

Abstract
Background
Hydrogen sulfide (H2S) exhibits protective effects in various disease models including cerebral ischemia–reperfusion (I/R) injury. Nonetheless, mechanisms and identity of molecules responsible for neuroprotective effects of H2S remain incompletely defined. In the current study, we observed that thiosulfate, an oxidation product of H2S, mediates protective effects of an H2S donor compound sodium sulfide (Na2S) against neuronal I/R injury.
Methods and Results
We observed that thiosulfate in cell culture medium is not only required but also sufficient to mediate cytoprotective effects of Na2S against oxygen glucose deprivation and reoxygenation of human neuroblastoma cell line (SH‐SY5Y) and murine primary cortical neurons. Systemic administration of sodium thiosulfate (STS) improved survival and neurological function of mice subjected to global cerebral I/R injury. Beneficial effects of STS, as well as Na2S, were associated with marked increase of thiosulfate, but not H2S, in plasma and brain tissues. These results suggest that thiosulfate is a circulating “carrier” molecule of beneficial effects of H2S. Protective effects of thiosulfate were associated with inhibition of caspase‐3 activity by persulfidation at Cys163 in caspase‐3. We discovered that an SLC13 family protein, sodium sulfate cotransporter 2 (SLC13A4, NaS‐2), facilitates transport of thiosulfate, but not sulfide, across the cell membrane, regulating intracellular concentrations and thus mediating cytoprotective effects of Na2S and STS.
Conclusions
The protective effects of H2S are mediated by thiosulfate that is transported across cell membrane by NaS‐2 and exerts antiapoptotic effects via persulfidation of caspase‐3. Given the established safety track record, thiosulfate may be therapeutic against ischemic brain injury.
Keywords: apoptosis, cerebral ischemia, hydrogen sulfide, sulfidation, thiosulfate
Introduction
Hydrogen sulfide (H2S) is produced by several enzymes in diverse tissues. H2S has a prominent role in vasorelaxation, neurotransmission, and inflammation. A number of studies suggest that H2S attenuates ischemia–reperfusion (I/R) injury in a variety of organs including brain, whether it is endogenously produced or exogenously administered as H2S gas or donor compounds.1, 2, 3, 4, 5, 6 Along these lines, we reported that administration of Na2S or cardiomyocyte‐specific overexpression of cystathionine gamma lyase, an enzyme that produces H2S, prevents neuronal death and improves long‐term neurological outcomes and survival after cardiac arrest in mice.4, 7 In a recent study, we also found that a novel H2S‐releasing compound improves outcomes after global cerebral I/R.5 Nevertheless, mechanisms responsible for the cytoprotective effects of H2S remain incompletely defined. In particular, how biological effects of H2S are transported to the brain is largely unknown.
Levels of free H2S in circulating blood or cell culture medium at steady state are very low (nmol/L≈low μmol/L).8, 9 Free H2S levels only transiently increase and quickly return to its baseline after administration of H2S donor compounds systemically or to cell culture medium.10 Therefore, for exogenously administered H2S to have pharmacological effects in remote organs or in cultured cells, H2S has to be converted to a metabolite that is stable in circulation or in culture medium, respectively. H2S is serially oxidized to persulfide, sulfite (SO3 2−), thiosulfate (S2O3 2), and sulfate (SO4 2−) in reactions catalyzed by several mitochondrial enzymes including sulfide quinone oxidoreductase, sulfur dioxygenase, thiosulfate sulfurtransferase (rhodanese), and sulfite oxidase (SO).10 H2S or its metabolites can also be serially converted to sulfane sulfur–containing compounds. Sulfane sulfur is a sulfur atom with 6 valence electrons but no charge (represented as S0).11 Biologically important sulfane sulfur–containing compounds include persulfides (R–S–SH), polysulfides (R–S–Sn–S–R), thiosulfate, and protein‐bound elemental sulfur. H2S and sulfane sulfur coexist, and recent reports suggest that sulfane sulfur–containing species may be the actual signaling molecules of the biological effects of H2S.11, 12, 13, 14, 15
In previous studies, we observed that beneficial effects of inhaled H2S during murine endotoxic shock were associated with markedly increased plasma thiosulfate levels. Because thiosulfate is relatively stable in circulation, these observations indicate that thiosulfate may mediate protective effects of H2S. However, thiosulfate is an anion that does not freely permeate across cell membranes. Therefore, for thiosulfate to exert any biological effects, it must be transported by some transport mechanisms into cells. Nonetheless, the role of thiosulfate transporter on the cytoprotective effects of H2S has not been examined.
In the current study, we observed that thiosulfate converted from H2S mediates the protective effects of Na2S against cerebral I/R injury. Administration of sodium thiosulfate (STS) prevented in vitro and in vivo cerebral I/R injury by increasing intracellular concentrations of thiosulfate per se without increasing levels of H2S and other H2S metabolites. The protective effects of STS were associated with inhibition of c‐jun N‐terminal kinase (JNK) and caspase‐3 and activation of extracellular signal‐regulated kinase (Erk) 1/2. Furthermore, we revealed that the transport of thiosulfate across cell membrane and the protective effect of Na2S and STS against oxygen glucose deprivation (OGD) and reoxygenation (OGD/R) were mediated by 1 of the SLC13 family proteins, sodium sulfate cotransporter 2 (SLC13A4, NaS‐2), which is expressed in human and murine brain.16
Materials and Methods
Cell Culture
Human neuroblastoma cell line SH‐SY5Y were cultured in Eagle's medium/Ham's F‐12 50:50 mix (DMEM/F12; Cellgro by Mediatech, Inc) supplemented with 10% fetal bovine serum (FBS), 100 IU/mL penicillin, and 100 μg/mL streptomycin.5 Primary neuronal cultures were prepared from the cortex of embryonic day 15 C57BL/6J mice as previously described.5
Measurement of Sulfide and Thiosulfate by High‐Performance Liquid Chromatography
Levels of sulfide and thiosulfate in the cell culture medium, SH‐SY5Y cells, plasma, or brain were measured by using monobromobimane‐based high‐performance liquid chromatography (HPLC) analysis as previously described.5, 17
Cell Viability Assays
Cell viabilities of SH‐SY5Y or primary cortical neurons were measured by using lactate dehydrogenase (LDH) assay or 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay as described previously.5
OGD/R
OGD for SH‐SY5Y or for primary cortical neurons was performed by placing cells in a hypoxia chamber (STEMCELL Technologies Inc) for 15 or 2.5 hours followed by 24 or 21 hours of reoxygenation, respectively, as described previously.5
Measurement of Sulfide, Thiols, Thiosulfate, and Persulfides by Liquid Chromatography–Tandem Mass Spectrometry
Intracellular sulfide, homocysteine (Hcys), cysteine (Cys), glutathione (GSH), thiosulfate, CysSSH, or GSSH levels were measured, as described previously.18 In brief, 24 hours after the addition of STS at 0.25 mmol/L to the medium, SH‐SY5Y cells were washed twice with PBS, scraped, collected, sonicated, and incubated in the methanol solution containing 5 mmol/L monobromobimane at 37°C for 30 minutes under dark conditions. After centrifugation, aliquots of the supernatants were diluted 10 to 100 times with distilled water containing known amounts of isotope‐labeled internal standards and analyzed by liquid chromatography–tandem mass spectrometry (LC‐MS/MS).
Western Blotting
Protein levels in SH‐SY5Y were determined by standard immunoblot techniques.5, 17 Primary antibodies were purchased from Cell Signaling Technology, Inc except the antibody against NaS‐2 from Santa Cruz Biotechnology, Inc.
Measurement of Caspase‐3 Activity
Caspase‐3 catalytic activity was determined based on measuring chromophore p‐nitroaniline (pNA) after cleavage from the labeled substrate DEVD‐pNA. Caspase‐3 activity in the cell lysate of SH‐SY5Y was measured by using a colorimetric assay kit (EMD Millipore Corp) according to the manufacturer's instructions. Inhibition of purified recombinant human caspase‐3 activity by Na2S or STS was examined by using the caspase‐3 inhibitor screening assay kit (EMD Millipore) according to the manufacturer's instructions with or without using (d,l)‐dithiothreitol (DTT) solution.
Persulfidation Assay (Modified Biotin Switch Assay)
The assay was carried out as described previously.19, 20 Briefly, SH‐SY5Y cells were lysed, scraped, collected, sonicated, and centrifuged at 14 000g for 5 minutes at 4°C to obtain supernatant of cell lysates. Supernatant was incubated with vehicle, Na2S, or STS at 37°C for 30 minutes or 19 hours followed by the procedure of modified biotin switch assay and subjected to Western blotting analysis.
Transfection of Plasmid DNA
DH5α that was transformed with Myc‐tagged pcDNA3‐Casp3 (wild‐type human caspase‐3, Addgene) or Myc‐tagged pcDNA3‐Casp3 C163A (mutant human caspase‐3, Addgene) was cultured in the LB medium containing 100 μg/mL ampicillin at 37°C for 2 days. Plasmids were purified by using QIAGEN plasmid kits. Transient transfections into SH‐SY5Y cells were performed by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were used 24 hours after transfection.
Gene Silencing by Transfection of siRNA
Transfection of siRNA (scrambled sequence, Invitrogen, Stealth RNAi, cat# 12935‐300, or NaS‐2, Invitrogen, Stealth RNAi, cat# HSS119981) into SH‐SY5Y cells were performed by using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) according to the manufacturer's protocol. Cells were used 48 hours after transfection. Gene silencing was confirmed by using immunoblotting (Figure S4).
Global Cerebral Ischemia and Reperfusion
After approval by the Massachusetts General Hospital Subcommittee on Research Animal Care, all animal experiments were performed in accordance with the guidelines of the National Institutes of Health. Male mice (C57BL/6J, 8 to 9 weeks old) purchased from the Jackson Laboratory were anesthetized and subjected to 40 minutes of bilateral common carotid artery occlusion (BCAO) with microsurgical clips as previously described.5 To ascertain that BCAO induces global cerebral ischemia, we measured cerebral blood flow of mice at middle cerebral artery region by using a laser Doppler flowmetry (VMS‐LDF; Moor Instrument Inc). STS at 10 mg/kg or vehicle was administered intraperitoneally (IP) 1 minute after the initiation of reperfusion. After reperfusion and recovery from anesthesia, mice were given IP 1 mL of 5% dextrose‐enriched lactated Ringer's solution with or without STS at 10 mg/kg daily for 1 week. Neurological function score (NFS) was evaluated as described previously.5, 21
Statistical Analysis
All data are presented as mean±SE. Data were analyzed by ANOVA by using Sigmastat 3.01a (Systat Software Inc) and the Prism 5 software package (GraphPad Software) unless otherwise described. Newman–Keuls multiple comparison post hoc test or Bonferroni post hoc test was performed for 1‐ or 2‐way ANOVA, respectively, as required. P values <0.05 were considered significant.
Results
Na2S Protects Neurons From OGD/R
We examined whether Na2S improves viability of SH‐SY5Y cells subjected to 15 hours of OGD followed by 24 hours of reoxygenation. Based on dose‐ (0.1, 0.2, or 0.5 mmol/L) and time‐ranging (0.5, 3, 5, or 8 hours after the end of OGD) studies, we determined that 0.5 mmol/L and at 5 hours after the end of OGD were the most effective dose and time point to add Na2S to improve viability of SH‐SY5Y cells after OGD/R (Figure 1).
Figure 1.
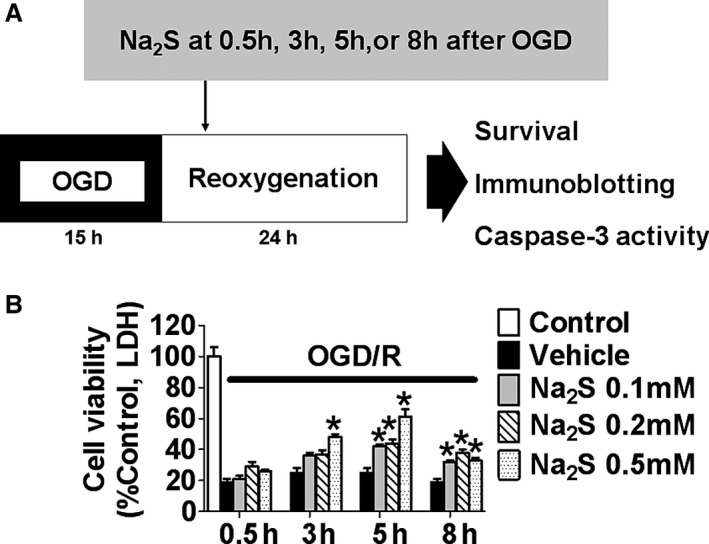
Effects of Na2S on cell viabilities of SH‐SY5Y after OGD/R. A, Protocol of OGD/R for SH‐SY5Y. B, Cell viabilities of SH‐SY5Y after OGD/R. Na2S at 0.1, 0.2, or 0.5 mmol/L was added at 0.5, 3, 5, or 8 h after the end of OGD. n=3 each; *P<0.05 vs control. LDH indicates lactate dehydrogenase; OGD/R, oxygen glucose deprivation and reoxygenation.
Thiosulfate Plays a Critical Role in the Protective Effect of Na2S
We measured extracellular and intracellular levels of sulfide (sum of H2S and HS−; an index of unbound H2S) and thiosulfate levels after the addition of Na2S or STS via an HPLC method (Figure 2A through D).5 The result showed that extracellular and intracellular sulfide levels increased rapidly and returned to the baseline by 3 hours after the addition of Na2S at 0.5 mmol/L to the cell culture medium. Extracellular thiosulfate levels increased to ≈0.25 mmol/L, corresponding with the timing when extracellular and intracellular sulfide levels returned to their respective baseline levels, suggesting a stoichiometric conversion of sulfide to thiosulfate. The addition of STS at 0.25 mmol/L to the medium increased extracellular and intracellular thiosulfate levels to the similar levels with what was achieved after the addition of 0.5 mmol/L Na2S. STS did not increase sulfide levels. We also observed that STS did not increase sulfide levels in medium and cells by using sulfide‐specific fluorescent probes HSip‐1 and HSip‐1 DA, respectively (Figure S1).22 Taken together, these results suggest that most, if not all, sulfide is converted to thiosulfate in culture medium and cells. Based on these observations, we examined whether the addition of thiosulfate itself has neuroprotective effects against OGD/R (Figure 3A). STS added 5 hours after the end of OGD improved cell viabilities of SH‐SY5Y in a dose‐dependent manner. Further, STS added 30 minutes before starting OGD or 30 minutes after the end of OGD improved cell viabilities of murine primary cortical neurons (Figure 3B). To determine the role of extracellular thiosulfate converted from H2S on the viability of SH‐SY5Y cells after OGD, we changed culture medium with fresh medium to remove thiosulfate in the medium 3 hours after the addition of Na2S at 0.5 mmol/L (Figure 3C and 3D). The time course for the medium‐change experiment is shown in Figure S2. The protective effect of Na2S was abolished by the medium change. On the other hand, replacing thiosulfate that was removed by the medium change with STS (0.25 mmol/L) restored neuroprotective effects. These results suggest that thiosulfate mediates the cytoprotective effects of Na2S after I/R injury.
Figure 2.
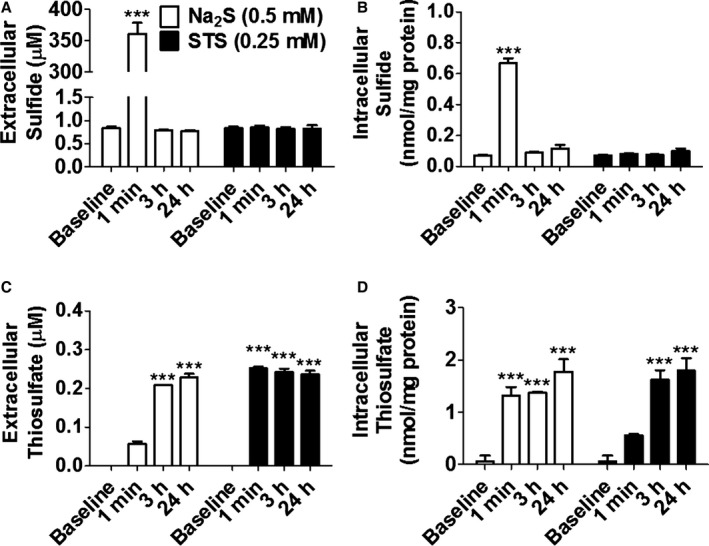
Na2S increased extracellular or intracellular sulfide levels and thiosulfate levels. A and B, Sulfide levels or (C and D) thiosulfate levels in culture medium or SH‐SY5Y cells incubated with or without Na2S or STS measured by HPLC. n=3 each; ***P<0.001 vs baseline at the same time point. HPLC indicates high‐performance liquid chromatography; STS, sodium thiosulfate.
Figure 3.
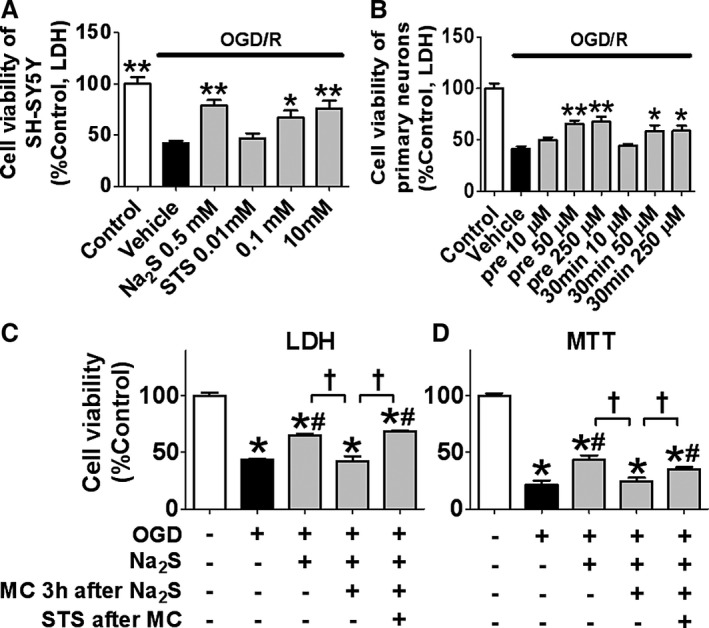
Effects of thiosulfate converted from H2S on cell viabilities after OGD/R. Cell viabilities of (A) SH‐SY5Y or (B) primary cortical neurons of mice after OGD/R. STS was added 5 h after the end of OGD (A) or 5 min before starting OGD or 30 min after the end of OGD (B). n=3 each; Horizontal bar indicates P<0.05 for control vs all other groups subjected to OGD/R. * or **P<0.05 or 0.01 vs vehicle. Cell viabilities of SH‐SY5Y 24 h after OGD/R measured by (C) LDH or (D) MTT method. Na2S at 0.5 mmol/L was added 5 h after the end of OGD. Medium was replaced with fresh 1 MC with or without STS at 0.25 mmol/L 3 h after the addition of Na2S. n=5 or 6 each; *P<0.05 vs control (left most column), # P<0.05 vs ODG/R alone and † P<0.05. LDH indicates lactate dehydrogenase; MC, medium change; MTT, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; OGD/R, oxygen glucose deprivation and reoxygenation; STS, sodium thiosulfate.
STS Increases Intracellular Levels of Thiosulfate but Not Sulfide and Persulfide
We measured levels of sulfide, Hcys, Cys, CysSSH, GSSH, thiosulfate, and GSH in the SH‐SY5Y cells 24 hours after the addition of STS at 0.25 mmol/L to the medium by using the LC‐MS/MS method. The result showed that the addition of STS to the medium did not alter the intracellular levels of sulfide, Hcys, Cys, and major antioxidative persulfides (CysSSH and GSSH),18 indicating that the protective effect of STS was not mediated via production of sulfide or antioxidative persulfides (Figure 4A through 4E). The addition of STS markedly increased intracellular levels of thiosulfate and modestly increased GSH in SH‐SY5Y cells (Figure 4F and 4G).
Figure 4.
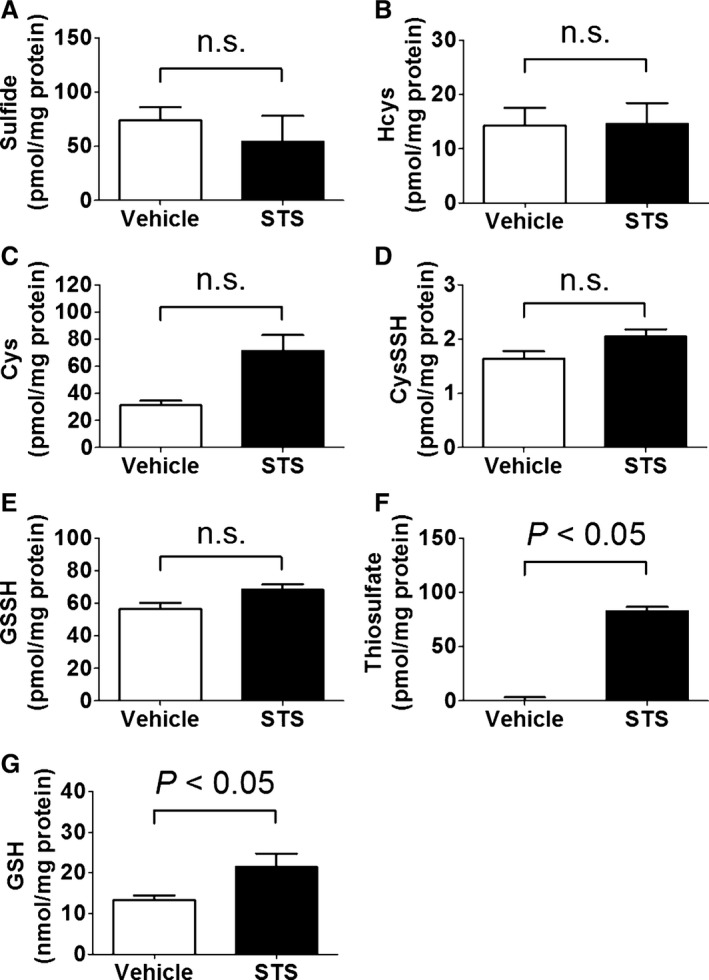
Intracellular levels of (A) sulfide, (B) Hcys, (C) Cys, (D) CysSSH, (E) GSSH, (F) thiosulfate, and (G) GSH in SH‐SY5Y cells 24 h after the addition of vehicle (medium) or STS at 0.25 mmol/L to the medium measured by LC‐MS/MS. Data were analyzed by using the Mann–Whitney test. n=3 each. CysSSH indicates cysteine persulfide; GSH, glutathione; GSSH, glutathione persulfide 2; LC‐MS/MS, liquid chromatography–tandem mass spectrometry; n.s., not significant; STS, sodium thiosulfate.
Na2S or STS Inhibits Activation of the Mitochondrial Pathway of Apoptosis
To elucidate molecular mechanisms responsible for the improvement of cell viability by Na2S or STS, we examined protein levels of phosphorylated JNK, phosphorylated Erk1/2, phosphorylated Bad, Bcl‐2, and cleaved caspase‐3 in SH‐SY5Y after OGD/R. Na2S at 0.5 mmol/L or STS at 0.25 mmol/L added 5 hours after the end of OGD attenuated phosphorylation of JNK and dephosphorylation of Erk1/2 and Bad (Figure 5A through 5C). The addition of Na2S or STS prevented the reduction of Bcl‐2 and the increase in caspase‐3 cleavage (Figure 5D and 5E). We also confirmed that Na2S or STS added 5 hours after the end of OGD inhibited caspase‐3 activity in SH‐SY5Y cells (Figure 5F). The medium change 3 hours after the addition of Na2S abolished the effects of Na2S on the changes of phosphorylation of JNK and Erk1/2. The addition of STS (0.25 mmol/L) after the medium change restored effects of Na2S on the phosphorylation of JNK and Erk1/2 (Figure 5G and 5H). These observations suggest that thiosulfate mediates the inhibitory effects of Na2S on the mitochondrial pathway of apoptosis.23, 24, 25
Figure 5.

H2S or STS inhibits mitochondrial pathway of apoptosis via modulating JNK pathway and Erk1/2 pathway in SH‐SY5Y cells. Na2S at 0.5 mmol/L or STS at 0.25 mmol/L were added 5 h after the end of OGD. Protein expression levels were determined by immunoblotting with rabbit polyclonal antibody against (A) phosphorylated (Thr183/Tyr185) or total JNK, n=4 each, (B) phosphorylated (Thr202/Tyr204) or total Erk1/2, n=3 each, (C) phosphorylated (Ser112) or total Bad, n=3 each, (D) Bcl‐2, n=3 each, or vinculin, and (E) cleaved or total caspase‐3. n=7 each; *, **, or ***P<0.05, 0.01, or 0.001 vs control, † or †† P<0.05 or 0.01 vs vehicle, and ‡ or ‡‡ P<0.05 or 0.01. (F) Caspase‐3 activity in SH‐SY5Y cells subjected to OGD/R. Na2S or STS was added 5 h after the end of OGD. Cells were lysed and used for caspase‐3 activity assay after reoxygenation. n=5 each; ** or ***P<0.01 or 0.001 vs control, and †† P<0.01 vs vehicle. Protein expression levels were determined by immunoblotting with rabbit polyclonal antibody against (G) phosphorylated (Thr183/Tyr185) or total JNK, n=4 each, and (H) phosphorylated (Thr202/Tyr204) or total Erk1/2, n=4 each. * or ***P<0.5 or 0.001 vs control, and †, †† or ††† P<0.05, 0.1, or 0.001 vs vehicle. JNK indicates c‐jun N‐terminal kinase Na; MC, medium change; OGD/R, oxygen glucose deprivation and reoxygenation; STS, sodium thiosulfate.
Na2S or STS Inhibits Human Recombinant Caspase‐3 Activity in Cell‐Free System
To examine whether Na2S or STS inhibits activated caspase‐3 after cleavage, we measured proteolytic activity of human recombinant caspase‐3, which was cleaved and preactivated in a cell‐free system. Na2S or STS dose‐dependently inhibited activity of recombinant caspase‐3. Of note, the inhibitory effects of Na2S and STS were abolished by DTT, suggesting that the inhibition of caspase‐3 was mediated by the reversible modification of protein thiols including persulfidation (Figure 6A).
Figure 6.
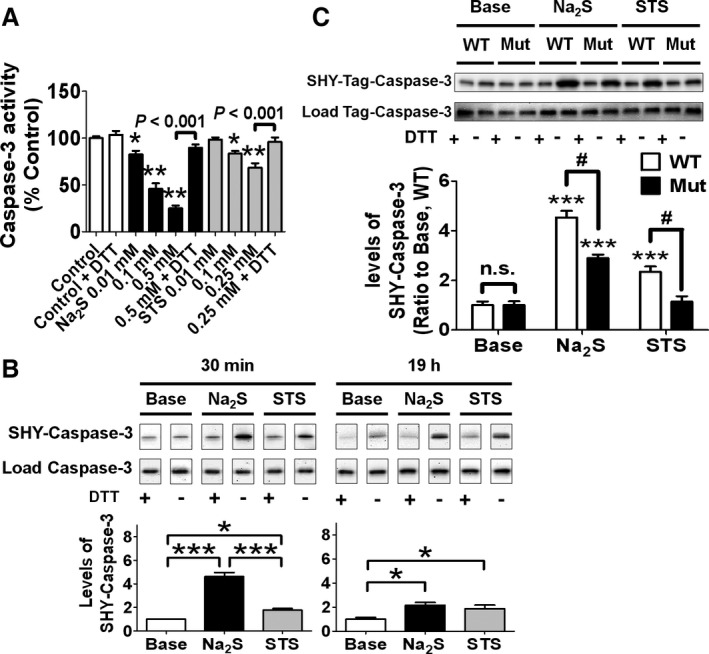
Effects of sulfhydration on cleavage of caspase‐3 or activity of cleaved caspase‐3. A, Activity of human recombinant caspase‐3 incubated with or without Na2S or STS with or without 2 mmol/L DTT at 37°C for 30 min. n=6 each; * or **P<0.05 or 0.001 vs control. B, Levels of sulfhydrated (SHY) caspase‐3 in cell lysates of SH‐SY5Y incubated with or without Na2S at 0.5 mmol/L or STS 0.25 mmol/L with or without 2 mmol/L DTT at 37°C for 30 min or 19 h. n=4 each; * or ***P<0.05 or 0.001 and #P<0.05. C, Levels of sulfhydrated caspase‐3 in cell lysates of SH‐SY5Y transfected with WT or Mut caspase‐3. Lysates were incubated with Na2S at 0.5 mmol/L or STS at 0.25 mmol/L at 37°C for 30 min. n=7, 7, 6, 7, 7 or 7 from left; ***P<0.001 vs baseline (Base) in the same genotype. DTT indicates dithiothreitol; STS, sodium thiosulfate; WT, wild‐type.
Na2S or STS Persulfidate Caspase‐3 at Cysteine 163
We found that incubating SH‐SY5Y cell lysates with Na2S at 0.5 mmol/L or STS at 0.25 mmol/L for 30 minutes persulfidated caspase‐3. Levels of caspase‐3 persulfidation were greater after incubation with Na2S than with STS at 30 minutes after addition. However, levels of persulfidated caspase‐3 were similar 19 hours after the addition of Na2S or STS (Figure 6B). To examine whether Na2S or STS causes persulfidation at cysteine 163 (Cys163), which is the catalytically active site of caspase‐3,26 we made SH‐SY5Y cells transfected with wild‐type human caspase‐3 (WT caspase‐3) or mutant human caspase‐3 in which Cys163 was substituted with alanine (C163A). We examined whether persulfidation levels of mutant caspase‐3C163A were lower than those of WT caspase‐3 after the addition of Na2S or STS to cell lysates. The successful transfection was confirmed by immunoblotting by using primary antibody against Myc‐tag. As expected, cells transfected with mutant caspase‐3C163A exhibited lower caspase‐3 activity (Figure S3A and S3B). Protein levels of Myc‐caspse‐3 in SH‐SY5Y transfected with WT caspae‐3 were comparable with those in SH‐SY5Y transfected with mutant caspase‐3C163A (Figure S3C). The result showed that mutation at Cys163 markedly decreased persulfidation levels of caspase‐3 at 30 minutes after the addition of Na2S or STS to cell lysates (Figure 6C). This result indicates that Na2S or STS causes persulfidation at Cys163 in caspase‐3.
Protective Effect of Thiosulfate Is Mediated by Sodium Sulfate Cotransporter 2 (NaS‐2, SLC13A4)
To elucidate how thiosulfate is transported across the cell membrane and accumulates in the cells, we compared thiosulfate levels in the SH‐SY5Y cells transfected with siRNA of NaS‐2 or scrambled sequence. The result showed that NaS‐2 silencing markedly attenuated accumulation of thiosulfate, but not sulfide, 3 hours after the addition of Na2S or STS to the medium (Figure 7A and 7B). Moreover, NaS‐2 silencing inhibited the improvement of cell viabilities of SH‐SY5Y cells after OGD/R by the addition of Na2S or STS, indicating that the protective effects of Na2S or STS were directly mediated by thiosulfate, but not sulfide, transported into cells via NaS‐2 (Figure 7C).
Figure 7.
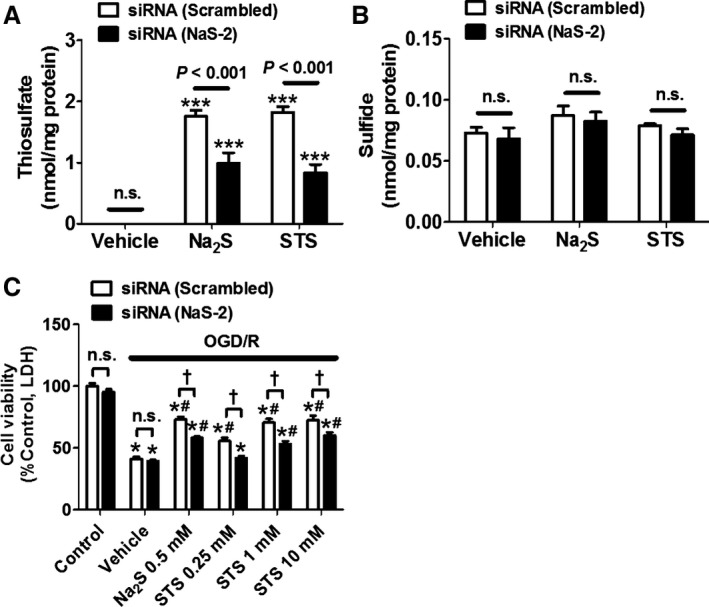
Effects of NaS‐2 silencing on the protective effect of Na2S or STS. Intracellular levels of thiosulfate (A) and sulfide (B) after addition of Na2S or STS to SH‐SY5Y transfected with siRNA (NaS‐2 or scrambled sequence). n=6 each. ***P<0.001 vs vehicle. (C) Cell viabilities of SH‐SY5Y transfected with siRNA (NaS‐2 or scrambled sequence) after OGD/R. Na2S or STS was added 5 h after the end of OGD. n=5 each; *P<0.001 vs control, # P<0.05 vs vehicle, or † P<0.05. LDH indicates lactate dehydrogenase; n.s., not significant; OGD/R, oxygen glucose deprivation and reoxygenation; STS, sodium thiosulfate.
Thiosulfate Plays a Critical Role in the Protective Effect of H2S Against Global Cerebral I/R Injury
To determine the neuroprotective effects of Na2S and STS in vivo, we measured levels of sulfide and thiosulfate in mice plasma and brain tissues 90 minutes after IP administration of Na2S at 25 μmol/kg or STS at 10 mg/kg (40 μmol/kg). The administration of Na2S at 25 μmol/kg was reported to protect mice from brain I/R injury.27 The results showed that thiosulfate, but not sulfide, levels were increased 90 minutes after the administration of Na2S in plasma and brain (Figure 8A). Similarly, thiosulfate, but not sulfide, levels were increased 90 minutes after the administration of STS (Figure 8A). At these respective doses of Na2S and STS, plasma and brain thiosulfate levels were similar 90 minutes after the administration of Na2S or STS. Next, we examined protective effects of STS on outcomes after global cerebral I/R induced by BCAO (Figure 8B). Cerebral blood flow was decreased to <4% of the baseline within 5 minutes of BCAO (Figure S5). The single or daily administration of STS improved the 20‐day survival rate of mice subjected to BCAO. The single administration of STS also improved NFS 24 hours after reperfusion and the 20‐day survival rate of mice. These results suggest that STS exerts neuroprotective effects against cerebral I/R.
Figure 8.

Effects of thiosulfate against brain I/R injury. A, Plasma or brain levels of sulfide or thiosulfate 90 min after IP administration of Na2S at 25 μmol/kg, STS at 10 mg/kg (40 μmol/kg), or vehicle. n=4 (brain) or 5 (plasma); *P <0.05 vs vehicle. B, Percent survival for 20 days or NFS at 24 h after reperfusion of mice subjected to 40 min of BCAO. Mice were given saline or STS at 10 mg/kg IP 1 min after reperfusion or 1 min plus every 24 h after reperfusion for 7 days. n=6 or 7, ** or ***P<0.01 or 0.001 vs sham. # or ### P<0.05 or 0.001. BCAO indicates bilateral common carotid artery occlusion; IP, intraperitoneal; I/R, ischemia–reperfusion; n.s., not significant; NFS, neurological function score; STS, sodium thiosulfate.
Discussion
The current study revealed that thiosulfate oxidized from H2S is essential and sufficient for the cytoprotective effects of Na2S in cultured neurons subjected to OGD/R. Incubation with Na2S stoichiometrically increased the levels of thiosulfate, but not H2S, in the cells, suggesting Na2S were almost completely converted to thiosulfate (Figure 2C). Administration of STS markedly increased cellular levels of thiosulfate, but not other sulfide metabolites. We observed that thiosulfate inhibits the mitochondrial apoptosis cascade and caspase‐3 activity. The cytoprotective effects of thiosulfate were associated with increased persulfidation of cleaved caspase‐3 at Cys163. The protective effect of Na2S or STS was facilitated by NaS‐2–mediated transportation of thiosulfate, but not sulfide, across the cell membrane. Last, administration of STS markedly prevented brain injury and improved survival in mice subjected to whole brain I/R. Taken together, these results suggest that thiosulfate mediates cytoprotective effects of H2S.
Numerous studies reported cytoprotective effects of H2S in vitro and in vivo. In the majority of studies, H2S was administered by using simple sulfide salts, Na2S or sodium hydrosulfide (NaHS). Once dissolved in water, both sulfide salts produce “hydrogen sulfide,” which exists as the equilibrium of H2S and HS− (in 2:8 ratio) at physiological pH.28 However, because H2S (and HS−) has a very short half‐life in biological fluids including cell culture medium and blood, how H2S reaches its presumed targets in the cells, and in the target tissues in the body when given in vivo, has been poorly understood. Our studies showed that H2S is converted to thiosulfate in vitro and in vivo. While removal of thiosulfate from the medium abolished the protective effects of Na2S, replacement of thiosulfate restored the protection. These results suggest that thiosulfate is not only required but also sufficient for the cytoprotective effects of Na2S. The addition of STS increased intracellular concentrations of thiosulfate and GSH but not sulfide, Hcys, Cys, CysSSH, and GSSH in SH‐SY5Y cells. Because GSH is an important intracellular reducing agent, it is possible that part of the beneficial effects of STS after OGD/R were mediated by increasing intracellular GSH. However, the percent increase of intracellular thiosulfate concentration was much greater than that of GSH after the addition of STS. Therefore, it is likely that thiosulfate directly exerted cytopotective effects. The administration of Na2S or STS markedly increased concentrations of thiosulfate, but not sulfide, in plasma and brain in mice. Taken together, our current observations suggest that thiosulfate is the “carrier” molecule responsible for the cytoprotective effects of H2S.
It has been suggested that the phosphorylation of JNK and the dephosphorylation of Erk1/2 mediate cerebral I/R injury by promoting the dephosphorylation of Bad and the downregulation of antiapoptotic Bcl‐2, which lead to activation of the mitochondrial pathway of apoptosis.24, 25, 29 In the present study, we observed that the cytoprotective effect of Na2S or STS in SH‐SY5Y cells subjected to OGD/R was associated with the prevention of the phosphorylation of JNK and the dephosphorylation of Erk1/2. It has been reported that H2S attenuates DNA damage in human endotherial cells and fibroblasts via persulfidation of mitogen‐activated protein kinase‐Erk kinase 1 at Cys341, which leads to Erk1/2 phosphorylation.30 Therefore, it is conceivable that Na2S or STS prevents dephosphorylation of Erk1/2 via persulfidation of mitogen‐activated protein kinase‐Erk kinase 1.
It has been suggested that H2S exerts its effects via protein persulfidation. Analogous to nitric oxide (NO)–dependent protein S–nitrosylation, persulfidation is another form of oxidative cysteine modification.20 It has been reported that nitrite, an oxidative metabolite of NO, causes S‐nitrosylation of caspase‐3 at Cys163 and inhibits caspase‐3 activity in primary human umbilical vein endothelial cells.31 Similarly, in the current study, we observed that Na2S and STS dose‐dependently inhibited activity of cleaved caspase‐3 in vitro. Interestingly, caspase‐3 activity of SH‐SY5Y cells subjected to OGD/R was completely inhibited by Na2S although caspase‐3 cleavage was incompletely prevented by Na2S, suggesting that Na2S directly inhibited caspase‐3 activity after cleavage (Figure 5E and 5F). The inhibition of cleaved caspase‐3 by Na2S and STS were associated with persulfidation of caspase‐3 at Cys163 (Figure 6A and 6B). Replacement of Cys163 of caspase‐3 with alanine attenuated, but did not completely prevent, the persulfidation of caspse‐3 induced by Na2S at 0.5 mmol/L, indicating that Na2S at 0.5 mmol/L might cause persulfidation at Cys163 and other cysteine residues (Figure 6C). We also observed that STS at 0.25 mmol/L caused persulfidation of WT caspase‐3 but not mutant caspase‐3C163A (Figure 6C), suggesting that STS at 0.25 mmol/L persulfidated only Cys163 and no other cysteine residues of caspase‐3. It has been suggested that sulfurtransferases such as thiosulfate sulfurtransferase mediate thiosulfate‐induced protein persulfidation.32 These results suggest that Na2S or STS inhibits caspase‐3 activity via thiosulfate‐mediated persulfidation of casepse‐3 at Cys163.
It has been reported that STS increases thiosulfate levels in the brain, choroid plexus, and cerebrospinal fluid.33 We observed that the administration of Na2S or STS increased the intracellular levels of thiosulfate in cultured neurons as well as in brain tissues. However, the mechanism responsible for the ability of Na2S or STS to increase intracellular thiosulfate levels in the brain was unclear. Thiosulfate anion cannot easily pass across the cell membrane unless ion transporters or ion channels for thiosulfate exist on the cell membrane. Sodium/sulfate cotransporter 2 is a member of the SLC13 transporter protein family that mediates the intake of a wide variety of molecules including thiosulfate with the concomitant uptake of sodium ions.16 We observed that knockdown of NaS‐2 markedly inhibited the accumulation of thiosulfate, but not sulfide, in SH‐SY5Y cells incubated with Na2S or STS and attenuated the cytoprotective effects of Na2S and STS after OGD/R. These observations unambiguously indicate that thiosulfate mediates the cytoprotective effects of H2S and thiosulfate. Furthermore, these results suggest that NaS‐2 plays an important role in neuroprotective effects of H2S or thiosulfate against cerebral I/R injury.
Although we observed neuroprotective effects of STS in a BCAO‐induced global cerebral ischemia model of mice, effects of STS were studied only in young healthy male mice. Because a majority of patients who experience ischemic brain injury have multiple comorbidities, including vascular insufficiency, our observations may not readily translatable to the clinical situation. Further, we did not examine the impact of STS on histological brain injury in the current study, although the severity of brain injury correlates with the deterioration of neurological function score in this model.5 Neuroprotective effects of STS remains to be determined in more clinically relevant model of ischemic brain injury in future studies.
In summary, the current study uncovered, for the first time, that thiosulfate is necessary and sufficient to exert cytoprotective effects of H2S in vitro and in vivo in the setting of I/R. We also identified a transport mechanism responsible for the uptake of thiosulfate into neurons, where it exerts antiapoptotic effects via persulfidation‐mediated inhibition of caspase‐3. The observed neuroprotective effects of thiosulfate against ischemic brain injury may be highly clinically relevant because STS has been clinically used for the detoxification of cyanide and other disorders in patients.34, 35, 36, 37 In addition, toxicity of STS is very low compared with Na2S or other sulfide donor compounds. Therapeutic potential of STS against ischemic neuronal injury warrants further studies.
Sources of Funding
This work was supported by National Institutes of Health grant HL101930 to Dr Ichinose.
Disclosures
None.
Supporting information
Figure S1. Na2S, but not STS, increased extracellular or intracellularsulfide levels. Fluorescent intensity of (A) medium containing HSip‐1 or (B) SH‐SY5Y cells loaded with HSip‐1 DA was measured after incubation with Na2S or STS at 37°C for 5 min or 24 h. Intracellular sulfide levels later than 3 h after incubation could not be measured since loaded HSip‐1 DA in cells leaked out into the medium. n=4 each; ***P<0.001 vs baseline at the same time point.
Figure S2. Time course for the medium‐change experiment in Figure 3 (C and D).
Figure S3. A, Protein levels of Myc‐tag or β‐tubilin in SH‐SY5Y transfected with vector alone, WT caspase‐3, or C163A caspase‐3 plasmid. B, Caspase‐3 activity in transfected SH‐SY5Y. n=5 each *P<0.01 vs WT by t‐test. C, Representative immunoblot of Caspase‐3 in SH‐SY5Y transfected with vector alone, WT, or C163A casase‐3 plasmid.
Figure S4. Representative immunoblot of NaS‐2 in SH‐SY5Y 48 h after transfection with siRNA (scrambled sequence or NaS‐2).
Figure S5. Average cerebral blood flow at middle cerebral artery region of mice subjected to BCAO for 40 min and reperfusion for 60 min. n=3 at each time point.
(J Am Heart Assoc. 2015;4:e002125 doi: 10.1161/JAHA.115.002125)
Accompanying Figures S1 through S5 are available at http://jaha.ahajournals.org/content/4/11/e002125/suppl/DC1
References
- 1. King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao Y‐X, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci USA. 2014;111:3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Salloum FN, Chau VQ, Hoke NN, Abbate A, Varma A, Ockaili RA, Toldo S, Kukreja RC. Phosphodiesterase‐5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein‐kinase g–dependent generation of hydrogen sulfide. Circulation. 2009;120:S31–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. George TJ, Arnaoutakis GJ, Beaty CA, Jandu SK, Santhanam L, Berkowitz DE, Shah AS. Inhaled hydrogen sulfide improves graft function in an experimental model of lung transplantation. J Surg Res. 2012;178:593–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Minamishima S, Bougaki M, Sips PY, De Yu J, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD, Ichinose F. Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3–dependent mechanism in mice. Circulation. 2009;120:888–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marutani E, Kosugi S, Tokuda K, Khatri A, Nguyen R, Atochin DN, Kida K, Van Leyen K, Arai K, Ichinose F. A novel hydrogen sulfide‐releasing N‐methyl‐D‐aspartate receptor antagonist prevents ischemic neuronal death. J Biol Chem. 2012;287:32124–32135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, Kimura H, Chow C‐W, Lefer DJ. Hydrogen sulfide attenuates myocardial ischemia‐reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104:15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kida K, Minamishima S, Wang H, Ren J, Yigitkanli K, Nozari A, Mandeville JB, Liu PK, Liu CH, Ichinose F. Sodium sulfide prevents water diffusion abnormality in the brain and improves long term outcome after cardiac arrest in mice. Resuscitation. 2012;83:1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, VandenEkart E, Toombs CF, Szabo C. A monobromobimane‐based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen X, Peter EA, Bir S, Wang R, Kevil CG. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic Biol Med. 2012;52:2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olson KR. The therapeutic potential of hydrogen sulfide: separating hype from hope. Am J Physiol Regul Integr Comp Physiol. 2011;301:R297–R312. [DOI] [PubMed] [Google Scholar]
- 11. Toohey JI. Sulfur signaling: is the agent sulfide or sulfane? Anal Biochem. 2011;413:1–7. [DOI] [PubMed] [Google Scholar]
- 12. Jacob C, Anwar A, Burkholz T. Perspective on recent developments on sulfur‐containing agents and hydrogen sulfide signaling. Planta Med. 2008;74:1580–1592. [DOI] [PubMed] [Google Scholar]
- 13. Francoleon NE, Carrington SJ, Fukuto JM. The reaction of H2S with oxidized thiols: generation of persulfides and implications to H2S biology. Arch Biochem Biophys. 2011;516:146–153. [DOI] [PubMed] [Google Scholar]
- 14. Kimura Y, Mikami Y, Osumi K, Tsugane M, Oka J‐I, Kimura H. Polysulfides are possible H2S‐derived signaling molecules in rat brain. FASEB J. 2013;27:2451–2457. [DOI] [PubMed] [Google Scholar]
- 15. Marutani E, Sakaguchi M, Chen W, Sasakura K, Liu J, Xian M, Hanaoka K, Nagano T, Ichinose F. Cytoprotective effects of hydrogen sulfide‐releasing N‐methyl‐D‐aspartate receptor antagonists mediated by intracellular sulfane sulfur. MedChemComm. 2014;5:1577–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bergeron MJ, Clémençon B, Hediger MA, Markovich D. SLC13 family of Na+‐coupled di‐ and tri‐carboxylate/sulfate transporters. Mol Aspects Med. 2013;34:299–312. [DOI] [PubMed] [Google Scholar]
- 17. Tokuda K, Kida K, Marutani E, Crimi E, Bougaki M, Khatri A, Kimura H, Ichinose F. Inhaled hydrogen sulfide prevents endotoxin‐induced systemic inflammation and improves survival by altering sulfide metabolism in mice. Antioxid Redox Signal. 2012;17:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie‐Baez NO, Xian M, Fukuto JM, Akaike T. Reactive cysteine persulfides and s‐polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci USA. 2014;111:7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ‐lyase. Science. 2008;322:587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S‐sulfhydration. Sci Signal. 2009;2:ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thal SC, Thal SE, Plesnila N. Characterization of a 3‐vessel occlusion model for the induction of complete global cerebral ischemia in mice. J Neurosci Methods. 2010;192:219–227. [DOI] [PubMed] [Google Scholar]
- 22. Sasakura K, Hanaoka K, Shibuya N, Mikami Y, Kimura Y, Komatsu T, Ueno T, Terai T, Kimura H, Nagano T. Development of a highly selective fluorescence probe for hydrogen sulfide. J Am Chem Soc. 2011;133:18003–18005. [DOI] [PubMed] [Google Scholar]
- 23. Dodson M, Darley‐Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med. 2013;63:207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ohtaki H, Nakamachi T, Dohi K, Aizawa Y, Takaki A, Hodoyama K, Yofu S, Hashimoto H, Shintani N, Baba A, Kopf M, Iwakura Y, Matsuda K, Arimura A, Shioda S. Pituitary adenylate cyclase‐activating polypeptide (PACAP) decreases ischemic neuronal cell death in association with IL‐6. Proc Natl Acad Sci USA. 2006;103:7488–7493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benakis C, Bonny C, Hirt L. JNK inhibition and inflammation after cerebral ischemia. Brain Behav Immun. 2010;24:800–811. [DOI] [PubMed] [Google Scholar]
- 26. Rössig L, Fichtlscherer B, Breitschopf K, Haendeler J, Zeiher AM, Mülsch A, Dimmeler S. Nitric oxide inhibits caspase‐3 by S‐nitrosation in vivo. J Biol Chem. 1999;274:6823–6826. [DOI] [PubMed] [Google Scholar]
- 27. Ren C, Du A, Li D, Sui J, Mayhan WG, Zhao H. Dynamic change of hydrogen sulfide during global cerebral ischemia–reperfusion and its effect in rats. Brain Res. 2010;1345:197–205. [DOI] [PubMed] [Google Scholar]
- 28. Kimura Y, Goto Y‐I, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2009;12:1–13. [DOI] [PubMed] [Google Scholar]
- 29. Nijboer CH, Bonestroo HJC, Zijlstra J, Kavelaars A, Heijnen CJ. Mitochondrial JNK phosphorylation as a novel therapeutic target to inhibit neuroinflammation and apoptosis after neonatal ischemic brain damage. Neurobiol Dis. 2013;54:432–444. [DOI] [PubMed] [Google Scholar]
- 30. Zhao K, Ju Y, Li S, Altaany Z, Wang R, Yang G. S‐sulfhydration of MEK1 leads to PARP‐1 activation and DNA damage repair. EMBO Rep. 2014;15:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lai Y‐C, Pan K‐T, Chang G‐F, Hsu C‐H, Khoo K‐H, Hung C‐H, Jiang Y‐J, Ho F‐M, Meng T‐C. Nitrite‐mediated S‐nitrosylation of caspase‐3 prevents hypoxia‐induced endothelial barrier dysfunction. Circ Res. 2011;109:1375–1386. [DOI] [PubMed] [Google Scholar]
- 32. Mishanina TV, Libiad M, Banerjee R. Biogenesis of reactive sulfur species for signaling by hydrogen sulfide oxidation pathways. Nat Chem Biol. 2015;11:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pollay M, Kaplan RJ. The movement of sulfate and thiosulfate into in vivo choroid plexus. J Neurobiol. 1971;2:221–230. [DOI] [PubMed] [Google Scholar]
- 34. De La Calzada‐Jeanlouie M, Coombs J, Shaukat N, Olsen D. Utility of sodium thiosulfate in acute cyanide toxicity. Ann Emerg Med. 2013;61:124–125. [DOI] [PubMed] [Google Scholar]
- 35. Ratsimbazafy V, Bahans C, Guigonis V. Dramatic diminution of a large calcification treated with topical sodium thiosulfate. Arthritis Rheum. 2012;64:3826. [DOI] [PubMed] [Google Scholar]
- 36. Nigwekar SU, Brunelli SM, Meade D, Wang W, Hymes J, Lacson E. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013;8:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Muldoon LL, Pagel MA, Kroll RA, Brummett RE, Doolittle ND, Zuhowski EG, Egorin MJ, Neuwelt EA. Delayed administration of sodium thiosulfate in animal models reduces platinum ototoxicity without reduction of antitumor activity. Clin Cancer Res. 2000;6:309–315. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Na2S, but not STS, increased extracellular or intracellularsulfide levels. Fluorescent intensity of (A) medium containing HSip‐1 or (B) SH‐SY5Y cells loaded with HSip‐1 DA was measured after incubation with Na2S or STS at 37°C for 5 min or 24 h. Intracellular sulfide levels later than 3 h after incubation could not be measured since loaded HSip‐1 DA in cells leaked out into the medium. n=4 each; ***P<0.001 vs baseline at the same time point.
Figure S2. Time course for the medium‐change experiment in Figure 3 (C and D).
Figure S3. A, Protein levels of Myc‐tag or β‐tubilin in SH‐SY5Y transfected with vector alone, WT caspase‐3, or C163A caspase‐3 plasmid. B, Caspase‐3 activity in transfected SH‐SY5Y. n=5 each *P<0.01 vs WT by t‐test. C, Representative immunoblot of Caspase‐3 in SH‐SY5Y transfected with vector alone, WT, or C163A casase‐3 plasmid.
Figure S4. Representative immunoblot of NaS‐2 in SH‐SY5Y 48 h after transfection with siRNA (scrambled sequence or NaS‐2).
Figure S5. Average cerebral blood flow at middle cerebral artery region of mice subjected to BCAO for 40 min and reperfusion for 60 min. n=3 at each time point.