

ABSTRACT
Cancer-associated fibroblasts (CAFs) are major participants in the crosstalk between tumor cells and their microenvironment. CAFs provide not only multiple soluble factors but also metabolic fuels to promote tumor growth, invasion, and metastasis. We discuss recent developments delineating the effects of metabolic symbiosis between CAFs and tumor cells on tumor growth.
KEYWORDS: Alpha-ketoglutarate, cancer-associated, fibroblast, IDH, metabolic symbiosis, metastasis, tumor microenvironment
Abbreviations
- bFGF
basic fibroblast growth factor
- CAF
cancer-associated fibroblast
- NAT
normal fibroblast
Although genetic mutations in tumor cells are the primary drivers of tumorigenesis, stromal cells in the tumor microenvironment, including fibroblasts, immune cells, and endothelial cells, are active conspirators in promoting tumor growth and metastasis. Oncogenic signaling within tumors frequently drives the recruitment of normal fibroblasts (NAFs) and reprograms them into cancer-associated fibroblasts (CAFs).1 CAFs are activated fibroblasts that share similarities with fibroblasts and are stimulated by inflammatory conditions or activated during wound healing.2 Hypoxia and reactive oxygen species (ROS) can also promote activation of CAFs. They constitute a significant component of the stroma and mediate changes in the composition of extracellular matrix to one with an increased amount of collagens (desmoplastic response).3,4 CAFs are phenotypically and functionally distinct from NAFs and can be identified based on the expression of several markers including α smooth muscle actin (α-SMA), fibroblast activation protein (FAP); fibroblast-specific protein 1 (FSP1), and platelet-derived growth factor receptor (PDGFR). Transforming growth factor β (TGF-β), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF), interleukin 6 (IL-6), and lysophosphatidic acid (LPA), which can induce desmoplastic reactions in tumors, also stimulate the differentiation and proliferation of NAFs to CAFs.
CAFs are functionally distinct from NAFs and significantly promote tumor growth in xenograft models when mixed with tumor cells. The tumor-promoting effects of CAFs are mainly mediated through a paracrine mechanism involving multiple secreted factors, including hepatocyte growth factor (HGF), connective tissue growth factor (CTGF), epidermal growth factor (EGF), insulin growth factor (IGF), nerve growth factor (NGF), bFGF, Wingless/integrase-1 (Wnt) ligands, and matrix metalloproteinase. In addition to these paracrine events, CAFs secrete many chemokines (such as C-X-C motif chemokine 12 [CXCL12], CXCL14, and CCL7) and vascular endothelial growth factor (VEGF-A) to regulate angiogenesis. For instance, CAFs secrete CXCL12, which binds and activates CXCR4 in tumor cells, to induce migration and proliferation of tumor cells.5 In addition, CAFs can change their metabolic profile and provide metabolic intermediates that enhance tumor growth. For example, phosphoglycerate kinase-1 (PGK1), a component of the glycolytic pathway, is dramatically upregulated in CAFs compared with NAFs. Overexpression of PGK1 reprograms NAFs into CAFs, causing a proliferation of CAFs, and promotes tumor growth when co-implanted in vivo.6 Interestingly, reports suggest that CAFs use aerobic glycolysis as an energy source. The current hypothesis is that a switch to aerobic glycolysis in CAFs would generate lactate and ketones, which, when secrete into the intracellular space, act as paracrine oncometabolites to fuel oxidative mitochondrial metabolism in tumor cells. The metabolic switch to aerobic glycolysis, referred to as the “reverse Warburg effect”7 (Fig. 1), occurs in CAFs through molecular mechanisms that are largely undefined. A recent paper by Zhang et al. showed that downregulation of isocitrate dehydrogenase 3α (IDH3α) plays a critical role in the metabolic reprogramming of CAFs.8 Downregulation of IDH3α decreases the effective level of α-ketoglutaric acid (α-KG) by reducing the ratio of α-KG to fumurate and succinate; this results in inhibition of prolyl hydroxylase domain-containing protein 2 (PHD2) and stabilization of hypoxia-inducible factor 1-α (HIF1α) protein. The accumulation of HIF1α, in turn, promotes glycolysis by increasing the uptake of glucose, upregulating expression of glycolytic enzymes under normoxic conditions, and inhibits oxidative phosphorylation by upregulating NADH dehydrogenase 1 α subcomplex 4-like 2 (NDUFA4L2). CAFs from tumor samples exhibit low levels of IDH3α, and overexpression of IDH3α prevents transformation of fibroblasts into CAFs. Together, these findings reveal that IDH3α is a critical metabolic switch in CAFs.
Figure 1.
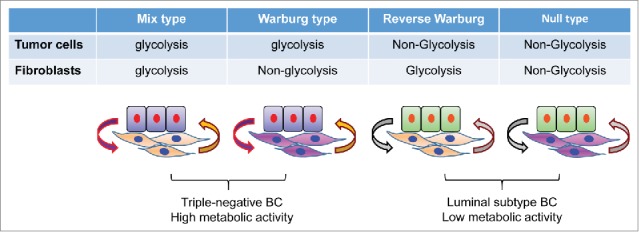
Metabolic symbiosis between breast cancer cells and cancer-associated fibroblasts. When tumors develop, their microenvironments evolve. This includes metabolic reprogramming in CAFs. In triple-negative breast cancer (BC), tumor cells are prone to glycolysis and the adjacent CAFs can also adapt to glycolysis. The acidic environment generated by this not only activates matrix metalloproteinase (MMP), but also prevents attack from immune cells. However, in the luminal subtype of breast cancer, tumor cells are not prone to glycolysis although their adjacent CAFs can adopt glycolysis. Metabolic intermediates, such as lactate, secreted by CAFs can be utilized by tumor cells for the biosynthesis of macromolecules. The metabolic symbiosis between tumor cells and CAFs provides a growth advance for the proliferation and metastatic dissemination of tumor cells.
The results of this study, together with others, have several implications. First, CAFs promote a dynamic metabolic interaction with tumor cells and generate a metabolic niche to nourish tumor growth. An example is breast cancer, a heterogeneous disease that can be divided into at least 4 different subtypes based on gene expression profiling: luminal A, luminal B, human epidermal growth factor receptor 2 (HER2), and basal-like. After examining markers of glycolysis, autophagy, and proliferation in 740 tumor tissues, Choi et al. identified 4 different metabolic phenotypes in tumor and stroma. These include Warburg type (glycolysis in tumor cells, non-glycolysis in stroma), reverse Warburg type (non-glycolysis in tumor cells, glycolysis in stroma), mixed type (glycolysis in both tumor cells and stroma), and null type (non-glycolysis in both tumor cells and stroma)9 (Fig. 1). Triple-negative (basal-like) breast cancer is enriched in mixed and Warburg types with high metabolic activity, whereas luminal breast cancer is enriched in null and reverse Warburg types with low metabolic activity. This study reveals the metabolic heterogeneity in breast cancer, and demonstrates a correlation between tumor subtypes and metabolic phenotypes. The findings also provide insight for targeting metabolic pathways in cancer. New therapeutic approaches that metabolically uncouple the “symbiosis” between tumor cells and CAFs may inhibit the development and growth of tumors.
Second, tumor cells co-evolve with NAFs during tumor progression and reprogram them into CAFs. This reprogramming converts NAFs from being tumor suppressive to being tumor supportive. Once reprogrammed, the CAF phenotype becomes “permanent” even in the absence of continued exposure to intratumoral stimuli.5 This stable phenotype suggests that epigenetic regulation and an autocrine signaling loop operate in the reprogramming of CAFs. Identification of the mechanisms that govern these inferred processes may provide a new approach to target CAFs.
Third, although CAFs are a key metabolic “fuel source” enabling tumor cell propagation, survival, and systemic dissemination, they are a heterogeneous cell population. Several cell types can be transdifferentiated into CAFs; the majority of CAFs are derived from resident tissue fibroblasts and mesenchymal stem cells. Stellate cells are predominantly responsible for the desmoplastic reaction seen in chronic pancreatitis and pancreatic cancer, as well as in liver fibrosis, and are categorized as CAFs when present in cancer tissue.1,10 In addition, epithelial and endothelial cells can be converted to CAFs through epithelial-mesenchymal transition and endothelial-mesenchymal transition, respectively.1 Thus, CAFs may be derived from multiple tissue types, reflecting local and distant cues that are sensed during tumorigenesis, and thus cannot be referred to as a single population of cells.
CAFs are a vital component of tumor progression. Although many exciting studies have been completed, the remaining challenge is to translate our knowledge into targeting tumor cells to alleviate side effects, drug resistance, metastasis, and recurrence.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We apologize to the many contributors to this field whose work is important but could not be cited due to space limitations. Our study is supported by the grants from NIH (RO1s CA125454 and CA188118), DOD Breakthrough Award (BC140733P1), the Mary Kay Ash Foundation (to B. P. Zhou), and the National Natural Science Foundation of China (81402432 to J. Liu).
Reference
- 1.Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med 2014; 211:1503-23; PMID:25071162; http://dx.doi.org/ 10.1084/jem.20140692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chang HY, et al.. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Bioly 2004; 2:E7; PMID:14737219; http://dx.doi.org/ 10.1371/journal.pbio.0020007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dotto GP. Multifocal epithelial tumors and field cancerization: stroma as a primary determinant. J Clin Invest 2014; 124:1446-53; PMID:24691479; http://dx.doi.org/ 10.1172/JCI72589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tripathi M, Billet S, Bhowmick NA. Understanding the role of stromal fibroblasts in cancer progression. Cell Adhe Migr 2012; 6:231-5; PMID:22568983; http://dx.doi.org/ 10.4161/cam.20419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orimo A, et al.. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005; 121:335-48; PMID:15882617; http://dx.doi.org/ 10.1016/j.cell.2005.02.034 [DOI] [PubMed] [Google Scholar]
- 6.Wang J, et al.. Characterization of phosphoglycerate kinase-1 expression of stromal cells derived from tumor microenvironment in prostate cancer progression. Cancer Res 2010; 70:471-80; PMID:20068185; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2863 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Pavlides S, et al.. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984-4001; PMID:19923890; http://dx.doi.org/ 10.4161/cc.8.23.10238 [DOI] [PubMed] [Google Scholar]
- 8.Zhang D, et al.. Metabolic Reprogramming of Cancer-Associated Fibroblasts by IDH3alpha Downregulation. Cell Rep 2015; 10:1335-48; PMID:25732824; http://dx.doi.org/ 10.1016/j.celrep.2015.02.006 [DOI] [PubMed] [Google Scholar]
- 9.Choi J, Kim H, Jung WH, Koo JS. Metabolic interaction between cancer cells and stromal cells according to breast cancer molecular subtype. Breast Cancer Res 2013; 15:R78; PMID:24020991; http://dx.doi.org/ 10.1186/bcr3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bachem MG, et al.. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998; 115:421-32; PMID:9679048; http://dx.doi.org/ 10.1016/S0016-5085(98)70209-4 [DOI] [PubMed] [Google Scholar]