

Abstract
Background
The titin gene (TTN) encodes the largest human protein, which plays a central role in sarcomere organization and passive myocyte stiffness. TTN truncating mutations cause dilated cardiomyopathy (DCM); however, the role of TTN missense variants in DCM has been difficult to elucidate because of the presence of background TTN variation.
Methods and Results
A cohort of 147 DCM index subjects underwent DNA sequencing for 313 TTN exons covering the N2B and N2BA cardiac isoforms of TTN. Of the 348 missense variants, we identified 44 “severe” rare variants by using a bioinformatic filtering process in 37 probands. Of these, 5 probands were double heterozygotes (additional variant in another DCM gene) and 7 were compound heterozygotes (2 TTN “severe” variants). Segregation analysis allowed the classification of the “severe” variants into 5 “likely” (cosegregating), 5 “unlikely” (noncosegregating), and 34 “possibly” (where family structure precluded segregation analysis) disease‐causing variants. Patients with DCM carrying “likely” or “possibly” pathogenic TTN “severe” variants did not show a different outcome compared with “unlikely” and noncarriers of a “severe” TTN variant. However, the “likely” and “possibly” disease‐causing variants were overrepresented in the C‐zone of the A‐band region of the sarcomere.
Conclusions
TTN missense variants are common and present a challenge for bioinformatic classification, especially when informative families are not available. Although DCM patients carrying bioinformatically “severe” TTN variants do not appear to have a worse clinical course than noncarriers, the nonrandom distribution of “likely” and “possibly” disease‐causing variants suggests a potential biological role for some TTN missense variants.
Keywords: cardiomyopathy, cardiovascular genetics, dilated cardiomyopathy, heart failure, missense variants
Introduction
Familial dilated cardiomyopathy (DCM) is a genetic heart muscle disease caused by mutations in genes encoding structural proteins of the cytoskeleton and sarcomere.1 Recent investigations have focused attention on the titin gene (TTN), which encodes the giant sarcomeric protein titin, the largest known human protein and key contributor to passive myocyte stiffness.2 TTN mutations were first associated with tibial muscular dystrophy, but truncation mutations of TTN have recently been implicated as the most frequent identifiable cause of DCM.3 Titin is composed of >33 000 amino acid residues, and >60 000 missense variants are reported in the 1000 Genomes Project.3, 4 This frequency is far above the expected frequency of disease‐causing mutations for the TTN gene. Indeed, Golbus et al analyzed the data of the 1000 Genomes Project and found a high frequency of predicted pathogenic protein altering variation in TTN.5 They suggested that many of these variants could be either benign or insufficient on their own to cause disease but could act as modifiers in genetically susceptible hosts. Additional evidence supporting a modifier role for TTN comes from Roncarati et al, who performed whole‐exome sequencing in a large family with DCM, and found a subset of relatives with both TTN missense and Lamin A/C (LMNA) variants showing a more severe early‐onset phenotype.6 The high prevalence of missense variants in TTN and potential modifier roles complicate interpretation of detected TTN missense variants in both research and clinical settings.
To further address the role of TTN missense variants, we sequenced TTN in a large cohort of 147 DCM index patients and sorted the variants by using a rigorous bioinformatic pipeline followed by segregation analysis to identify TTN rare missense variants likely to be pathogenic. We further investigated if carriers of TTN putative disease‐causing missense mutations display a distinct phenotype and survival compared with noncarriers.
Methods
Patient Population
Probands from 147 DCM families were selected from the International Familial Cardiomyopathy Registry, a multicenter, 3‐decade–long ongoing project studying human hereditary cardiomyopathies. The diagnosis of DCM was created on the basis of the 1999 consensus criteria of the Guidelines for Familial Dilated Cardiomyopathy (FDC) and all available living subjects were evaluated by the investigators.7 Clinical data collected included family history, physical examination, laboratory investigations, electrocardiogram, and echocardiogram. Medical records from deceased subjects were reviewed when available. Criteria for the diagnosis of DCM were the presence of left ventricular fractional shortening <25% and/or an ejection fraction <45%, and left ventricular end‐diastolic diameter >117% of the predicted value provided by the Henry formula.7 Exclusion criteria included any of the following conditions: blood pressure >160/110 mm Hg, obstruction >50% of a major coronary artery branch, alcohol intake >100 g/d, persistent high‐rate supraventricular arrhythmia, systemic diseases, pericardial diseases, congenital heart diseases, cor pulmonale, and myocarditis.7 Informed consent was obtained from living subjects, and local institutional review boards approved the protocol.
DNA Sequence Analysis
The University of Washington, Department of Genome Science, completed DNA resequencing of TTN in 2008 under National Heart, Lung, and Blood Institute (NHLBI) grant N01‐HV‐48194.3, 8 Exons and periexonic regions of titin isoform N2A (NM_133378) along with additional exons unique to the principal cardiac isoform N2B (NM_003319) were amplified from genomic DNA by using polymerase chain reaction (PCR). This covered 312 exons (311 expressed as titin protein) and the complete 3′‐untranslated region. The PCR primers were Tm matched and designed from a masked reference sequence. Each primer pair was “tailed” with a universal M13 forward and reverse sequencing primer for subsequent sequencing. Once the regions were amplified, each PCR product was sequenced from the forward and reverse direction to provide double‐stranded coverage. Sequencing was carried out with Sanger Big‐Dye Terminator sequencing on capillary‐based machines (AB 3730; Applied Biosystems).
Bioinformatic Filter Criteria
The single nucleotide polymorphisms (SNPs) identified from the sequence were annotated using by ANNOVAR9 and dbNSFP,10 the database for nonsynonymous SNP functional predictions, which provides scores from the widely used algorithms SIFT, PolyPhen2, LRT, Mutation Taster, and GERP. Variants were then scored as “severe” based on the following criteria (Table S1): SIFT score of zero,11 Polyphen2 HDVAR “possibly damaging” or “damaging”,12 GERP score >4.2,13 absence in the 1000 Genomes Project cohort,14 and an allele frequency of ≤0.04% in the NHLBI Exome‐Sequencing Project dataset (Figure S1 and Table S2).15 Variants were excluded and considered “nonsevere” if they (1) failed to meet at least 1 of these criteria or (2) they were nonsynonymous or intronic mutations predicted to have no effect on splicing. In families where samples from >1 affected family member were available, segregation analysis was performed by using Sanger sequencing (Table S3). In accordance with the scheme proposed by Hershberger et al, variants that showed concordant or discordant segregation among affected and unaffected relatives were classified as “likely” or “unlikely” to be disease causing, respectively; variants where no segregation analysis was achievable were classified as “possibly” disease causing; and probands with no bioinformatically “severe” variants were classified as “noncarriers” (Figure 1).16
Figure 1.
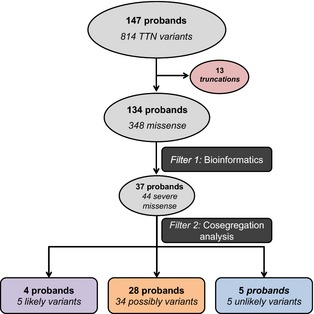
Bioinformatic and segregation analysis in 134 DCM Probands. TTN variants were filtered with bioinformatic algorithms as described and classified into “severe” or “nonsevere” categories. Mutations initially identified as “severe” were studied by segregation analysis when samples were available and further classified as “likely,” “possibly,” or “unlikely” to be disease causing. DCM indicates dilated cardiomyopathy; TTN, titin gene.
Statistical Analysis
To estimate the effect of TTN rare missense variants on the phenotype and natural history of DCM, we compared the clinical data and long‐term death/heart transplantation event‐free survival of the index DCM subjects among the different TTN variant groups and with the noncarriers. Summary statistics of clinical and instrumental variables at enrollment were expressed as median and IQR values or as counts and percentages, as appropriate. Comparison between rare variant carriers and noncarriers for continuous variables were analyzed by using ANOVA, with the Brown–Forsythe statistic when the assumption of equal variances did not hold, and the χ2 or Fisher exact test was used for discrete variables. Event‐free survival curves for death/heart transplantation were estimated and plotted by using the Kaplan–Meier method, and the log‐rank test was applied to investigate differences in long‐term survival. Statistical analyses were performed by using the IBM SPSS Statistical Package version 19.0.
Mapping the TTN Variants to Protein Domains
Severe variants in “likely” and “possibly” disease‐causing categories were mapped to their respective protein domains starting from their exon location by using the Ensembl database (http://uswest.ensembl.org) with the identifier Q8WZ42‐12 to locate the variants within the cDNA sequence, followed by mapping to amino acid position by referencing the Uniprot database (http://www.uniprot.org) to identify titin domains. Pearson's χ2 goodness‐of‐fit tests were used to test the random distribution of the “likely” and “possibly” disease‐causing variants, incorporating the size in amino acids of each protein region tested as one group and the remainder of titin protein as a second group (df=1).17
Results
Genetic Analysis of TTN Missense Variants
TTN was sequenced in 147 DCM probands. Truncating TTN variants were found in 13 of 147 and have previously been reported.3 Among the remaining 134 probands, we found 814 rare variants (≤0.04% in NHLBI Exome‐Sequencing Project dataset); of those, 466 variants were either synonymous or intronic and were not further studied. Of the remaining 348 missense variants, 44 were bioinformatically classified as “severe” and were present in 37 probands (Table 1). In 9 of 37 families, samples from other affected and unaffected family members were available to test for segregation analysis. Segregation of the rare TTN variants was confirmed by the presence in an affected or absence in an unaffected relative within the family. Among those 9 families, confirmation by Sanger sequencing revealed there were 5 TTN rare missense variants among 4 families.
Table 1.
TTN Rare Variants Classified as “Severe” by Bioinformatic Analyses
| DCM Gene Mutation | Disease Associated | AA Change (NP_001254479) | Exon | Domain | Nucleotide Change (NM_001267550) | RS Number | Non‐TTN Gene Mutation | LVEF% |
|---|---|---|---|---|---|---|---|---|
| per Bang, et al17 | ||||||||
| Z‐disk | ||||||||
| TTN | Possibly | Glu222Lys | 5 | Unique sequence | c.664 G>A | 72647844 | — | 30 |
| TTN | Possibly | Glu1039Gly | 19 | Unique sequence | c.3116 A>G | 72647867 | MYH6: Pro830Leu | 49 |
| I‐Band | ||||||||
| TTN a | Possibly | Ala2258Val | 29 | I2 Ig | c.6773 C>T | 72647881 | — | 17a |
| TTN a | Possibly | Ser7726Leu | 81 | I59 Ig | c.23177 C>T | 17452588 | — | 22b |
| TTN a | Possibly | Gly8886Arg | 93 | I71 Ig | c.26656 G>C | 72648991 | — | |
| TTN | Likely | Ser13702Pro | 225 | I84 Ig | c.41104 T>C | 72650078 | — | 35 |
| TTN | Possibly | Thr14183Ala | 231 | I89 Ig | c.42547 A>G | 72650081 | — | 45 |
| TTN a | Likely | Arg14640Cys | 237 | I94 Ig | c.43918 C>T | 72650088 | — | 43c |
| TTN a | Possibly | Ile15254Phe | 247 | I101 Ig | c.45760A>T | 72677226 | — | 25f |
| TTN | Possibly | Arg15484Met | 250 | I104 Ig | c. 46451 G>T | 72677229 | — | 59 |
| I‐Band/A‐Band Junction | ||||||||
| TTN | Possibly | Leu16469His | 263 | I114 FN3 | c.49406 T>A | 72677245 | — | 32 |
| TTN | Possibly | Glu16938Gln | 269 | I118 FN3 | c.50812 G>C | 72677250 | — | 45 |
| A‐Band (D‐Zone) | ||||||||
| TTN | Unlikely | Arg17086His | 271 | A2 FN3 | c.51257 G>A | 72632860 | SCN5A: Arg222Gln | 37 |
| TTN | Possibly | Arg19705Cys | 300 | A28 FN3 | c.59113 C>T | 72646839 | — | 25 |
| TTN b | Possibly | Ser20273Tyr | 304 | A34 FN3 | c.60818 C>T | 72646844 | — | 7g |
| Possibly | Ser20273Tyr | 304 | A34 FN3 | c.60818 C>T | 72646844 | — | 10 | |
| A‐Band (C‐Zone) | ||||||||
| TTN | Possibly | Leu21176Ser | 306 | A43 Ig | c.63527 T>C | 72646854 | — | 50 |
| TTN | Possibly | Pro21563Ala | 310 | A47 FN3 | c.64687 C>G | 72646860 | — | 44 |
| TTN | Unlikely | Arg22029His | 314 | A51 FN3 | c.66086 G>A | 72646868 | — | 40 |
| TTN | Unlikely | Phe22653Leu | 320 | A58 FN3 | c.67959 T>A | 72646877 | LMNA: Arg89Leu | NA |
| TTN | Possibly | Glu23217Gly | 325 | A63 FN3 | c.69650 A>G | 72646884 | — | 30 |
| TTN | Possibly | Tyr23494His | 326 | A66 FN3 | c.70480 T>C | 72646888 | — | 30 |
| TTN a | Possibly | Ala24343Thr | 326 | A75 FN3 | c.73027 G>A | 72646895 | — | 17a |
| TTN a | Possibly | Val24516Ile | 326 | A76 Ig | c.73546 G>A | 72646897 | — | 25d |
| TTN | Possibly | Pro25207Arg | 326 | A84 FN3 | c.75620 C>G | 72646900 | — | 29 |
| TTN a | Possibly | Tyr27008Asp | 326 | A102 FN3 | c.81022 T>G | 72648211 | — | 55e |
| TTN a | Possibly | Arg27563Cys | 326 | A107 FN3 | c. 82687 C>T | 72648214 | — | 55e |
| TTN | Possibly | Ser27585Tyr | 326 | A108 FN3 | c.82754 C>A | 72648215 | — | 15 |
| TTN | Likely | Arg28118His | 326 | A113 FN3 | c.84353 G>A | 72648220 | — | 23 |
| TTN | Likely | Ser29303Gly | 329 | A125 FN3 | c.87907 A>G | 72648231 | LMNA: c.936 G>A | 17 |
| TTN | Possibly | Leu29499Arg | 331 | A127 Ig | c. 88496 T>G | 72648234 | — | 25 |
| TTN a | Possibly | Gly29562Asp | 332 | A128 FN3 | c.88685 G>A | 72648235 | — | 25f |
| TTN a | Likely | Glu29590Gln | 332 | A128 FN3 | c.88768 G>C | 72648236 | — | 43c |
| TTN | Possibly | Gly30358Glu | 335 | A136 FN3 | c.91073 G>A | 72648243 | — | 38 |
| TTN | Possibly | Trp30667Arg | 338 | A139 FN3 | c.91999 T>A | 72648246 | — | 29 |
| TTN b | Possibly | Ile31757Thr | 343 | A150 FN3 | c.95270 T>C | 72648259 | — | 25d |
| Possibly | Ile31757Thr | 343 | A150 FN3 | c.95270 T>C | 72648259 | — | 37 | |
| TTN | Possibly | Arg31856Gly | 344 | A151 FN3 | c.95566 C>G | 72648261 | — | 28 |
| TTN a | Possibly | Arg33052His | 354 | A163 FN3 | c.99155 G>A | 72648276 | — | 10g |
| C‐Zone/M‐Band Junction | ||||||||
| TTN | Unlikely | Gly33319Arg | 356 | A166 FN3 | c.99955 G>A | 72648279 | — | 28 |
| M‐Band | ||||||||
| TTN | Possibly | Arg33903Leu | 358 | Kinase | c.101708 G>T | 72629782 | — | 50 |
| TTN | Possibly | Lys34293Glu | 358 | M2 Ig | c.102877 A>G | 72629783 | — | 15 |
| TTN | Possibly | Ile34411Asn | 358 | M3 Ig | c.103232 T>A | 72629784 | — | 25 |
| TTN | Unlikely | Arg34653Leu | 358 | is2 between M3 and M4 | c.103958 G>T | 72629786 | LMNA: Arg166Pro | 25 |
Variants classified by bioinformatic analysis as “severe” are shown along with categorization as “likely,” “possibly,” or “unlikely” disease causing based on whether segregation with DCM was confirmed. DCM indicates dilated cardiomyopathy; LMNA, Lamin A/C; LVEF, left ventricular ejection fraction; NA, not available; TTN, titin gene.
Compound heterozygous TTN variants: a, b, c, d, e, f, g.
TTN variant shared by 2 nuclear families.
Two variants (TTN‐251996 and TTN‐247572) in 2 different families (DNFDC014 and DNFDC131, respectively), 2 TTN variants (TTN‐178217 and TTN‐254772) in the same family TSFDC044, and 1 variant (TTN‐172724) in family DNFDC074 segregated with the DCM phenotype and were classified as “likely” disease‐causing (Figure 2 and Table 2). The remaining 5 tested variants failed to segregate with the DCM phenotype and were classified as “unlikely” disease causing. The 34 variants present in 28 families that could not be examined for segregation because of lack of additional samples for testing were classified as “possibly” disease causing.18 Seven probands harbored compound heterozygous “severe” TTN variants. Among the 4 families with “likely” TTN variants, 1 also harbored an LMNA mutation (Table 2).
Figure 2.
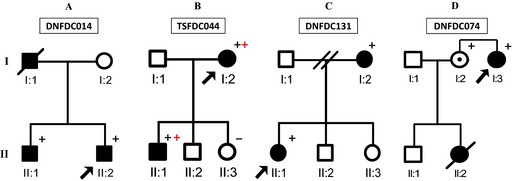
Pedigrees for “likely” TTN families harboring disease‐causing pathogenic variants. Squares, circles, plus signs, and arrows indicate males, females, positive TTN variant status, and probands, respectively. An obligate carrier is indicated by a dotted circle. Black shading indicates individuals affected with DCM. Double plus symbols mean double heterozygous variants. DNFDC014 has variant rs72648231, TSFDC044 has variants rs72648236 and rs72650088, DNFDC131 has variant rs72648220, and DNFDC074 has variant rs72650078. DCM indicates dilated cardiomyopathy; TTN, titin gene.
Table 2.
Clinical Characteristics of “Likely” TTN Rare Variant Carriers
| Family | DNFDC014 | DNFDC131 | DNFDC074 | TSFDC044 | ||||
|---|---|---|---|---|---|---|---|---|
| TTN variant | Ser29303Gly | Arg28118His | Ser13702Pro | Arg14640Cys Glu29590Gln | ||||
| Chr. 2; Exon(s) | 2; 329 | 2; 326 | 2; 225 | 2; | 237 332 | |||
| Individual | II‐1 | II‐2 | II‐1 | I‐2 | I:2a | I:3 | I‐2 | II‐1 |
| Sex | M | M | F | F | F | F | F | M |
| NYHA | 3 | 1.5 | 2 | 3 | 1 | 1 | 1 | 1 |
| LVEDD, mm | 50.3 | 53 | 64.2 | 50 | 45 | 58 | 60 | 56 |
| LVEF, % | 17 | 46 | 23 | 40 | 62 | 35 | 42 | 43 |
| Non‐TTN gene mutation |
LMNA c.936 G>A |
LMNA c.936 G>A |
||||||
| Age of onset, y | 60 | 40 | 49 | 39 | 54 | 48 | 42 | 26 |
| Transplantation | Yes | No | No | No | No | No | No | No |
LMNA indicates Lamin A/C; LVEDD, left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association functional classification; TTN, titin gene.
DNFDC074, individual I:2 is an obligate carrier.
TTN Variant Association With Functional Domains
The “likely” and “possibly” disease‐causing TTN variants were nonrandomly distributed across the titin protein with overrepresentation of variants found in the C‐zone repeats of titin (P<0.01) (Figures 3 and 4) with a bias toward variants in the terminal 3 fibronectin type III (Fn‐III) domains.
Figure 3.
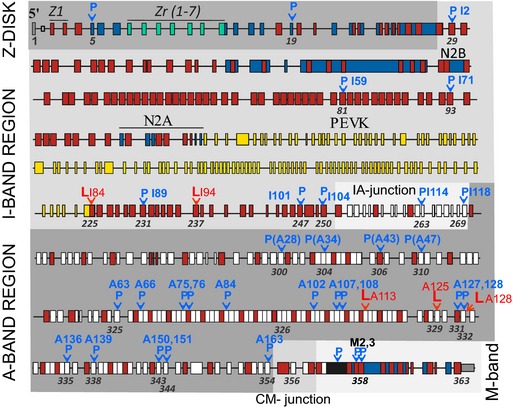
Exon structure of human TTN gene with identified DCM missense mutations. P indicates “possible” disease‐causing mutation; L, “likely” disease‐causing mutation. Indicated are also titin domain numbers (top) and exon numbers (bottom). Red rectangle represents immunoglobulin‐like domain; white, fibronectin type 3 domain; blue, unique sequence; green, z‐repeat domain; yellow, PEVK domain; black, titin kinase domain (gene structure based on Bang et al17). DCM indicates dilated cardiomyopathy; TTN, titin gene.
Figure 4.
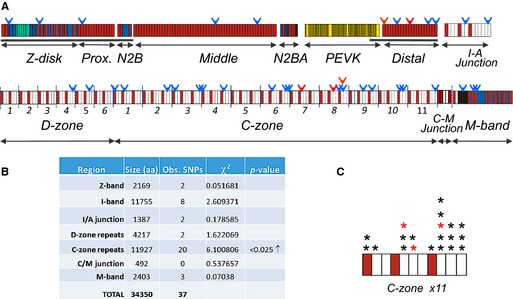
Missense variants mapped on TTN gene. A, Protein domain structure of largest full‐length titin isoform (UniProt entry Q8WZ42‐12, which includes all exons except for Novex‐3; this is the same as Ensembl transcript Ttn‐018Ensembl) with indicated locations of “possibly” (blue arrowheads) and “likely” (red arrowheads) SNPs. Top: Z‐disk and I‐band region of titin with tandem immunoglobulin segments (proximal [prox.], middle and distal subsegments), N2B and N2BA elements, and PEVK segment of I‐band indicated separately and I/A junction. Bottom: A‐band and M‐band regions of titin with D‐zone (6 super‐repeats each containing 7 domains) and C‐zone (11 super‐repeats with 11 domains each), C‐M junction, and M‐band regions indicated. I‐band region of titin expressed in N2B cardiac titin (the dominant isoform in the LV) is shown by the gray underline (the A‐band region is constitutively expressed in all full‐length isoforms). B, Pearson's χ2 goodness‐of‐fit tests were used to test if the distribution of the “likely” and “possibly” categorized nonsynonymous missense SNPs were random or not, incorporating the size in amino acids of each protein region tested as one group and the remainder of titin protein as a second group (df=1). Mutations in the C‐zone repeats are overrepresented (indicated by direction of arrow). C, The SNPs in the C‐zone are not randomly distributed over the 11 domains of the super‐repeats but appear to be biased toward the last 3 Fn‐III domains (the 11 super‐repeats were aligned and the number of SNPs in each of the 11 domains summed). LV indicates left ventricle; SNPs, single nucleotide polymorphisms; TTN, titin gene.
Genotype–Phenotype Analysis
No phenotypic differences were observed comparing the 32 probands of “likely” (n=4) or “possibly” (n=28) disease‐causing TTN variants with the 102 noncarriers (97 “noncarriers” plus 5 probands with “severe” variants subsequently classified as “unlikely” by segregation analysis) (Table 3). Moreover, during a mean follow‐up of 8 years (Figure 5), event free‐survival from cardiovascular death/transplant was not different between the 2 groups. Survival rates free from cardiovascular death/heart transplant at 40, 50, and 60 years of age were 96%, 77%, and 29% versus 87%, 77%, and 48% in carriers and noncarriers, respectively (P=NS). Including data from the 13 patients with truncating TTN variants previously reported3 did not identify any clinical or prognostic differences with respect to carriers of TTN “severe” missense or truncation variants compared with other DCM patients (Table 3 and Figure 5). No significant difference in clinical phenotype was noted between the group of subjects carrying a single TTN variant and the smaller groups carrying double or compound TTN variants. In addition, among the 5 “unlikely” families, the affected individuals who carried a specific TTN variant did not express a more severe phenotype than did affected individuals who did not carry the TTN variant, as measured by left ventricular ejection fraction, left ventricular end‐diastolic diameter, and New York Heart Association functional classifications (data not shown). These results would imply that there is no modifier effect within families.
Table 3.
Genotype–Phenotype Analysis by Variant Category
| Truncations | Likely+Possibly | Unlikely+Noncarriers | P Value | |
|---|---|---|---|---|
| Subjects, n | 13 | 32 | 102 | |
| Women, n | 4 (31%) | 13 (41%) | 40 (39%) | 0.817 |
| Age of onset, y | 40±14 | 44±11 | 41±14 | 0.555 |
| NYHA, median [IQR] | 1 [1;2] | 2 [1;2.5] | 2 [1;2.5] | 0.251 |
| LVEDD, mm | 64±11 | 64±9 | 64±11 | 0.997 |
| LVEF, % | 32±11 | 32±15 | 33±13 | 0.934 |
| Follow‐up (y), median [IQR] | 8 [1; 16] | 7 [4; 12] | 8 [2; 13] | 0.758 |
| Heart transplant, n | 3 (23%) | 5 (16%) | 21 (21%) | 0.787 |
| Cardiovascular death, n | 3 (23%) | 6 (19%) | 19 (19%) | 0.928 |
LVEDD indicates left ventricular end‐diastolic dimension; LVEF, left ventricular ejection fraction; NYHA, New York Heart Association functional classification.
Figure 5.
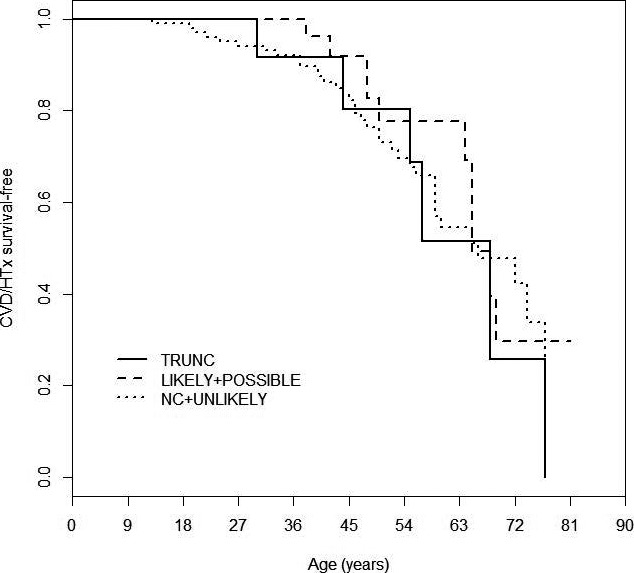
Long‐term survival curves in TTN variant carriers. Kaplan–Meier event‐free survival for CVD or HTx based on TTN variant categories: TRUNC, “likely” and “possibly”; NC, “unlikely.” CVD indicates cardiovascular death; HTx, heart transplant; NC, noncarrier; TRUNC, truncations; TTN indicates titin gene.
Interestingly, there was a trend toward a lower left ventricular ejection fraction for variants located farther from the Z‐disk: the mean left ventricular ejection fraction was 40% in Z‐disk, 35% in I‐band, and 29% in A‐band variants. We also noted that “likely” and “possibly” carriers of TTN variants with double or compound heterozygous mutations (Table 1) had lower left ventricular ejection fractions (29±16%) compared with those with single missense variants (34±14%), although because of the small number of observations the difference did not reach statistical significance.
Discussion
The TTN gene, encoding the giant protein titin, harbors a large array of amino acid sequence variation, rendering genetic analysis for cardiomyopathy diagnosis challenging. Our bioinformatic filtering process identified variants bioinformatically classified as “severe” in 12.6% (44/348) of TTN missense variants and 27.6% (37/134) of DCM subjects. The variant analysis used stringent filtering criteria as currently recommended in research and clinical settings.15, 19 Despite the filtering process, 5 of 9 families with TTN variants classified as “severe” demonstrated incompatible segregation with the affected phenotype, implying a significant false‐positive rate from the bioinformatics analysis alone. Four families harbored 5 “severe” TTN variants that segregated with the DCM phenotype, and 28 probands had “severe” variants that could not be assessed by segregation. Thus, in contrast to TTN truncation variants that account for up to 25% of DCM,3 pathogenic TTN missense variants are not easily resolved and likely contribute to a smaller fraction of DCM.
Similar to prior data on TTN truncations,3 the presence of a TTN missense variant did not correlate with clinical measures of disease severity or progression, indicating that the DCM phenotype because of TTN missense variants is indistinguishable from other forms of DCM. However, although many variants could not be assessed by segregation analysis, the clustering of severe “possibly” variants in the A‐band regions is suggestive of a biological consequence of A‐band variation and aligns with previously noted overrepresentation of TTN truncating variants within this region.3 We also observed a progression of lower ejection fractions with greater distance from the Z‐disc; this trend toward lower ejection fractions in A‐band variants is consistent with prior observations that A‐band truncation variants have worse clinical outcomes.20 The reason for these outcomes is unclear, but we speculate the likelihood that the mutant protein incorporates in the sarcomere exerts in a dominant negative effect that varies with the location of the mutation.
Interestingly, the distribution of bioinformatically “severe” TTN missense variants across titin domains was nonrandom and similar to what has been shown previously with TTN truncation variants. Indeed, TTN variants were overrepresented in the A‐band region of titin,3 specifically in the C‐zone of the A‐band that consists of a super‐repeat of 11 immunoglobulin‐like domains and Fn‐III domains, that are organized in a consistent pattern repeated 11 times (Figure 4B and 4C). Interestingly, the Fn‐III domains of the C‐zone are overrepresented (Figure 4C). These Fn‐III domains have previously been shown to bind myosin subfragment‐1 and to be essential for the length dependency of force development (they depress calcium sensitivity at short sarcomere length).21 Although the biological explanation for the clustering of A‐band of variants and whether any modifying effects exist remain to be critically evaluated in future studies, we currently speculate that some A‐band missense mutations may have a functional detrimental effect on contractility.
Our study was limited by lack of functional assays performed on the large number of TTN variants detected and by the possibility that some variants that did not meet criteria for “severe” could have effects on DCM risk. Cardiomyopathy gene mutations were not fully characterized in this cohort because samples were collected over 2 decades and gene screening was done throughout this period; therefore, some samples were not screened for all possible genes. Small family sizes and lack of additional DNA samples from affected relatives limited our ability to test for segregation in all cases. Given that instances of nonsegregation of “severe” variants did occur in our study, the interpretation of bioinformatically “severe” TTN variants in isolated patients remains challenging. Finally, this study considered only index patients in the outcome analysis.
In conclusion, TTN missense variants are common and the interpretation of TTN resequencing findings is challenging even when stringent bioinformatic and segregation criteria are used.5, 19 Using such criteria, missense variants bioinformatically classified as “severe” have a frequency comparable to that of TTN truncations in DCM. However, in contrast to truncation variants, our study did not demonstrate a strong association of missense variants with a risk of DCM, and when segregation analysis was possible, 13.5% of the “severe” variants were ultimately classified as “unlikely” because of incongruous segregation. These data argue that TTN missense mutations should not currently be interpreted as disease causing in most situations, urge caution in the interpretation of missense mutations in clinical practice, and support the need for the development of functional assays that better assess pathogenicity. Not surprisingly, the “severe” TTN missense variants did not associate with differences in clinical phenotypes in our DCM population, further arguing that many missense TTN variants are not disease‐causing variants. Last, large‐scale TTN sequencing and functional investigations on TTN variant domains will allow a better understanding of the role of TTN variants in DCM and pave the way to the development of diagnostic and therapeutic strategies.
Sources of Funding
This study was supported by National Institutes of Health grants UL1 RR025780, UL1 TR001082 N01‐HV‐48194, R01 HL69071, and R01 116906 to Dr Mestroni; K23 JL067915 and R01HL109209, 3R01HL109209‐01A1S, and 3R01HL109209‐01A1S1 to MT; HL062881 (to Dr Granzier); Leducq Foundation 14‐CVD 03; Fondazione CRTrieste and Fondazione GENERALI to Sinagra.
Disclosures
None.
Supporting information
Figure S1. Venn Diagram of SIFT/PolyPhen2/GERP Relationships
Table S1. Sequential Bioinformatic Filter Criteria of TTN Variants
Table S2. Bioinformatic Data for the 44 “severe” TTN Rare Variants
Table S3. Primer Sequences and PCR Conditions
Acknowledgments
The authors are grateful to the patients and their families for their participation in the study.
(J Am Heart Assoc. 2015;4:e002645 doi: 10.1161/JAHA.115.002645)
Accompanying Figure S1 and Tables S1 through S3 are available at http://jaha.ahajournals.org/content/4/11/e002645/suppl/DC1
References
- 1. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752–762. [DOI] [PubMed] [Google Scholar]
- 2. LeWinter MM, Granzier HL. Titin is a major human disease gene. Circulation. 2013;127:938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Merlo M, Sinagra G, Carniel E, Slavov D, Zhu X, Barbati G, Spezzacatene A, Ramani F, Salcedo E, Di Lenarda A, Mestroni L, Taylor MR. Poor prognosis of rare sarcomeric gene variants in patients with dilated cardiomyopathy. Clin Transl Sci. 2013;6:424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave‐Castillo LM, Wolfgeher D, McNally EM. Population‐based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, di Pasquale E, Papa L, Portararo P, Columbaro M, Forni A, Faggian G, Condorelli G, Robinson PN. Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet. 2013;21:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J. 1999;20:93–102. [DOI] [PubMed] [Google Scholar]
- 8. Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S, Anderson B, Simon B, Bogomolovas J, Labeit S, Granzier H, Mestroni L. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy‐overlap syndromes. Circulation. 2011;124:876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat. 2011;32:894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acid Res. 2003;31:3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol. 2010;6:e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Norton N, Li D, Hershberger RE. Next‐generation sequencing to identify genetic causes of cardiomyopathies. Curr Opin Cardiol. 2012;27:214–220. [DOI] [PubMed] [Google Scholar]
- 16. Hershberger RE, Parks SB, Kushner JD, Li D, Ludwigsen S, Jakobs P, Nauman D, Burgess D, Partain J, Litt M. Coding sequence mutations identified in MYH7, TNNT2, SCN5A, CSRP3, LBD3, and TCAP from 313 patients with familial or idiopathic dilated cardiomyopathy. Clin Transl Sci. 2008;1:21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700‐kDa titin isoform, and its interaction with obscurin identify a novel Z‐line to I‐band linking system. Circ Res. 2001;89:1065–1072. [DOI] [PubMed] [Google Scholar]
- 18. MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S, Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA, Hershberger RE. Exome sequencing and genome‐wide linkage analysis in 17 families illustrate the complex contribution of TTN truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O'Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O'Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA. Integrate allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7: 270ra276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Muhle‐Goll C, Habeck M, Cazorla O, Nilges M, Labeit S, Granzier H. Structural and functional studies of titin's fn3 modules reveal conserved surface patterns and binding to myosin S1–a possible role in the Frank‐Starling mechanism of the heart. J Mol Biol. 2001;313:431–447. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Venn Diagram of SIFT/PolyPhen2/GERP Relationships
Table S1. Sequential Bioinformatic Filter Criteria of TTN Variants
Table S2. Bioinformatic Data for the 44 “severe” TTN Rare Variants
Table S3. Primer Sequences and PCR Conditions