptor (Ldlr), which mediates endocytic uptake of cholesterol-rich LDL (Fig. 1). Ldlr transcripts are selectively overexpressed in both well-differentiated and de-differentiated cancer cells that express epithelial-to-mesenchymal transition markers.7 The fold-increase of Ldlr protein in PDAC compared to control pancreas is largely superior to that of the rate-limiting enzyme in cholesterol synthesis, 3-hydroxy-3-methylglutaryl coenzyme A reductase (Hmgcr), suggesting that cholesterol uptake prevails over the cholesterol synthesis pathway (Fig. 1).7 Therefore, the massive accumulation of cholesterol in PDAC, revealing the marked dependency of pancreatic tumor cells on cholesterol, is mainly the result of an increase in exogenous cholesterol supply. We next demonstrated that disruption of Ldlr-dependent cholesterol entry through Ldlr knockdown induces an imbalance in cholesterol homeostasis by inverting the ratio of free cholesterol (FC; i.e., membrane constituent cholesterol) and cholesteryl ester (CE; i.e., stored form in lipid droplets). Indeed, the FC content overcomes the CE stores.7 As a consequence, FC overload drastically impairs the proliferation and tumorigenic capacities of pancreatic cancer cells and inhibits the ERK-dependent survival pathway.
Figure 1.
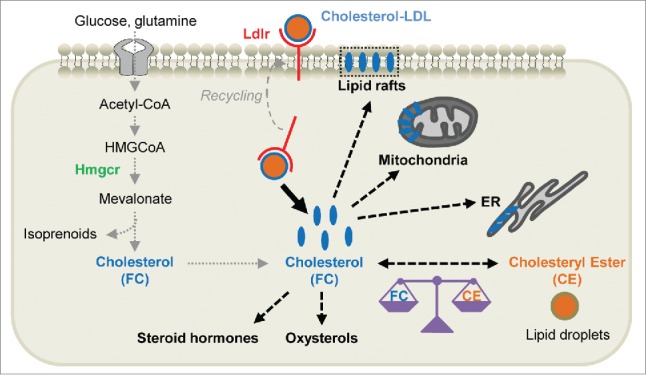
Cholesterol uptake and metabolism in pancreatic cancer cells. The excessive demand for cholesterol by pancreatic ductal adenocarcinoma (PDAC) cells is mainly met through activation of low-density lipoprotein receptor (Ldlr)-dependent uptake, which prevails over the de novo synthesis pathway. First, cholesterol-rich LDLs are internalized via Ldlr-mediated uptake and reach the endocytic circuits, from where the receptor is recycled to the cell surface. LDLs are hydrolyzed and the resultant free cholesterol (FC) is either stored in lipid droplets after being esterified (i.e., cholesteryl esters or CE), transferred into plasma membranes where it resides in lipid rafts, or accumulated in mitochondria and/or endoplasmic reticulum (ER) membranes. Moreover, cholesterol may be converted into oxysterols or used as a precursor for steroid hormone synthesis. Secondly, glucose and glutamine that are taken up by PDAC cells are metabolized into acetyl-CoA, which generates precursors for cholesterol synthesis through the tricarboxylic acid cycle. 3-Hydroxy-3-methylglutaryl coenzyme A reductase (Hmgcr) constitutes the rate-limiting enzyme of this pathway, which, in addition to ultimately leading to cholesterol formation, promotes isoprenoid production.
One of the current clinical challenges is to improve the poor efficacy of commonly used chemotherapeutic agents (i.e., gemcitabine) in the treatment of PDAC. Using preclinical models, we proved that metabolic weakening of pancreatic tumor cells through disruption of the physiologic balance between FC and CE content potentiates the tumor regression induced by gemcitabine.7 From a clinical aspect, we present LDLR as a promising metabolic marker for diagnosis of patients with a high risk of recurrence, since elevated LDLR expression is correlated with an increased rate of relapse in patients with PDAC.7 Hence, treatments targeting LDLR in this patient group could delay the time to the first relapse occurrence and consequently improve patient survival. Recent evidence from a clinical meta-analysis showed that a high-cholesterol diet might increase the risk of pancreatic cancer, illustrating the marked contribution of exogenous cholesterol supply to pancreatic cancer development.10
In summary, identifying the contribution of cholesterol uptake and its tumoral repartition into FC and CE in the context of PDAC pinpointed Ldlr as the main driver of cholesterol metabolic rewiring in pancreatic cancer cells. Moreover, mapping the numerous lipid pathways that are upregulated in PDAC will help to unveil new, exciting, and promising combined therapies that may be successful in the treatment of patients with PDAC.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371:1039-49; PMID:25207767; http://dx.doi.org/ 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 2.Mayers JR, Vander Heiden MG. Famine versus feast: understanding the metabolism of tumors in vivo. Trends Biochem Sci 2015; 40:130-40; PMID:25639751; http://dx.doi.org/ 10.1016/j.tibs.2015.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olivares O, Vasseur S. Metabolic rewiring of pancreatic ductal adenocarcinoma: New routes to follow within the maze. Int J Cancer 2015; PMID:25732227. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 4.Guillaumond F, Leca J, Olivares O, Lavaut MN, Vidal N, Berthezene P, Dusetti NJ, Loncle C, Calvo E, Turrini O, et al.. Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 2013; 110:3919-24; PMID:23407165; http://dx.doi.org/ 10.1073/pnas.1219555110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, Vocadlo DJ, Seagroves TN, Reginato MJ. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol Cell 2014; 54:820-31; PMID:24857547; http://dx.doi.org/ 10.1016/j.molcel.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012; 337:975-80; PMID:22923583; http://dx.doi.org/ 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guillaumond F, Bidaut G, Ouaissi M, Servais S, Gouirand V, Olivares O, Lac S, Borge L, Roques J, Gayet O, et al.. Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A 2015; 112:2473-8; PMID:25675507; http://dx.doi.org/ 10.1073/pnas.1421601112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greenhill C. Pancreatic cancer: Disrupted lipid metabolic pathways in PDAC identified. Nat Rev Gastroenterol Hepatol 2015; 12(4):188; PMID:25708047 [DOI] [PubMed] [Google Scholar]
- 9.Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res 2013; 52:585-9; PMID:24001676; http://dx.doi.org/ 10.1016/j.plipres.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen H, Qin S, Wang M, Zhang T, Zhang S. Association between cholesterol intake and pancreatic cancer risk: Evidence from a meta-analysis. Sci Rep 2015; 5:8243; PMID:25649888; http://dx.doi.org/ 10.1038/srep08243. [DOI] [PMC free article] [PubMed] [Google Scholar]