

ABSTRACT
Ataxia telangiectasia mutated (ATM) is an important signaling molecule in the DNA damage response and inhibitors of ATM are under clinical development. We identified a synthetic lethal interaction between ATM inhibition and phosphatase and tensin homolog (PTEN) loss that was the result of increased oxidative stress. Inhibition of ATM therefore represents a novel strategy to target PTEN-associated cancers.
KEYWORDS: ATM, DNA damage, DSB, PTEN, ROS
The genomes of all living organisms constantly suffer deleterious attacks. Of the many types of DNA lesion, one of the most harmful is the double-stranded break (DSB). DSBs are caused by exogenous damage, such as ionizing radiation (IR), or by endogenous damage, mainly oxidative stress.1 DSBs are also generated during the normal processes of meiotic recombination and V(D)J recombination.2 DSBs are therefore a frequent event in the cell and, if left unchecked, represent potentially lethal lesions that may result in cell death or in chromosomal rearrangements, which in turn may promote neoplastic transformation. In order to respond to this damage, cells activate a complex network called the DNA damage response (DDR), which coordinates the activation of cell cycle checkpoints, appropriate DNA repair pathways, and numerous other responses.2
The primary transducer following the detection of DSBs is the serine–threonine protein kinase ataxia telangiectasia mutated (ATM), which phosphorylates numerous key proteins in various branches of the DDR.2 ATM is recruited to DSBs as a homodimer through interaction with the MRE11/RAD50/Nijmegen breakage syndrome 1 (MRN) complex at these sites. Autophosphorylation of ATM generates an active monomer that phosphorylates several other proteins required for an appropriate response, including cell cycle regulation, repair, and, in the event of failed repair, activation of senescence3 or programmed cell death2, thereby guarding against malignancy.
Loss of tumor suppressor gene (TSG) function is an important step in malignant progression. Loss of TSGs can result in DSBs either through loss of repair processes or through increased production of toxic metabolic by-products.4 In the early phase of cancer development following loss of TSGs these DSBs activate ATM, which prevents replication of damaged cells.3 Later in malignant progression, however, the DNA damage is detected and repaired to allow cancer cell survival and proliferation. Previously, ATM has been shown to be important for cellular viability in the absence of the Fanconi anemia (FA)/breast cancer susceptibility gene 1/2 (BRCA1/2) pathway, a DDR mechanism that is frequently lost in cancer.5 Inhibition of ATM together with loss of TSGs associated with this pathway results in cell death. Importantly, this toxic effect is specific to the cancer cell in that loss of ATM function or the FA/BRCA pathway alone is not lethal, a phenomenon referred to as “synthetic lethality.”6
In our current publication7 we have extended this work to look for synthetic lethal interactions between ATM and other TSGs. We used siRNA screening to individually knockdown 178 TSGs in paired ATM functional and non-functional cell lines and identified potential synthetic lethal interactions. One of the top hits was phosphatase and tensin homolog (PTEN), a phosphatase important in regulating the phosphoinositide 3-kinase (PI3K)/v-akt murine thymoma viral oncogene homolog (AKT) pathway. PTEN is one of the most commonly lost tumor suppressor genes in cancer. For example, approximately 50% of advanced prostate cancers have lost function in this gene through either mutation or epigenetic silencing,8 making a synthetically lethal treatment approach attractive for this group of patients.
Consistent with the screening results, small interfering RNA (siRNA)-mediated knockdown and chemical inhibition of ATM resulted in selective toxicity in a range of PTEN-deficient cell line models. PTEN-deficient cells were found to arrest in the mitotic part of the cell cycle and demonstrated DNA aberrations consistent with catastrophic DNA damage7 that led to apoptotic cell death (Fig. 1). This synthetic lethal interaction may be clinically important as ATM inhibitors are under clinical development.9 We therefore introduced a tetracycline inducible PTEN prostate cancer cell line model (PC3 cells +/− PTEN) into a mouse system. The PTEN-deficient cell line was aggressive and rapidly grew into a tumor mass, but was significantly inhibited by administration of an ATM inhibitor compared to cells expressing wild-type PTEN.7
Figure 1.
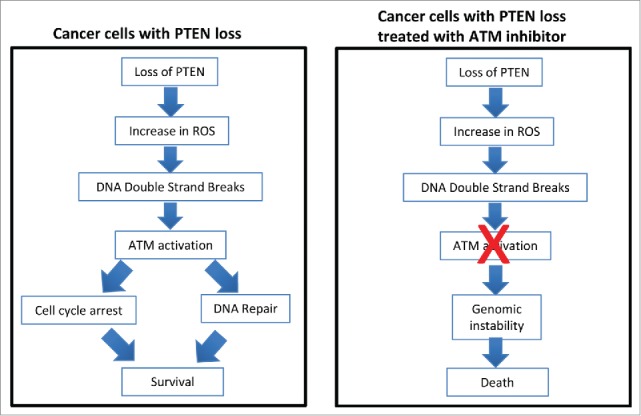
Synthetic lethality between PTEN loss and ATM inhibition. Loss of phosphatase and tensin homolog (PTEN) results in increased oxidative stress (reactive oxygen species [ROS]) and DNA damage, resulting in checkpoint activation, DNA repair, and cell survival. In the absence of ataxia telangiectasia mutated (ATM), loss of checkpoints and further DNA damage result in genomic instability and death.
From a mechanistic perspective, PTEN-deficient cells were found to have elevated levels of endogenous DNA damage and activation of ATM compared to PTEN wild-type cells. RAD51 is an important component of DSB repair through a process termed homologous recombination (HR). Loss of RAD51 has previously been observed in PTEN-deficient cell lines and could potentially explain the increased DNA damage and requirement for ATM that we had observed. However, RAD51 expression and function was entirely normal in our PTEN-deficient model systems.7 We therefore asked whether the observed increase in DNA damage occurred through another mechanism. Metabolism is the major cause of endogenous DNA damage in the cell. Oxidative species are particularly toxic as they can oxidize DNA bases, eventually leading to DSBs.4 PTEN loss was correlated to increased oxidative stress and DNA damage in our model systems indicating that this was probably the underlying mechanism for the synthetic lethal interaction observed between PTEN loss and ATM inhibition (Fig. 1). Consistent with this notion, antioxidants were found to attenuate the effects of ATM inhibition in PTEN-deficient cells.7
Interestingly, ATM has been reported to prevent malignant progression of premalignant conditions through the signaling of oncogene-related DNA damage.3 Our study suggests that loss of tumor suppressors such as PTEN during malignant progression may result in a dependency on ATM to maintain DNA integrity.7 Rather than being protective, ATM has become complicit in the malignant process. This change in roles, however, may provide an opportunity for a novel approach to cancer treatment. Cancers with a poor outcome, such as advanced prostate cancer or glioblastoma, are often PTEN deficient and may respond to therapeutic ATM inhibition. Both of these cancer types are often treated with localized radiotherapy, which may also be more effective following administration of a therapeutic ATM inhibitor.10 ATM inhibitors may therefore provide a novel dual approach to cancer treatment by improving both localized response to radiotherapy and specifically targeting metastatic PTEN-deficient tumors. With the development of ATM inhibitors for clinical use, a clinical study to test this hypothesis should be relatively straightforward to perform.
Disclosure of potential conflicts of interest
Nuala McCabe, Steven Walker and Richard Kennedy are all employees of Almac Group, Craigavon, UK
References
- 1.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330:517-21; PMID:20966255; http://dx.doi.org/ 10.1126/science.1192912 [DOI] [PubMed] [Google Scholar]
- 2.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; PMID:23847781; http://dx.doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- 3.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al.. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633-7; PMID:17136093; http://dx.doi.org/ 10.1038/nature05268 [DOI] [PubMed] [Google Scholar]
- 4.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 2013; 12:931-47; PMID:24287781; http://dx.doi.org/ 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- 5.Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, Shimamura A, D'Andrea AD. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest 2007; 117:1440-9; PMID:17431503; http://dx.doi.org/ 10.1172/JCI31245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaelin WG, Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 2005; 5:689-98; PMID:16110319; http://dx.doi.org/ 10.1038/nrc1691 [DOI] [PubMed] [Google Scholar]
- 7.McCabe N, Hanna C Dr, Walker SM, Gonda D, Li J, Wikstrom K, Savage KI, Butterworth KT, Chen C, Harkin DP, et al.. Mechanistic rationale to target PTEN-deficient tumour cells with inhibitors of the DNA damage response kinase ATM. Cancer Res 2015; 75(11):2159-65; PMID:25870146 [DOI] [PubMed] [Google Scholar]
- 8.Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, et al.. PTEN: multiple functions in human malignant tumors. Front Oncol 2015; 5:24; PMID:25688333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basu B, Yap TA, Molife LR, de Bono JS. Targeting the DNA damage response in oncology: past, present and future perspectives. Curr Opin Oncol 2012; 24:316-24; PMID:22476188; http://dx.doi.org/ 10.1097/CCO.0b013e32835280c6 [DOI] [PubMed] [Google Scholar]
- 10.Carruthers R, Ahmed SU, Strathdee K, Gomez-Roman N, Amoah-Buahin E, Watts C, Chalmers AJ. Abrogation of radioresistance in glioblastoma stem-like cells by inhibition of ATM kinase. Mol Oncol 2015; 9:192-203; PMID:25205037; http://dx.doi.org/ 10.1016/j.molonc.2014.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]