

Abstract
Recently, we reported that heme binds to tumor suppressor p53 protein (TP53, best known as p53) and promotes its nuclear export and cytosolic degradation, whereas iron chelation stabilizes p53 protein and suppresses tumors in a p53-dependent manner. This not only provides mechanistic insights into tumorigenesis associated with iron excess, but also helps guide the administration of chemotherapy based on iron deprivation in the clinic.
Keywords: heme, p53, iron deprivation, nuclear export, hemochromatosis, tumorigenesis
Numerous studies have established that iron excess caused by dietary, environmental, or genetic factors can significantly promote tumorigenesis.1,2 Hereditary hemochromatosis (HH), a genetic disorder that leads to iron overload, affects nearly 1 in 300–400 of the white population in the United States alone. Clinical complications in HH patients typically include liver cirrhosis and up to a 20- to 200-fold increased risk of hepatocellular carcinoma or other cancer types, although the underlying mechanisms remain elusive.3 Moreover, tumors reprogram iron metabolism to achieve advantages in growth, proliferation, and/or metastasis.2 Iron deprivation, through iron chelation or application of transferrin receptor (TFR)-neutralizing antibodies, has emerged as a major strategy for chemotherapy. However, iron deprivation only suppresses select types of human malignancies while exerting no effects on other types of cancer.4 We recently reported a link between iron metabolism and tumor suppressor p53 function, providing mechanistic insight into both the increased tumorigenesis associated with iron overload and the selective therapeutic efficacy of iron deprivation-based chemotherapy.
Tumor suppressor p53 (TP53, hereafter referred to as p53) suppresses tumorigenesis and regulates DNA damage repair, cell-cycle arrest, and tumor responses to chemotherapy, while also serving as a hub in the control of cellular metabolism.5 A few small molecules, such as NAD+ and ADP, have been identified as potential physiological ligands for p53 protein that can modulate changes in p53 signaling in response to perturbation in cell redox state and energy metabolism. It remains unclear whether and how other metabolites or signaling molecules might bind to p53 and modulate its function in vivo.
Heme, an iron polyporphyrin, is a major form of intracellular bio-iron that constitutes the prosthetic group for proteins that function in myriad fundamental biological processes, including respiration, energetic homeostasis, signal transduction, xenobiotic detoxification, iron metabolism, mRNA processing, control of circadian rhythm, and some protein degradation pathways.6-9 As more heme–protein interactions continue to be identified, it is reasonable to believe that we may be only just beginning to understand the influence of heme on biological functions.
In our recent report, we showed that levels of both intracellular iron and heme are elevated 6- to 9-fold in the livers of Hfe−/− mice compared to wild-type mice. The HFE gene, standing for high Fe, encodes a protein that functions in iron absorption by regulating the interaction of the transferrin receptor with transferrin; Hfe−/− mice are a mouse model for human HH, in which the static level of tumor suppressor p53 protein is significantly downregulated.10 Cell-based expe-riments further indicate that heme accelerates proteasome-dependent degradation of p53 protein, whereas deferoxamine (DFO), an iron chelator, has the opposite effect. We next demonstrated that heme directly binds to p53 protein, apparently through the heme regulatory motif in the DNA-binding domain of p53. Such binding interferes with the interactions between p53 and its target DNA. Heme binding may also induce conformational changes in p53 and expose its C-terminal nuclear exporting sequences, thus promoting interaction of p53 with chromosomal region maintenance 1 (CRM1)/exportin-1 and facilitating the nuclear export of p53 and its subsequent cytosolic degradation. As a result, cellular p53 signaling is downregulated during iron or heme excess.
Similarly, we found that heme also binds and destabilizes p63 and p73, the other 2 members of the p53 protein family. Thus, the p53 protein family members contain the heme regulatory motif, bind to heme, and undergo accelerated degradation upon hemin treatment. It is conceivable that heme-induced nuclear export and destabilization of p53 family proteins, along with the ensuing functional changes, also exerts effects at the organismal level. Iron overload and accumulation of heme in afflicted cells or tissues eventually leads to increased production of reactive oxygen species (ROS) and damage to intracellular structures including proteins and DNA,2 a situation in which normally functioning p53, the “guardian of the genome,” would be essential to mitigate these insults and promote cell survival. However, heme-induced nuclear export and destabilization of p53 and other proteins may exacerbate the insults, thus contributing to the pathogenesis of disorders associated with iron excess, such as the tendency for tumorigenesis observed in hemochromatosis. It is intriguing to ask whether heme-accelerated degradation of p53 might play a role in other pathogenic features of hemochromatosis.
Through experiments using isogenic HCT116 human colon cancer cells and their p53-null mutants in cell culture and a tumorigenicity model, we were able to show that the tumor-suppressing effect of DFO relies on wild-type p53 signaling. Furthermore, iron deprivation induced by DFO was found to suppress multiple types of human tumor cell lines with predominantly wild-type p53 signaling but not p53-null cell lines, suggesting that upregulation of wild-type p53 signaling might critically underlie the selective efficacy of iron deprivation. Therefore, this work not only offers mechanistic insights into tumorigenesis associated with iron or heme excess (see Fig. 1 for a schematic view of the proposed mechanism), but also provides a molecular basis for chemotherapy based on iron deprivation.
Figure 1.
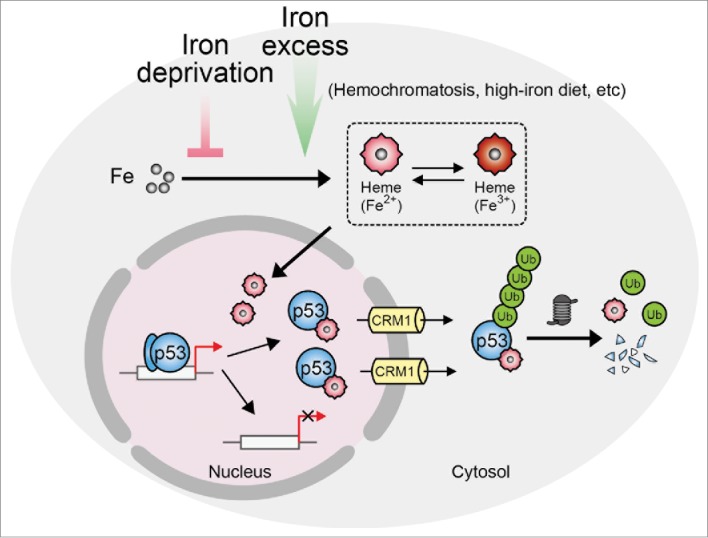
Schematic showing how iron metabolism might regulate p53 signaling and tumorigenesis. Heme directly binds to p53 protein and downregulates p53 signaling by interfering with p53–DNA interaction and promoting nuclear export of p53 and its subsequent cytosolic proteolysis. p53 refers to tumor suppressor p53 (TP53); CRM1, chromosomal region maintenance 1/exportin 1; Ub, ubiquitin.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The authors truly appreciate the support from all collaborators and funding from the Ministry of Science and Technology of China (2010CB912101, 2012CB910800, 2013CB910900, 2011CB915501), the National Natural Science Foundation of China (31270828, 31070678), Chinese Academy of Sciences (Instrument Developing Project YZ201339), Cancer Center of SIBS-Xuhui Central Hospital (CCR2012003), and Shanghai Institute of Neurosciences (SKLN-201206).
References
- 1. Weinberg ED. The role of iron in cancer. Eur J Cancer Prev 1996; 5:19-36; PMID:8664805 [PubMed] [Google Scholar]
- 2. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer 2013; 13:342-55; PMID:23594855; http://dx.doi.org/ 10.1038/nrc3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elmberg M, Hultcrantz R, Ekbom A, Brandt L, Olsson S, Olsson R, Lindgren S, Lööf L, Stål P, Wallerstedt S, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology 2003; 125:1733-41; PMID:14724826; http://dx.doi.org/ 10.1053/j.gastro.2003.09.035 [DOI] [PubMed] [Google Scholar]
- 4. Buss JL, Greene BT, Turner J, Torti FM, Torti SV. Iron chelators in cancer chemotherapy. Curr Top Med Chem 2004; 4:1623-35; PMID:15579100; http://dx.doi.org/ 10.2174/1568026043387269 [DOI] [PubMed] [Google Scholar]
- 5. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307-10; PMID:11099028; http://dx.doi.org/ 10.1038/35042675 [DOI] [PubMed] [Google Scholar]
- 6. Hu RG, Wang H, Xia Z, Varshavsky A. The N-end rule pathway is a sensor of heme. Proc Natl Acad Sci U S A 2008; 105:76-81; PMID:18162538; http://dx.doi.org/ 10.1073/pnas.0710568105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barr I, Smith AT, Senturia R, Chen Y, Scheidemantle BD, Burstyn JN, Guo F. DiGeorge critical region 8 (DGCR8) is a double-cysteine-ligated heme protein. J Biol Chem 2011; 286, 16716-25; PMID:21454614; http://dx.doi.org/ 10.1074/jbc.M110.180844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, et al. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science 2007; 318:1786-9; PMID:18006707; http://dx.doi.org/ 10.1126/science.1150179 [DOI] [PubMed] [Google Scholar]
- 9. Gilles-Gonzalez MA, Gonzalez G. Heme-based sensors: defining characteristics, recent developments, and regulatory hypotheses. J Inorg Biochem 2005; 99:1-22; PMID:15598487; http://dx.doi.org/ 10.1016/j.jinorgbio.2004.11.006 [DOI] [PubMed] [Google Scholar]
- 10. Shen J, Sheng X, Chang Z, Wu Q, Wang S, Xuan Z, Li D, Wu Y, Shang Y, Kong X, et al. Iron metabolism regulates p53 signaling through direct heme-p53 interaction and modulation of p53 localization, stability, and function. Cell Rep 2014; 7, 180-93; PMID:24685134; http://dx.doi.org/ 10.1016/j.celrep.2014.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]