

abstract
Analysis of cancer methylomes has dramatically changed our concept of the potential of diagnostic and prognostic methylation biomarkers in disease stratification. Through whole-genome methylation capture sequencing of triple-negative breast cancers (TNBCs) we recently identified differentially methylated regions with diagnostic and prognostic value that promise to stratify TNBCs for more personalized management.
Keywords: Breast cancer, DNA methylation, DNA methylation biomarkers, prognostic biomarkers, triple-negative breast cancer
Cancer arises through a multitude of disruptions of normal cellular control, with both genetic and epigenetic aberrations playing major roles in the acquisition of the cancer phenotype. DNA methylation is the most-studied epigenetic modification in mammalian cells and is characterized by the addition of a methyl group at the carbon-5 position of cytosine residues within CpG dinucleotides through the action of DNA methyltransferase enzymes,1 forming 5-methylcytosine (Fig. 1A). During the initiation and progression of cancer, genome-wide changes in DNA methylation patterns are typically observed, characterized by DNA hypermethylation of gene regulatory regions associated with CpG islands and gene inactivation,2 in parallel with hypomethylation of the adjacent genic region and CpG-poor intergenic regions (Fig. 1B). The advancement of genome-wide DNA methylation sequencing technologies is allowing more comprehensive mapping of cancer DNA methylomes,3 providing greater insight into the underlying mechanisms and location of cancer-specific methylation changes.
Figure 1.
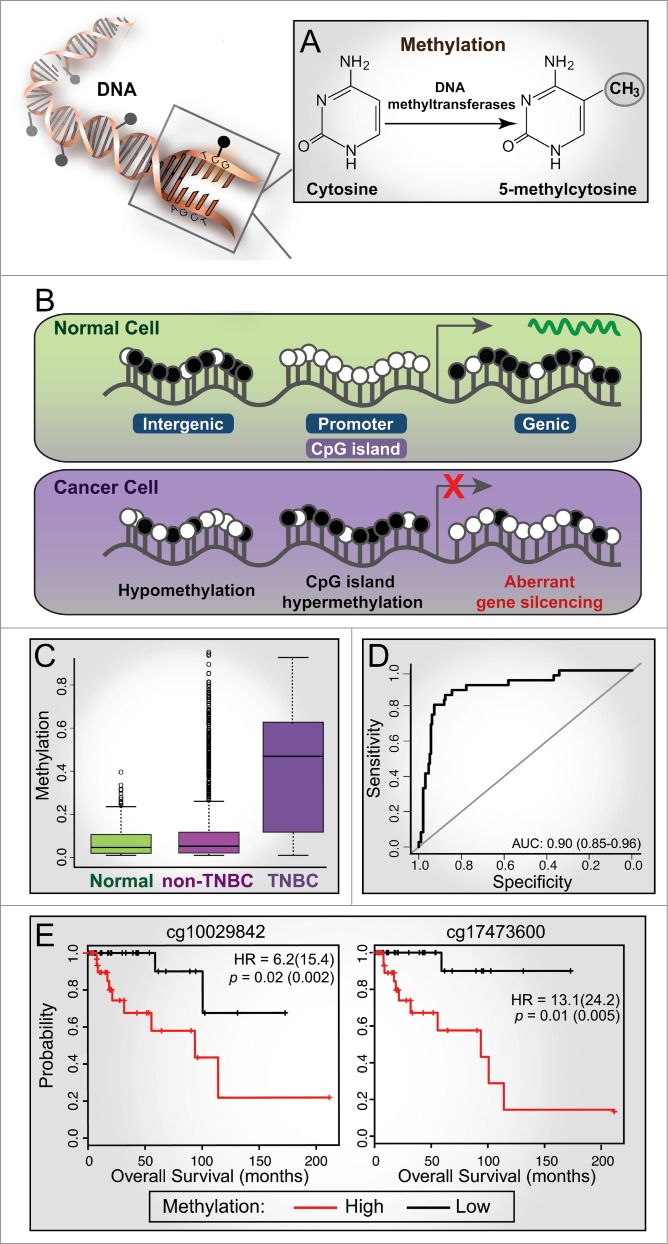
Aberrant methylation in cancer and its diagnostic and prognostic value in triple-negative breast cancer (TNBC). (A) DNA methylation is characterized by the addition of a methyl group to the cytosine base within CpG dinucleotides through the action of DNA methyltransferase enzymes, forming 5-methylcytosine. (B) A schematic representation of methylation changes that typically occur between normal and cancer cells. White circle, unmethylated CpG; black circle, methylated CpG. CpG islands are often associated with gene promoters and are unmethylated in normal cells; gene expression is associated with methylation of adjacent genic regions. In cancer cells, CpG islands are often hypermethylated, resulting in aberrant gene silencing (e.g., tumor suppressor genes) and hypomethylation of adjacent genic and intergenic regions. (C) Box plots showing the distribution of methylation levels for Illumina Human Methylation 450 BeadChip (HM450K) probes across a differentially methylated region (DMR) on chr19-46114 (exon of PPFIA3) showing hypermethylation in TNBC tumors (n = 73) compared to normal (n = 83) and non-TNBC samples (n = 354) from The Cancer Genome Atlas (TCGA) breast cancer cohort. (D) ROC curve analysis based on methylation values of the TNBC-specific probes (n = 282) classifies TCGA HM450K tumor samples into TNBC and non-TNBC with high specificity and sensitivity (AUC of 0.9; sensitivity of 0.72 and specificity of 0.94). (E) Kaplan-Meier plots showing survival curves for TNBC patients stratified based on median β methylation values of 2 HM450K probes (chr1-47207; exon of LHX8); hazard ratios (HR) and p-values from Cox proportional hazard model are shown for univariate analyses (with multivariate values in parentheses).
Breast cancer is a disease with diverse tumor subtypes and outcomes. In particular, triple-negative breast cancers (TNBCs) are a heterogeneous group of cancers with varying prognoses that present a challenge for effective clinical management. The TNBC subtype is clinically defined by the absence of estrogen receptor (ER) and progesterone receptor (PR) expression, and neither overexpression nor amplification of human epidermal growth factor receptor 2 (HER2).4 TNBC represents approximately 15-20% of newly diagnosed cases and is generally associated with a higher degree of disease recurrence and shorter overall survival compared to non-TNBC. The prognostic stratification of TNBC patients remains a significant challenge in breast cancer research. Current efforts to stratify early breast cancer prognosis primarily focus on multi-gene expression signatures.5 Such signatures are most effective at assigning recurrence risk to early-stage hormone receptor-positive breast cancer, in which the rate of proliferation is closely associated with overall prognosis. However, the majority of TNBCs are highly proliferative, and therefore cannot be stratified using these multi-gene classifiers. The potential use of DNA methylation signatures as biomarkers of disease is now being assessed, with a number of studies documenting aberrant methylation events in breast carcinogenesis, as well as identifying specific DNA methylation biomarkers that have significant diagnostic and prognostic potential.6 To date, several studies have described DNA methylation signatures that can distinguish between ER-positive and ER-negative tumors, and some of these are also predictive of outcome, but no study to date has focused on TNBC methylation.
Our study
In our recently published study,6 we were motivated to specifically investigate the DNA methylome of TNBC to identify epigenetic changes unique to TNBC and any association with disease outcome. To this end, we performed whole-genome methyl-CpG binding domain (MBD)-based capture sequencing (MBDCap-seq) on formalin-fixed paraffin-embedded (FFPE) triple-negative clinical tumors and matched normal DNA samples with known clinical outcome data. This high-resolution technique combines affinity capture of methylated DNA by MBD-based proteins with next-generation sequencing. Using this approach, we identified 865 differentially methylated regions (DMRs) in TNBC tumors compared to matched normal samples, and showed that these regions were enriched in promoters associated with transcription factor binding sites and DNA hypersensitive sites. Importantly, we found 36 DMRs that were specific to TNBC, which we validated on a larger cohort of clinical samples (n = 542) using Illumina Human Methylation 450 BeadChip (HM450K) methylation data extracted from The Cancer Genome Atlas (TCGA) breast cancer cohort.7 The TNBC-specific methylated regions were significantly hypermethylated in TNBCs compared to normal and non-TNBC samples (Fig. 1C). Specifically, we were able to classify tumor samples in the TCGA HM450K cohort into TNBCs and non-TNBCs with sensitivity of 0.72, specificity of 0.94, and area under the curve (receiver operating characteristic) of 0.90 (Fig. 1D), highlighting the excellent potential diagnostic value of these DMRs.
Strikingly, in this study we also showed that DMRs stratified TNBC patients into 3 distinct methylation clusters. Survival analysis revealed that the largely hypomethylated cluster was associated with better prognosis, whereas the other 2 more methylated clusters were associated with worse prognoses. Additional survival analysis identified 17 DMRs in the TCGA breast cancer data, each harbouring 3 or more HM450K probes associated with survival in TNBC samples: 14 genomic regions were associated with poor survival and 3 regions with longer survival. Kaplan-Meier plots of individual CpG sites in each region showed very good survival separation, highlighting their potential value as prognostic biomarkers (Fig. 1E). Interestingly, most of these regions overlap with DNase I hypersensitive sites and contain many transcription factor binding sites, suggesting that they may harbor important regulatory functions. Our study has therefore provided the first evidence that DNA methylation profiling can be used to classify breast cancer subtypes and stratify TNBCs according to patient outcome.
Future perspectives
Advances in genome-wide DNA methylation sequencing technologies are leading to the identification of DNA methylation cancer biomarkers that have significant diagnostic and prognostic potential, in addition to revealing epigenetic signatures that can distinguish tumor molecular subtypes and predict therapeutic response. Already, the measurement of promoter hypermethylation of individual genes has been successfully implemented in the clinic for early cancer detection in tissue samples and body fluids8 and as a predictor of tumor response to treatment.9,10 These examples highlight the promise of translating epigenetic markers into the clinic for early detection of cancer, especially given that deregulation of cellular epigenetic patterns is one of the initial events in carcinogenesis. To distinguish ‘driver’ DNA methylation events from ‘passenger’ roles is more challenging but important to further enable personalized therapeutic programs, and future analyses will reveal specific methylation signatures that are either associated with, or drive, the survival capacity of cancer cells.
Cumulatively, our findings demonstrate the feasibility of profiling the cancer methylome with limited archival tissue and how epigenetic profiling can aid in diagnostic classification and identification of potential prognostic biomarkers. We believe that, after further validation in larger independent cohorts, methylation signatures will become valuable tools in the future clinical management of TNBC.
Acknowledgments
We thank Dr Brigid O'Gorman for careful reading of the manuscript and design of the figure. The work is supported by NHMRC project grants (1029579 and 1070418) and National Breast Cancer Foundation (NBCF) program grant. S.J.C. is supported by a NHMRC Fellowship.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Bestor TH. Cloning of a mammalian DNA methyltransferase. Gene 1988; 74:9-12; PMID:3248734; http://dx.doi.org/ 10.1016/0378-1119(88)90238-7 [DOI] [PubMed] [Google Scholar]
- 2.Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007; 128:683-92; PMID:17320506; http://dx.doi.org/ 10.1016/j.cell.2007.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet 2014; 30:75-84; PMID:24368016; http://dx.doi.org/ 10.1016/j.tig.2013.11.004 [DOI] [PubMed] [Google Scholar]
- 4.Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist 2011; 16(Suppl 1):61-70; PMID:21278442; http://dx.doi.org/ 10.1634/theoncologist.2011-S1-61 [DOI] [PubMed] [Google Scholar]
- 5.Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet 2011; 378:1812-23; PMID:22098854; http://dx.doi.org/ 10.1016/S0140-6736(11)61539-0 [DOI] [PubMed] [Google Scholar]
- 6.Stirzaker C, Zotenko E, Song JZ, Qu W, Nair SS, Locke WJ, Stone A, Armstong NJ, Robinson MD, Dobrovic A, et al.. Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commu 2015; 6:5899; PMID:25641231; http://dx.doi.org/ 10.1038/ncomms6899 [DOI] [PubMed] [Google Scholar]
- 7.TCGA . Comprehensive molecular portraits of human breast tumours. Nature 2012; 490:61-70; PMID:23000897; http://dx.doi.org/ 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ladabaum U, Allen J, Wandell M, Ramsey S. Colorectal cancer screening with blood-based biomarkers: cost-effectiveness of methylated septin 9 DNA versus current strategies. Cancer Epidemiol Biomarkers Prev 2013; 22:1567-76; PMID:23796793; http://dx.doi.org/ 10.1158/1055-9965.EPI-13-0204 [DOI] [PubMed] [Google Scholar]
- 9.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000; 343:1350-4; PMID:11070098; http://dx.doi.org/ 10.1056/NEJM200011093431901 [DOI] [PubMed] [Google Scholar]
- 10.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, et al.. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005; 352:997-1003; PMID:15758010; http://dx.doi.org/ 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]