

Abstract
Background
Myocardial fibrosis (MF) in noninfarcted myocardium may be an interstitial disease pathway that confers vulnerability to hospitalization for heart failure, death, or both across the spectrum of heart failure and ejection fraction. Hospitalization for heart failure is an epidemic that is difficult to predict and prevent and requires potential therapeutic targets associated with outcomes.
Method and Results
We quantified MF with cardiovascular magnetic resonance extracellular volume fraction (ECV) measures in 1172 consecutive patients without amyloidosis or hypertrophic or stress cardiomyopathy and assessed associations with outcomes using Cox regression. ECV ranged from 16.6% to 47.8%. Over a median of 1.7 years, 111 patients experienced events after cardiovascular magnetic resonance, 55 had hospitalization for heart failure events, and there were 74 deaths. ECV was more strongly associated with outcomes than “nonischemic” MF observed with late gadolinium enhancement, thus ECV quantified MF in multivariable models. Adjusting for age, sex, renal function, myocardial infarction size, ejection fraction, hospitalization status, and heart failure stage, higher ECV was associated with hospitalization for heart failure (hazard ratio 1.77; 95% CI 1.32 to 2.36 for every 5% increase in ECV), death (hazard ratio 1.87 95% CI 1.45 to 2.40) or both (hazard ratio 1.85, 95% CI 1.50 to 2.27). ECV improved classification of persons at risk and improved model discrimination for outcomes (eg, hospitalization for heart failure: continuous net reclassification improvement 0.33, 95% CI 0.05 to 0.66; P=0.02; 0.16, 95% CI 0.01 to 0.33; P=0.02; integrated discrimination improvement 0.037, 95% CI 0.008 to 0.073; P<0.01).
Conclusion
MF measured by ECV is associated with hospitalization for heart failure, death, or both. MF may represent a principal phenotype of cardiac vulnerability that improves risk stratification. Because MF can be reversible, cells and enzymes regulating collagen could be potential therapeutic targets.
Keywords: cardiovascular magnetic resonance, extracellular matrix, extracellular volume fraction, heart failure, myocardial fibrosis, T1 mapping
Subject Categories: Heart Failure, Magnetic Resonance Imaging (MRI), Fibrosis, Mortality/Survival
Introduction
Myocardial fibrosis (MF) in noninfarcted myocardium (regardless of whether any myocardial infarction [MI] is present) may be an identifiable and potentially treatable interstitial disease pathway that confers vulnerability to adverse outcomes including hospitalization for heart failure (HHF), death, or both. If this vulnerability occurs across the spectrum of left ventricular ejection fraction (EF) and heart failure stage (ie, heart failure evolution and progression) in a dose‐response fashion,1 it would emphasize the biological importance of MF and its candidacy as a potential therapeutic target. MF occurs in a wide variety of conditions including heart failure with reduced or preserved EF (with similar elevations in collagen)2 and diabetic3 and hypertensive3, 4, 5, 6 heart disease with7 or without8 heart failure and ischemic9 and nonischemic cardiomyopathy.10, 11 MF distorts myocardial architecture, culminating in mechanical,4, 5, 10 coronary vasomotor,6 and electrical12 dysfunction. Given the dynamic nature of MF, in which collagen fibril synthesis predominates over degradation, MF can reverse to restore collagen homeostasis.4, 5, 6, 10, 13 If associations between outcomes and MF truly exist across the spectrum of EF and heart failure stage, then cells (eg, fibroblasts or others) and enzymes promoting collagen synthesis (eg, procollagen proteinases and lysyl oxidases) and degradation (eg, matrix metalloproteinases) may be attractive therapeutic targets among other abnormalities involving the microcirculation, myocyte metabolism, and myocyte dysfunction.
HHF is an enormous problem in contemporary medicine but remains incompletely understood and difficult to predict.14 HHF is a sentinel event representing both decreased health status and disease progression with higher mortality than outpatient heart failure.14, 15 The HHF epidemic incurs enormous costs and is increasing in prevalence. Despite initial improvement with hospitalization, mortality and rehospitalization after discharge remain high16; however, these outcomes have not improved despite >20 phase III pharmacological trials targeting mostly non–extracellular matrix disease pathways16 and increased adherence to performance measures.17 Potentially, fundamental structural myocardial derangements underlying HHF, such as MF, may remain uncorrected, may adversely affect cardiac function, and may confer persistent vulnerability to HHF and mortality.15
To investigate the association of MF with HHF, death, or both across the entire spectrum of EF and heart failure stage, we enrolled a large consecutive cohort of patients referred for clinical cardiovascular magnetic resonance (CMR) at a single center. We hypothesized (1) that MF in noninfarcted myocardium quantified by the extracellular volume fraction (ECV) measured with CMR18 would be associated with HHF, death, or both in a “dose‐response” fashion, even after adjusting for important clinical conditions, and (2) that ECV measures would improve risk prediction and the classification of individual patients at risk using contemporary statistical metrics. Because MF has been shown to be reversible,4, 5, 6, 10, 13 these data could suggest that MF may represent a principal phenotype of cardiac vulnerability and highlight the cells and enzymes that regulate MF as potential therapeutic targets.
Methods
Patient Population
After institutional review board approval, we recruited 1765 consecutive adult patients at the time of clinical CMR at the UPMC Cardiovascular Magnetic Resonance Center from December 16, 2009, to May 13, 2013, and followed patients until July 23, 2013. The study complied with the Declaration of Helsinki. This cohort was formed to examine a priori whether novel CMR measures of MF are associated with outcomes. Inclusion criteria were written informed consent and completion of gadolinium contrast‐enhanced CMR. Exclusion criteria included (1) any evidence at baseline CMR or during follow‐up for cardiac amyloidosis (n=27), a disorder with marked interstitial expansion independent of collagen; (2) hypertrophic cardiomyopathy (n=133), a unique genetic disorder; (3) stress‐induced cardiomyopathy (n=10), which may have interstitial expansion from edema rather than MF; (4) adult congenital heart disease (n=195); and (5) inadequate image quality (n=4). We also excluded 228 patients who did not follow up within the integrated UPMC health system network because that may have compromised our ability to retrieve medical records specific to HHF and could potentially bias hazard ratios (HRs). To maximize generalizability, we included those with MI (1) because MI size can vary greatly; (2) because we limited ECV measure to remote noninfarcted myocardium; and (3) because MF in remote myocardium occurs in ischemic cardiomyopathy, which can contain more total collagen than the infarct itself9 if widely distributed over left ventricular mass (with less density or concentration). The final cohort included 1172 patients.
Data Elements
Data were managed using REDCap (Research Electronic Data Capture), hosted at the University of Pittsburgh,19 which incorporated quality checks such as missing data alerts, branching logic, and data range constraints to minimize data entry error. Hospitalization status and baseline comorbidity data at the time of CMR were determined from the medical record. Medical record data reflect the actual data supporting medical decisions, and that is relevant for generalizability; therefore, prior heart failure diagnosis and adjudication for first HHF after CMR required documentation during admission from physicians responsible for the patient's care. Heart failure stage was defined by practice guidelines1: stage 0, not at risk for heart failure (ie, no diabetes, hypertension, obesity, or vascular disease); stage A, at risk without structural heart disease (normal mass and volumes); stage B, structural heart disease without heart failure; stage C, structural heart disease with heart failure signs and symptoms; and stage D, refractory heart failure, requiring specialized support.
First HHF after CMR included any HHF event after CMR scanning (regardless of any prior HHF) and was identified by medical record review using a definition from prior epidemiological studies.20 HHF required physician documentation and (1) documented symptoms (eg, shortness of breath, fatigue, orthopnea, paroxysmal nocturnal dyspnea) and physical signs (eg, edema, pulmonary rates) consistent with heart failure; (2) supporting clinical findings (eg, pulmonary edema on radiography); or (3) therapy for heart failure, including diuretics, digitalis, angiotensin‐converting enzyme inhibitors, or beta blockers.20 Vital status was ascertained by Social Security Death Index queries and medical record review. Two investigators confirmed HHF as true HHF events (eg, not exacerbations of primary lung disease) and were blinded to ECV and CMR data; there were no disagreements.
CMR Scans
Cine CMR
Patients received clinical CMR scans from dedicated CMR technologists with a 1.5‐T scanner (Magnetom Espree; Siemens Medical Solutions) and a 32‐channel phased‐array cardiovascular coil. Examinations included standard cine imaging with steady state free precession, as described previously.21, 22 Left ventricular volumes and EF were measured by experienced readers without geometric assumptions about an elliptical shape of the short‐axis left ventricular area measured from short‐axis stacks of cine frames that covered the ventricles (6 mm thick, 4 mm space).
Late gadolinium enhancement
Late gadolinium enhancement (LGE) imaging was performed 10 minutes after a 0.2‐mmol/kg intravenous gadoteridol bolus (ProHance; Bracco Diagnostics) with a phase‐sensitive inversion recovery pulse sequence. The left ventricle was imaged in the 2‐, 3‐, and 4‐chamber planes and volumetric short‐axis stacks (6 mm thick, 4 mm space) to cover the entire ventricle, matching the cine imaging planes. The extent of MI and LGE was assessed visually in terms of the extent of LGE (none, <25%, 26% to 50%, 51% to 75%, >75%), rendering 5 categories for each of the 17 segments and thus creating 85 potential levels of global left ventricular involvement.23 The percentage of left ventricular myocardial mass exhibiting LGE was computed by summating the extent of LGE for each of the 17 segments.22
Quantification of MF with ECV
We used reproducible24 and validated25, 26, 27, 28, 29 ECV measures after a gadolinium bolus, as described previously.30, 31 We did not exclude foci of nonischemic scar on LGE images (ie, atypical of MI) from ECV measures acquired in noninfarcted myocardium.18, 30, 31 Such practice would bias ECV measures, and we did not want spatial variation of MF (the key feature rendering it detectable by LGE) to confound its quantification.18, 26, 32 LGE for quantifying MF in noninfarcted myocardium is not well validated for this purpose and lacks reproducibility.18 For ECV measures, we excluded myocardium with MI and carefully avoided myocardium in the vicinity of infarcted myocardium and traced the middle third of myocardium to avoid partial volume effects (Figure 1). We identified MI when LGE involved the subendocardium in a typical coronary distribution, a strategy that yields sensitivities and specificities >90% for MI detection.33
Figure 1.
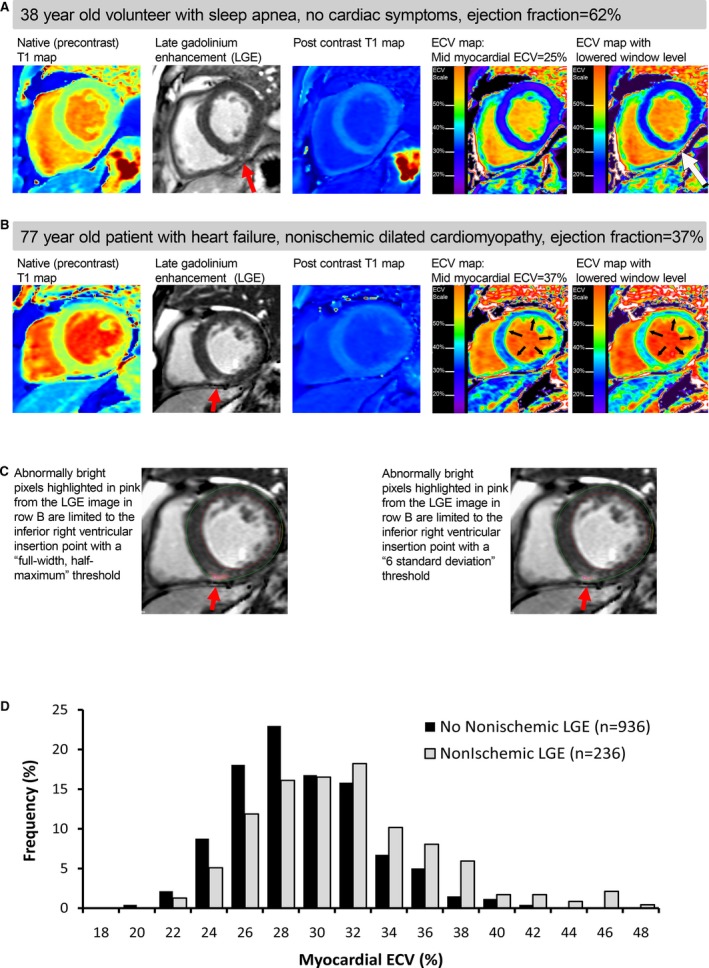
ECV maps generated from T1 maps can display normal myocardium (A) as well as severe diffuse MF (B) that is not detectable with LGE imaging (C), and there was overlap in the distributions of ECV in those with and without evident LGE (D). Semiautomated quantitative LGE thresholding techniques (C) using 2 common methods failed to identify the severe diffuse MF present in null myocardium (row B). The upward shift of ECV distributions for those with focal LGE was small compared with the spectrum of ECV (D). Midmyocardial ECV was measured to avoid contamination from partial volume effects from limited spatial resolution and/or misregistration errors, depicted by the green‐colored pixels along the blood pool and myocardium interface. ECV indicates extracellular volume fraction; LGE, late gadolinium enhancement; MF, myocardial fibrosis.
We quantified MF with ECV with the following definition:
The following definition was used for λ before and after gadolinium contrast, in which R1=1/T1 (spin‐lattice relaxation time)18:
Each ECV measurement for a short‐axis slice location was derived from a single precontrast and postcontrast T1 (spin‐lattice relaxation time) occurring after clinical LGE images (usually 15 to 20 minutes after contrast bolus). Hematocrit measures were acquired on the day of scanning and measured in the clinical laboratory. We averaged ECV measures from basal and midventricular short‐axis slices to yield final measurements. Apical slices were avoided due to concerns about error related to partial volume averaging.18 ECV measures acquired previously in 16 healthy volunteers (median age 23 years, interquartile range 21 to 33 years) that were not part of this cohort study yielded a mean ECV of 24.1% (SD 2.0%), which agreed well with the mean (SD) ECV values from prior reports.26, 34, 35, 36
Statistical Analysis
The χ2 or Fisher exact tests were used to compare categorical variables between patients with low and high ECV relative to the median. Some variables exhibited skewed non‐normal distributions, so Wilcoxon rank sum tests compared continuous variables between patients with low and high ECV. Survival analyses were performed separately to examine (1) time to first HHF after CMR (right censoring for mortality), (2) time to death, and (3) time to either HHF or death. Statistical tests were 2 sided, and P<0.05 was considered significant.
The log‐rank test with ECV (categorized arbitrarily in 5% increments that were considered clinically relevant intervals) and Cox regression (ECV expressed as a continuous percentage variable) examined associations between ECV and outcomes. To benchmark ECV against EF or other LGE‐based metrics, we compared univariable Cox regression χ2 values. Initial multivariable Cox regression models constrained the number of covariates to yield ≈10 events per predictor variable and stratified for hospitalization status and heart failure stage. These disease severity variables do not provide insight into etiology.14, 16 Because we were not interested in quantifying their association with outcomes, their use as stratification variables still permitted risk adjustment for hospitalization and baseline heart failure stage. We performed further risk adjustment for key clinical covariates including EF (which governs eligibility for treatments), age (frailty marker), MI size (determines extent of irreversible myocardial damage), and renal function (important for volume homeostasis), all of which are important risk markers.14 We also adjusted for sex because ECV appears to be higher in women.36 These models with the specific risk adjustment described above were labeled as model A. All Cox regression models included ECV as a continuous variable and expressed HRs for 5% ECV increments, which we thought were clinically meaningful intervals for ease of interpretation.
We tested for interactions between variables by adding terms into the model that were their product. Nonsignificant time interaction terms for ECV confirmed the proportional hazards assumption. ECV did not interact with MI size or the presence of nonischemic scar evident on LGE; however, we found a significant interaction between EF and ECV for all outcomes. To formally address this interaction, we created additional Cox models (termed model B for each outcome) stratifying by hospitalization status and clinically relevant EF categories (EF <30%, 30% to 50%, ≥50%) while adjusting for heart failure stage, age, MI size, and renal function. Because we also detected a significant interaction between age and EF (P<0.001) as continuous variables (but not for age and ECV), we further stratified the cohort according to whether age was above or below the median of 56 years. We used the integrated discrimination improvement and net reclassification improvement (NRI) indices to evaluate the added predictive ability of survival models with the introduction of the ECV variable.37 Integrated discrimination improvement measured the new model's improvement in average sensitivity without sacrificing average specificity (analogous to the change in receiver operating characteristic curves). The NRI measured the correctness of reclassification of individual participants based on their predicted probabilities of events using the new model.38 The NRI is the sum of (1) the net percentage of persons with events classified at higher risk with the addition of the new variable to the model and (2) the net percentage of persons without events classified at lower risk with the addition of the new variable to the model. Statistical analyses were performed using SAS 9.3 (SAS Institute).
Results
Patient Characteristics
Baseline characteristics of the cohort are summarized in Table 1. ECV ranged from 16.6% to 47.8%. Participants with ECV in noninfarcted myocardium above the median of 28.1% were more often female, were hospitalized, and had comorbidity. ECV was also significantly higher in those with focal nonischemic MF evident on LGE; however, there was substantial overlap in the ECV distributions among those with and without focal nonischemic MF evident on LGE (Figure 1).
Table 1.
Patient Characteristics (n=1172) According to Whether ECV in Noninfarcted Myocardium was High or Low (ie, Above or Below, Respectively, the Median ECV of 28.1%)
| Variable | Low ECV (n=586) | High ECV (n=586) | P Value |
|---|---|---|---|
| Demographics | |||
| Age, median (Q1 to Q3), y | 55 (42 to 64) | 57 (46 to 67) | <0.001 |
| Female, n (%) | 174 (27) | 310 (53) | <0.001 |
| White race, n (%) | 521 (89) | 512 (87) | 0.41 |
| Black race, n (%) | 52 (9) | 60 (10) | 0.43 |
| General indication for CMR exama | |||
| Known or suspected cardiomyopathy, n (%) | 246 (42) | 308 (53) | <0.001 |
| Possible coronary disease/viability/vasodilator stress testing, n (%) | 246 (42) | 285 (49) | 0.022 |
| Vasodilator stress testing, n (%) | 147 (25) | 166 (29) | 0.210 |
| Viability assessment, n (%) | 99 (17) | 119 (20) | 0.133 |
| Evaluation for arrhythmia substratea, n (%) | 183 (31) | 178 (30) | 0.752 |
| Post cardiac arrest evaluation | 6 (1%) | 15 (3%) | 0.048 |
| Rule out ARVD evaluation | 30 (5%) | 25 (4%) | 0.490 |
| Atrial fibrillation or flutter evaluation | 76 (13%) | 63 (11%) | 0.240 |
| Syncope | 32 (5%) | 39 (7%) | 0.391 |
| Ventricular ectopy | 28 (5%) | 35 (6%) | 0.365 |
| Atrial arrhythmia other than fibrillation/flutter | 13 (2%) | 10 (2%) | 0.528 |
| Palpitations | 46 (8%) | 37 (6%) | 0.305 |
| Sarcoidosis | 37 (6%) | 24 (4%) | 0.087 |
| Assessment for myocardial iron (siderosis) | 9 (2%) | 8 (1%) | 0.807 |
| Valve disease assessment | 40 (7%) | 35 (6%) | 0.551 |
| Pericardial disease assessment, n (%) | 23 (4) | 40 (7) | 0.028 |
| Possible mass or thrombus, n (%) | 19 (3) | 31 (5) | 0.083 |
| Thoracic aorta assessment, n (%) | 37 (6) | 16 (3) | 0.003 |
| Comorbidity | |||
| Diabetes, n (%) | 91 (16) | 148 (25) | <0.001 |
| Hypertension, n (%) | 294 (50) | 291 (50) | 0.861 |
| Dyslipidemia, n (%) | 234 (40) | 214 (37) | 0.229 |
| Current cigarette smoking, n (%) | 60 (10) | 112 (19) | <0.001 |
| Atrial fibrillation or flutter, n (%) | 65 (11) | 56 (10) | 0.388 |
| Hospitalized/inpatient status, n (%) | 144 (25) | 266 (45) | <0.001 |
| Prior coronary revascularization, n (%) | 109 (33) | 108 (6) | 0.940 |
| Body mass index, median (Q1 to Q3), kg/m2 | 29 (26 to 34) | 28 (23 to 33) | <0.001 |
| Baseline heart failure, n (%) | 60 (10) | 177 (30) | <0.001 |
| Heart failure stage, n (%) | |||
| 0 | 103 (18) | 95 (16) | |
| A | 186 (32) | 111 (19) | <0.001 (for trend) |
| B | 237 (40) | 203 (35) | |
| C or D | 60 (10) | 177 (30) | |
| Medications | |||
| ACEI, angiotensin receptor blocker, or mineralocorticoid antagonist | 239 (41%) | 267 (46%) | 0.099 |
| Beta blockers | 271 (46%) | 311 (53%) | 0.0194 |
| Aspirin or other antiplatelet | 291 (50%) | 295 (50%) | 0.815 |
| Statin | 231 (39%) | 225 (38%) | 0.719 |
| Loop diuretic | 71 (12%) | 175 (30%) | <0.001 |
| Laboratory and CMR characteristics | |||
| Creatinine, median (Q1 to Q3), mg/dL | 1.0 (0.8–1.1) | 0.9 (0.8–1.1) | 0.023 |
| Glomerular filtration rate, median (Q1 to Q3), mL/min per 1.73 m2 | 90 (73–92) | 88 (67–97) | 0.271 |
| Hematocrit, % | 41.0 (38.3–43.5) | 36.8 (33.2–39.8) | <0.001 |
| Ejection fraction, median (Q1 to Q3), % | 59 (51–64) | 55 (36–63) | <0.001 |
| Left ventricular mass index, median (Q1 to Q3), g/m2 | 57 (47–68) | 58 (45–74) | 0.200 |
| End diastolic volume index, median (Q1 to Q3), mL/m2 | 75 (66–95) | 85 (68–110) | <0.001 |
| End systolic volume index, median (Q1 to Q3), mL/m2 | 32 (24–44) | 37 (27–66) | <0.001 |
| Moderate or severe mitral regurgitation by cine CMR, n (%) | 12 (2) | 32 (5) | 0.002 |
| Myocardial infarction, n (%) | 113 (19) | 135 (23) | 0.116 |
| Nonischemic fibrosis evident on LGE images, n (%) | 84 (14) | 152 (26) | <0.001 |
ACEI indicates angiotensin‐converting enzyme inhibitor; ARVD, arrhythmogenic right ventricular dysplasia; CMR indicates cardiovascular magnetic resonance; LGE, late gadolinium enhancement; Q, quartile.
The categories for CMR indication were not exclusive, thus patients could have multiple indications for CMR, and there may be overlap in the classification of indications.
Baseline ECV Measures are Associated With Subsequent HHF and Death
Over a median of 1.7 years (quartiles 1 to 3, 1.0 to 2.4 years), there were 55 HHF events and 74 deaths after the baseline CMR scan among 111 participants who experienced adverse events in the cohort (18 participants experiencing HHF subsequently died). ECV in noninfarcted myocardium was significantly higher in those who experienced adverse events (Figure 2). We observed a graded dose‐response relationship, in which higher ECV was associated with significantly higher event rates (Figure 3). Similarly, those with low ECV composed a low‐risk group that demonstrated low event rates.
Figure 2.
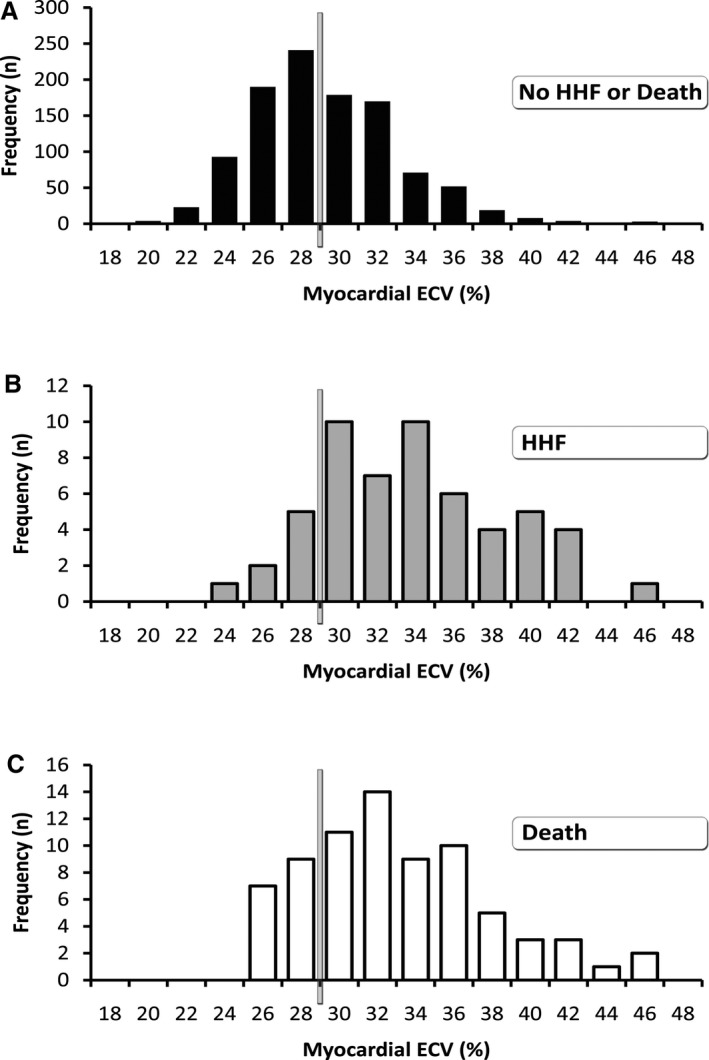
Compared with participants who did not experience adverse events (n=1061, median ECV 27.8) (A), the distribution of ECV in noninfarcted myocardium was significantly higher (P<0.001) in those experiencing HHF (n=55, median ECV 32.8) (B) or all‐cause death (n=74, median ECV 31.8) (C). The vertical bars estimate the upper limit of normal, in which an ECV of ≈29% approximates the upper 95th percentile based on 16 healthy volunteers (ie, mean ECV of 24.1%, SD 2.0%). ECV indicates extracellular volume fraction; HHF, hospitalization for heart failure.
Figure 3.
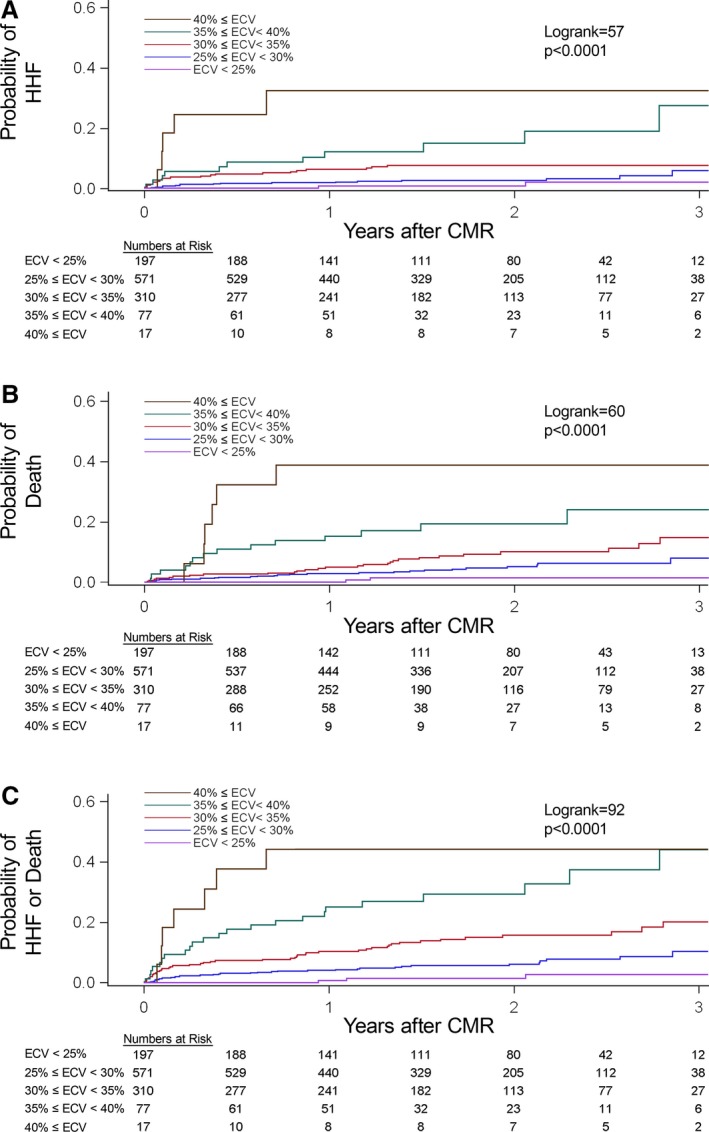
Among 1172 participants, increasing degrees of extracellular matrix expansion in noninfarcted myocardium quantified by the myocardial ECV was significantly associated with increased risks of adverse events following CMR scanning: first HHF after CMR (n=55) (A), all‐cause death (n=74) (B), or either HHF or death (n=111) (C). CMR indicates cardiovascular magnetic resonance; ECV, extracellular volume fraction; HHF, hospitalization for heart failure.
ECV Compared With Nonischemic LGE
ECV was more strongly associated with outcomes than nonischemic LGE (expressed as a dichotomous or continuous variable) using χ2 values to compare their strength of association in univariable models (Table 2). When ECV was added to a model with nonischemic LGE expressed as a binary variable as the sole covariate, ECV improved the classification of participants at risk for HHF (continuous NRI 0.80, 95% CI 0.50 to 1.07; categorical NRI 0.17, 95% CI 0.01 to 0.31 with 0.05 and 0.35 risk categories, selected arbitrarily) and improved the model's discrimination (integrated discrimination improvement 0.047, 95% CI 0.025 to 0.075). Similarly significant results were obtained when expressing nonischemic LGE as a continuous variable (continuous NRI 0.81, 95% CI 0.53 to 1.08; categorical NRI 0.30, 95% CI 0.15 to 0.44 with 0.05 and 0.35 risk categories selected arbitrarily) and improved the model's discrimination (integrated discrimination improvement 0.043, 95% CI 0.024 to 0.068). Consequently, we chose to use ECV rather than nonischemic LGE to quantify MF in noninfarcted myocardium in final multivariable models.
Table 2.
Among CMR Parameters, ECV and EF Expressed as Continuous Variables Were Comparable in Their Univariable Association With HHF, as Shown by χ2 Values, But ECV was More Strongly Associated With Mortality
| Outcome | Univariable Cox Regression Model Covariate | HR (95% CI) | χ2 | P Value |
|---|---|---|---|---|
| HHF (n=55) | ECV (5% increase) | 2.41 (1.93–3.02) | 59.6 | <0.001 |
| High ECV (above median) | 5.25 (2.57–10.72) | 20.7 | <0.001 | |
| EF (5% decrease) | 1.33 (1.23–1.43) | 54.3 | <0.001 | |
| Presence of nonischemic LGE (dichotomous variable) | 3.69 (2.17–6.27) | 23.3 | <0.001 | |
| Percentage of left ventricular mass exhibiting nonischemic LGE (5% increase) | 1.44 (1.22–1.70) | 18.3 | <0.001 | |
| Myocardial infarction size tertile on LGE images | 1.57 (1.25–1.97) | 15.3 | <0.001 | |
| Death (n=74) | ECV (5% increase) | 2.13 (1.74–2.61) | 53.4 | <0.001 |
| High ECV (above median) | 3.60 (2.07–6.26) | 20.5 | <0.001 | |
| EF (5% decrease) | 1.21 (1.16–1.29) | 32.0 | <0.001 | |
| Presence of nonischemic LGE (dichotomous variable) | 2.57 (1.61–4.09) | 15.6 | <0.001 | |
| Percentage of left ventricular mass exhibiting nonischemic LGE (5% increase) | 1.28 (1.07–1.52) | 7.7 | 0.006 | |
| Myocardial infarction size tertile on LGE images | 1.55 (1.28–1.89) | 19.4 | <0.001 | |
| HHF or death (n=111) | ECV (5% increase) | 2.25 (1.91–2.64) | 96.0 | <0.001 |
| High ECV (above median) | 4.41 (2.74–7.08) | 37.4 | <0.001 | |
| EF (5% decrease) | 1.26 (1.19–1.33) | 71.7 | <0.001 | |
| Presence of nonischemic LGE (dichotomous variable) | 3.15 (2.16–4.58) | 35.7 | <0.001 | |
| Percentage of left ventricular mass exhibiting nonischemic LGE (5% increase) | 1.39 (1.23–1.58) | 26.2 | <0.001 | |
| Myocardial infarction size tertile on LGE images | 1.55 (1.32–1.82) | 28.5 | <0.001 |
ECV ranged from 16.6% to 47.8%, EF ranged from 7% to 79%, and percentage of left ventricular mass exhibiting nonischemic LGE range from 0% to 42%. Note that the χ2 value is the parameter that describes the strength of association between variables and outcomes in the Cox models from which P values are derived. HRs reflect the increased hazards per increment. For continuous variables, HRs are “scaled” to the selected increment. Therefore, the specific HR for a continuous variable can increase as the increment increases, but the χ2 value remains constant. CMR indicates cardiovascular magnetic resonance; ECV, extracellular volume fraction; EF, ejection fraction; HHF, hospitalization for heart failure; HR, hazard ratio; LGE, late gadolinium enhancement.
ECV Compared With Ejection Fraction
Using χ2 values to compare strength of association in univariable models, ECV appeared at least as strongly associated with outcomes as EF, a clinically important benchmark for risk stratification because many decisions in contemporary cardiology revolve around EF (Table 2). EF and ECV displayed interactions in all multivariable models (P<0.001). Figure 4 shows that lower grade ECV elevations were more prognostically relevant in the setting of reduced EF (using the EF cut point of 45% also used in the TOPCAT trial39). When ECV was added to a model with EF as the sole covariate, ECV improved the classification of participants at risk for HHF (continuous NRI 0.44, 95% CI 0.11 to 0.76; categorical NRI 0.29, 95% CI 0.15 to 0.43 with 0.05 and 0.35 risk categories) and improved the model's discrimination (integrated discrimination improvement 0.038, 95% CI 0.019 to 0.068).
Figure 4.
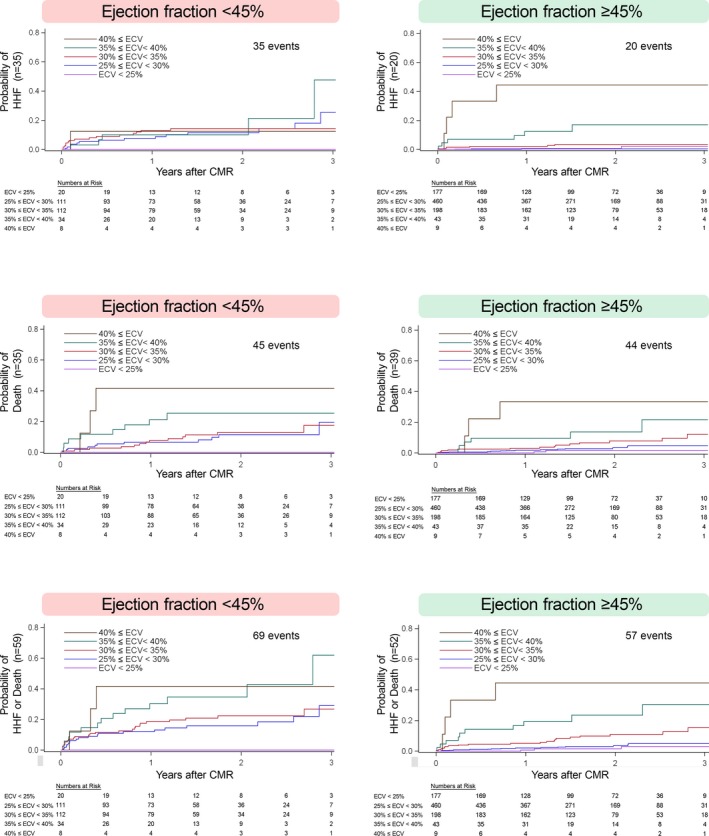
ECV was significantly associated with adverse outcomes in univariable Cox regression models (P<0.05 for all), whether EF was reduced (<45%) or preserved (EF ≥45%). Despite the decreased statistical power occurring with subgroup analysis, the basis for the statistically significant interactions between ECV and EF was evident qualitatively. Associations between ECV measures at the lower end of the ECV spectrum (eg, blue and red lines) and events appeared strengthened when EF was reduced. ECV indicates extracellular volume fraction; EF, ejection fraction.
ECV and EF Associations With Outcomes Across Age Strata
Because variables such as EF may vary in their association with outcomes as a function of age, we stratified the sample according to whether age was above or below the median of 56 years. In older patients (n=566), we found associations between the composite outcome of death and heart failure (n=75) for both ECV (HR 2.00, 95% CI 1.58 to 2.53, for every 5% increase, χ2=33) and EF (HR 1.12, 95% CI 1.05 to 1.20, for every 5% decrease, χ2=12) without further risk adjustment in multivariable Cox regression models. We found similar associations in younger patients (n=606) between the composite outcome of death and heart failure (n=36) for both ECV (HR 1.82, 95% CI 1.37 to 2.42, for every 5% increase, χ2=17) and EF (HR 1.25, 95% CI 1.13 to 1.37, for every 5% increase, χ2=20).
Baseline ECV Is Associated With Subsequent HHF and Death in Multivariable Models
ECV remained associated with HHF, death, or both after risk adjustment in final multivariable Cox regression models, as shown in Table 3. We also obtained significant HRs for ECV when adding diabetes, active smoking, and prior smoking covariates to these fully adjusted models (for HHF in model A: HR 1.70, 95% CI 1.28 to 2.27, for every 5% increase in ECV; for death in model A: HR 1.80, 95% CI 1.39 to 2.32, for every 5% increase in ECV; for HHF or death in model A: HR 1.77, 95% CI 1.43 to 2.18, for every 5% increase in ECV). Supporting its merit as an MF metric, ECV remained associated with outcomes even when adding nonischemic LGE (expressed as a binary or continuous variable) to the multivariable models (Tables S1 and S2). Similarly, without ECV in the final multivariable models, LGE was not consistently associated with outcomes, and LGE did not consistently provide added prognostic value (Tables S3 and S4). In contrast, ECV reclassified participants at risk and improved model discrimination for all final models. Similar results were obtained regardless of whether the interaction between ECV and EF was addressed (ie, Cox regression model A or B).
Table 3.
In Multivariable Models ECV in Noninfarcted Myocardium Remained Associated With HHF, Death, or the Combined Endpoint of HHF/Death and Improved the Classification of Individuals at Risk (NRI) and the of the Model Discrimination (IDI)
| Multivariable Cox Regression Model | Hazard Ratio for Every 5% Increase in ECV (95% CI; P Value) | Category Free NRI (95% CI; P Value) | Categorical NRI 0.05, 0.35 Risk Categories (95% CI; P Value) | IDI (95% CI; P Value) |
|---|---|---|---|---|
| HHF (model A) | 1.77 (1.32–2.36; χ2=14.9; P<0.01) | 0.33 (0.05–0.66; P=0.02) | 0.16 (0.01–0.33; P=0.02) | 0.037 (0.008–0.073; P<0.01) |
| HHF (model B) | 1.85 (1.39–2.48; χ2=17.2; P<0.01) | 0.40 (0.08–0.70; P=0.02) | 0.26 (0.10–0.45; P<0.01) | 0.059 (0.026–0.097; P<0.01) |
| Death (model A) | 1.87 (1.45–2.40; χ2=23.9; P<0.01) | 0.53 (0.22–0.87; P<0.01) | 0.21 (0.12–0.37; P=0.02 | 0.026 (0.009–0.058; P<0.01) |
| Death (model B) | 1.85 (1.44–2.38; χ2=23.2; P<0.01) | 0.47 (0.25–0.78; P<0.01 | 0.15 (0.04–0.30; P<0.01) | 0.027 (0.003–0.066; P<0.01) |
| HHF or death (model A) | 1.85 (1.50–2.27; χ2=33.9; P<0.001) | 0.44 (0.21–0.68; P<0.01) | 0.20 (0.06–0.34; P=0.02) | 0.033 (0.010–0.063; P<0.01) |
| HHF or death (model B) | 1.88 (1.53–2.31; χ2=35.9; P<0.001) | 0.45 (0.23–0.69; P<0.01) | 0.21 (0.10–0.32; P<0.01) | 0.039 (0.009–0.072; P<0.01 |
Model A ignored EF–ECV interactions, stratified by heart failure stage and hospitalization status and adjusted for EF, age, glomerular filtration rate, myocardial infarction size, and sex. Model B stratified by EF categories (to address ECV–EF interactions) and hospitalization status and adjusted for heart failure stage, age, glomerular filtration rate, myocardial infarction size, and sex. ECV, extracellular volume fraction; HHF, hospitalization for heart failure; IDI, integrated discrimination improvement; NRI, net reclassification improvement.
Discussion
These data from a large consecutive cohort of patients referred for CMR demonstrate that MF in noninfarcted myocardium quantified by ECV is associated with subsequent HHF, all‐cause mortality, or both across the spectrum of EF and heart failure stage in a dose‐response fashion; MF quantified by LGE was not. We also used novel statistical metrics to demonstrate, for the first time, that ECV can add incremental prognostic value, improving the classification of patients at risk and improving model prediction beyond several relevant covariates including EF, renal function, heart failure stage, and age. Underscoring the prognostic relevance of MF, associations between ECV and outcomes appear at least as strong as EF, a clinically important prognostic benchmark that guides decision making in cardiology. We observed MF–outcomes associations in those with reduced or preserved EF.
This work expands our preliminary research showing associations between short‐term mortality and ECV in 793 participants.30 We now report data from a larger cohort (n=1172), and we used a discrete HHF end point, longer median follow‐up (1.7 years versus 0.8 years), greater numbers of events (55 HHF events, 74 deaths, 111 events total versus 39 deaths in earlier work30), and greater risk adjustment for potential confounders. We also demonstrated stronger associations with outcomes for ECV compared with nonischemic LGE, and we report a statistical interaction between ECV and EF in which MF may be less well tolerated when EF is reduced.
There is an urgent need to improve our understanding of the development of heart failure and the vulnerability to HHF and mortality. Given the ongoing epidemic of HHF15 and the series of interventions that have not improved HHF outcomes,16 identifying the robust associations between outcomes and MF across the spectrum of EF and heart failure stage may provide a foundation to reverse these trends. Extending prior work from others showing the adverse functional consequences of MF,4, 5, 6, 10, 12 we now show that MF may be a principal phenotype of cardiac vulnerability to subsequent adverse events across the spectrum of EF and heart failure stage. These data mirror prior proof‐of‐concept work in rodents, in which isolated fibroblast activation to create MF culminated in eventual heart failure.40 Theoretically, excess collagen might perturb cardiomyocyte energetics if interposing excess collagen isolates cardiomyocytes from capillaries and increases local afterload from its stiffening effects.15 Interstitial expansion from MF might also affect the bioavailability of growth factors, hormones, cytokines, and drugs and impair communication between cells. Further work is needed to understand MF disease mechanisms, especially relationships with contractility, microcirculation, and metabolism, which may influence one another.
The dynamic nature of MF, in which collagen synthesis predominates over degradation, provides opportunities for pharmacological treatment targeting the cells (eg, fibroblasts or others) and enzymes that regulate collagen metabolism. Antifibrotic medications modulating the renin–angiotensin–aldosterone system can reverse diffuse MF and restore collagen homeostasis.4, 5, 6, 10, 13 These agents reduce HHF and mortality in large trials in the setting of reduced EF. The heightened vulnerability of MF occurring in the setting of reduced EF may partly explain the higher success rates of trials using these medications compared with preserved EF. We hypothesize that these same medications still may be useful in heart failure with preserved EF if also accompanied by MF; however, the greater etiologic heterogeneity in heart failure with preserved EF, the interaction between EF and ECV (in which MF appears better tolerated with preserved EF), and limited antifibrotic efficacy may have obscured detection of potential benefits in trials of heart failure with preserved EF. Noninvasive ECV measures to quantify MF are validated25, 26, 27, 28, 29 and reproducible15, 24 and are well suited to identify heart failure with preserved EF subpopulations with MF. Consequently, ECV may facilitate pharmaceutical development of novel antifibrotic therapies by demonstrating myocardial extracellular matrix regression without prohibitively expensive large‐scale outcome trials. Serum MF biomarkers are less expensive than CMR and easier to acquire but lack the myocardial specificity of ECV.
Limitations
Our study has limitations. First, with observational data, associations do not establish causality and may reflect unmeasured confounders. Limited numbers of events constrained risk adjustment and statistical power, and we could not adjust for every difference in baseline characteristics or subsequent treatment differences. Nevertheless, we adjusted for several clinically relevant variables encountered in practice that also relate to unmeasured confounders, and ECV appeared at least as strongly associated with outcomes as EF measured by CMR.21 ECV values were also not available to clinicians to bias their treatment. Second, although we studied a large cohort to maximize generalizability, our data reflect only single‐center experience. Third, we lacked histological validation of ECV in our cohort, but ECV as a metric of fibrosis has been validated repeatedly. Fourth, the cause of death was not always clear (eg, heart failure or malignant arrhythmia). Still, adjudication for cause of death can be challenging, controversial, and biased,41 whereas all‐cause mortality remains objective and inherently relevant. We believe cardiac causes are likely in our cohort based on the limited documentation available describing circumstances of death. Excess noncardiac mortality would only bias our results toward the null. Fifth, the probability‐of‐event curves in Figures 3 and 4 cross somewhat, although minimally, we believe. This finding may indicate that the proportional hazards assumption is violated and that the hazard may vary with time. In this case, the HRs simply summarize the overall “averaged” hazard for the follow‐up period,42 and we believe that remains relevant to the clinician and the patient. Sixth, LGE was assessed visually as opposed to being quantified by more labor‐intensive thresholding techniques, which may be more precise; however, we note that LGE expressed as a binary variable yielded higher χ2 values in multivariable Cox models than LGE expressed as a percentage of left ventricular mass (Tables S3 and S4).
Finally, HHF and death may not be independent events but rather could be considered competing risks. The effect of ECV on the cause‐specific hazard model used in our study (ie, the instantaneous rate of occurrence of a given event among the patients who are still event free) can be different from its effect on the cumulative incidence model (ie, probability of occurrence of a given event [or the proportion of patients with a certain event] by time) of the corresponding cause. Indeed, the Kaplan–Meier estimator used in a cause‐specific hazard model disregards censoring from a competing event and cannot estimate the cumulative incidence in the presence of competing events.43 In the absence of final consensus on how to analyze competing risks end points,43 we note that we observed similar associations between ECV and all 3 of the outcomes (HHF, death, or both).
Conclusions
MF in noninfarcted myocardium measured by ECV is associated with HHF, death, or both across the spectrum of EF and heart failure stage in a dose‐response fashion. MF may represent a principal phenotype of cardiac vulnerability that could improve risk stratification by providing added prognostic value. Given these associations and the dynamic nature of MF, its reversibility, the epidemic of HHF, and the slow progress in identifying strategies to improve outcomes, MF and the cells and enzymes that regulate collagen homeostasis may be attractive therapeutic targets for novel therapeutics.
Sources of Funding
This work was supported by a grant from The Pittsburgh Foundation (PA), Grant M2009‐0068; and an American Heart Association Scientist Development grant (09SDG2180083) including a T. Franklin Williams Scholarship Award; funding provided by: Atlantic Philanthropies, Inc, the John A. Hartford Foundation, the Association of Specialty Professors, and the American Heart Association (Dallas, TX). This work was also supported by Grant Number UL1 RR024153 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
Disclosures
Dr Schelbert has accepted contrast material from Bracco Diagnostics for research purposes beyond the scope of this work. Dr Wong was supported by a grant K12 HS19461‐01 from the Agency for Healthcare Research and Quality. Dr Shroff's research efforts were partially supported by the McGinnis Endowed Chair funds (Pittsburgh, PA). Dr Gheorghiade is a consultant for Abbott Laboratories, Astellas, Astra‐Zeneca, Bayer Schering Pharma AG, Cardiorentis Ltd, CorThera, Inc, Cytokinetics, Inc, DebioPharm SA, Errekappa Terapeutici (Milan, Italy), GlaxoSmithKline, Ikaria, Intersection Medical, Inc, Johnson & Johnson, Medtronic, Merck, Novartis Pharma AG, Ono Pharmaceuticals USA, Otsuka Pharmaceuticals, Palatin Technologies, Pericor Therapeutics, Protein Design Laboratories, Sanofi‐Aventis, Sigma Tau, Solvay Pharmaceuticals Sticares Inter‐ACT, Takeda Pharmaceuticals North America, Inc, and Trevena Therapeutics. The remaining authors have no relationships relevant to the contents of this paper to disclose.
Supporting information
Table S1. In Multivariable Models, Extracellular Volume Fraction in Noninfarcted Myocardium Remained Associated With Hospitalization for Heart Failure (HHF), Death, or the Combined End Point of HHF and Death, Even When Adding the Presence of Nonischemic Late Gadolinium Enhancement to the Models in Table 3
Table S2. In Multivariable Models Extracellular Volume Fraction in Noninfarcted Myocardium Remained Associated With Hospitalization for Heart Failure (HHF), Death, or the Combined End Point of HHF and Death, Even When Adding the Extent of Nonischemic Late Gadolinium Enhancement (Expressed as a Continuous Variable) to the Models in Table 3
Table S3. For Comparison to Extracellular Volume Fraction (ECV), the Presence of Nonischemic Late Gadolinium Enhancement Without ECV in the Multivariable Model was Also Associated With Hospitalization for Heart Failure (HHF), Death, or the Combined End Point of HHF and Death But Less Consistently and to a Lesser Extent Than the Models With ECV Based on the χ2 Values, Net Reclassification Improvement, and Integrated Discrimination Improvement Data in Table 3, Which Used ECV Exclusively to Quantify Myocardial Fibrosis
Table S4. For Comparison to Extracellular Volume Fraction (ECV), the Percentage of Left Ventricular Mass Exhibiting Nonischemic Late Gadolinium Enhancement (With the Hazard Ratio Reflecting a 5% Increase) Without ECV in the Multivariable Model was Also Associated With Outcomes But Less Consistently and to a Lesser Extent Than the Models With ECV Based on the χ2 Values, Net Reclassification Improvement, and Integrated Discrimination Improvement Data in Table 3, Which Used ECV Exclusively to Quantify Myocardial Fibrosis
Acknowledgments
We thank the patients who volunteered to participate in this work. This work would not have been possible without the support of Drs Joan Lacomis, Christopher Deible, Glenn Andrews, Joao Cavalcante, and Elliott Gozansky from the Department of Radiology, as well as the contributions of Kathy Puntil, BS, RN, Marie‐Therese Najjar, BS, RT(R)(MR), Steven Mancuso, BS, RT(R)(MR), Deborah Yasko, RT(R)(MR), and Elizabeth Ruhl, BS, RN.
(J Am Heart Assoc. 2015;4:e002613 doi: 10.1161/JAHA.115.002613)
Accompanying Tables S1 through S4 are available at http://jaha.ahajournals.org/content/4/12/e002613/suppl/DC1
References
- 1. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–e239. [DOI] [PubMed] [Google Scholar]
- 2. van Heerebeek L, Borbely A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. [DOI] [PubMed] [Google Scholar]
- 3. van Hoeven KH, Factor SM. A comparison of the pathological spectrum of hypertensive, diabetic, and hypertensive‐diabetic heart disease. Circulation. 1990;82:848–855. [DOI] [PubMed] [Google Scholar]
- 4. Diez J, Querejeta R, Lopez B, Gonzalez A, Larman M, Martinez Ubago JL. Losartan‐dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation. 2002;105:2512–2517. [DOI] [PubMed] [Google Scholar]
- 5. Brilla CG, Funck RC, Rupp H. Lisinopril‐mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation. 2000;102:1388–1393. [DOI] [PubMed] [Google Scholar]
- 6. Schwartzkopff B, Brehm M, Mundhenke M, Strauer BE. Repair of coronary arterioles after treatment with perindopril in hypertensive heart disease. Hypertension. 2000;36:220–225. [DOI] [PubMed] [Google Scholar]
- 7. Querejeta R, Lopez B, Gonzalez A, Sanchez E, Larman M, Martinez Ubago JL, Diez J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation. 2004;110:1263–1268. [DOI] [PubMed] [Google Scholar]
- 8. Querejeta R, Varo N, Lopez B, Larman M, Artinano E, Etayo JC, Martinez Ubago JL, Gutierrez‐Stampa M, Emparanza JI, Gil MJ, Monreal I, Mindan JP, Diez J. Serum carboxy‐terminal propeptide of procollagen type I is a marker of myocardial fibrosis in hypertensive heart disease. Circulation. 2000;101:1729–1735. [DOI] [PubMed] [Google Scholar]
- 9. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end‐stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. [DOI] [PubMed] [Google Scholar]
- 10. Izawa H, Murohara T, Nagata K, Isobe S, Asano H, Amano T, Ichihara S, Kato T, Ohshima S, Murase Y, Iino S, Obata K, Noda A, Okumura K, Yokota M. Mineralocorticoid receptor antagonism ameliorates left ventricular diastolic dysfunction and myocardial fibrosis in mildly symptomatic patients with idiopathic dilated cardiomyopathy: a pilot study. Circulation. 2005;112:2940–2945. [DOI] [PubMed] [Google Scholar]
- 11. Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Sonnenblick EH, Olivetti G, Anversa P. The cellular basis of dilated cardiomyopathy in humans. J Mol Cell Cardiol. 1995;27:291–305. [DOI] [PubMed] [Google Scholar]
- 12. Tamarappoo BK, John BT, Reinier K, Teodorescu C, Uy‐Evanado A, Gunson K, Jui J, Chugh SS. Vulnerable myocardial interstitium in patients with isolated left ventricular hypertrophy and sudden cardiac death: a postmortem histological evaluation. J Am Heart Assoc. 2012;1:e001511 doi: 10.1161/JAHA.112.001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lopez B, Querejeta R, Gonzalez A, Sanchez E, Larman M, Diez J. Effects of loop diuretics on myocardial fibrosis and collagen type I turnover in chronic heart failure. J Am Coll Cardiol. 2004;43:2028–2035. [DOI] [PubMed] [Google Scholar]
- 14. Giamouzis G, Kalogeropoulos A, Georgiopoulou V, Laskar S, Smith AL, Dunbar S, Triposkiadis F, Butler J. Hospitalization epidemic in patients with heart failure: risk factors, risk prediction, knowledge gaps, and future directions. J Card Fail. 2011;17:54–75. [DOI] [PubMed] [Google Scholar]
- 15. Schelbert EB, Fonarow GC, Bonow RO, Butler J, Gheorghiade M. Therapeutic targets in heart failure: refocusing on the myocardial interstitium. J Am Coll Cardiol. 2014;63:2188–2198. [DOI] [PubMed] [Google Scholar]
- 16. Butler J, Fonarow GC, Gheorghiade M. Strategies and opportunities for drug development in heart failure. JAMA. 2013;309:1593–1594. [DOI] [PubMed] [Google Scholar]
- 17. Fonarow GC, Peterson ED. Heart failure performance measures and outcomes: real or illusory gains. JAMA. 2009;302:792–794. [DOI] [PubMed] [Google Scholar]
- 18. Moon JC, Messroghli DR, Kellman P, Piechnik SK, Robson MD, Ugander M, Gatehouse PD, Arai AE, Friedrich MG, Neubauer S, Schulz‐Menger J, Schelbert EB. Myocardial T1 mapping and extracellular volume quantification: a Society for Cardiovascular Magnetic Resonance (SCMR) and CMR Working Group of the European Society of Cardiology consensus statement. J Cardiovasc Magn Reson. 2013;15:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata‐driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalogeropoulos A, Psaty BM, Vasan RS, Georgiopoulou V, Smith AL, Smith NL, Kritchevsky SB, Wilson PW, Newman AB, Harris TB, Butler J. Validation of the health ABC heart failure model for incident heart failure risk prediction: the Cardiovascular Health Study. Circ Heart Fail. 2010;3:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong TC, Piehler K, Puntil KS, Moguillansky D, Meier CG, Lacomis JL, Kellman P, Cook SC, Schwartzman DS, Simon MA, Mulukutla SR, Schelbert EB. Effectiveness of late gadolinium enhancement to improve outcomes prediction in patients referred for cardiovascular magnetic resonance after echocardiography. J Cardiovasc Magn Reson. 2013;15:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Piehler KM, Wong TC, Puntil KS, Zareba KM, Lin K, Harris DM, Deible CR, Lacomis JM, Czeyda‐Pommersheim F, Cook SC, Kellman P, Schelbert EB. Free‐breathing, motion‐corrected late gadolinium enhancement is robust and extends risk stratification to vulnerable patients. Circ Cardiovasc Imaging. 2013;6:423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wagner A, Mahrholdt H, Holly TA, Elliott MD, Regenfus M, Parker M, Klocke FJ, Bonow RO, Kim RJ, Judd RM. Contrast‐enhanced MRI and routine single photon emission computed tomography (SPECT) perfusion imaging for detection of subendocardial myocardial infarcts: an imaging study. Lancet. 2003;361:374–379. [DOI] [PubMed] [Google Scholar]
- 24. Schelbert EB, Testa SM, Meier CG, Ceyrolles WJ, Levenson JE, Blair AJ, Kellman P, Jones BL, Ludwig DR, Schwartzman D, Shroff SG, Wong TC. Myocardial extravascular extracellular volume fraction measurement by gadolinium cardiovascular magnetic resonance in humans: slow infusion versus bolus. J Cardiovasc Magn Reson. 2011;13:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flett AS, Hayward MP, Ashworth MT, Hansen MS, Taylor AM, Elliott PM, McGregor C, Moon JC. Equilibrium contrast cardiovascular magnetic resonance for the measurement of diffuse myocardial fibrosis: preliminary validation in humans. Circulation. 2010;122:138–144. [DOI] [PubMed] [Google Scholar]
- 26. Miller CA, Naish J, Bishop P, Coutts G, Clark D, Zhao S, Ray SG, Yonan N, Williams SG, Flett AS, Moon JC, Greiser A, Parker GJ, Schmitt M. Comprehensive validation of cardiovascular magnetic resonance techniques for the assessment of myocardial extracellular volume. Circ Cardiovasc Imaging. 2013;6:373–383. [DOI] [PubMed] [Google Scholar]
- 27. Fontana M, White SK, Banypersad SM, Sado DM, Maestrini V, Flett AS, Piechnik SK, Neubauer S, Roberts N, Moon J. Comparison of T1 mapping techniques for ECV quantification. Histological validation and reproducibility of ShMOLLI versus multibreath‐hold T1 quantification equilibrium contrast CMR. J Cardiovasc Magn Reson. 2012;14:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. White SK, Sado DM, Fontana M, Banypersad SM, Maestrini V, Flett AS, Piechnik SK, Robson MD, Hausenloy DJ, Sheikh AM, Hawkins PN, Moon JC. T1 mapping for myocardial extracellular volume measurement by CMR: bolus only versus primed infusion technique. JACC Cardiovasc Imaging. 2013;6:955–962. [DOI] [PubMed] [Google Scholar]
- 29. Schelbert EB, Hsu LY, Anderson SA, Mohanty BD, Karim SM, Kellman P, Aletras AH, Arai AE. Late gadolinium‐enhancement cardiac magnetic resonance identifies postinfarction myocardial fibrosis and the border zone at the near cellular level in ex vivo rat heart. Circ Cardiovasc Imaging. 2010;3:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, Aneizi AA, Shakesprere J, Kellman P, Shroff SG, Schwartzman DS, Mulukutla SR, Simon MA, Schelbert EB. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short‐term mortality. Circulation. 2012;126:1206–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wong TC, Piehler K, Kang IA, Kadakkal A, Kellman P, Schwartzman DS, Mulukutla SR, Simon MA, Shroff SG, Kuller LH, Schelbert EB. Myocardial extracellular volume fraction quantified by cardiovascular magnetic resonance is increased in diabetes and associated with mortality and incident heart failure admission. Eur Heart J. 2014;35:657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kellman P, Wilson JR, Xue H, Bandettini WP, Shanbhag SM, Druey KM, Ugander M, Arai AE. Extracellular volume fraction mapping in the myocardium, part 2: initial clinical experience. J Cardiovasc Magn Reson. 2012;14:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim HW, Farzaneh‐Far A, Kim RJ. Cardiovascular magnetic resonance in patients with myocardial infarction: current and emerging applications. J Am Coll Cardiol. 2010;55:1–16. [DOI] [PubMed] [Google Scholar]
- 34. Singh A, Horsfield MA, Bekele S, Khan J, Greiser A, McCann GP. Myocardial T1 and extracellular volume fraction measurement in asymptomatic patients with aortic stenosis: reproducibility and comparison with age‐matched controls. Eur Heart J Cardiovasc Imaging. 2015;16:763–770. [DOI] [PubMed] [Google Scholar]
- 35. Aus dem Siepen F, Buss SJ, Messroghli D, Andre F, Lossnitzer D, Seitz S, Keller M, Schnabel PA, Giannitsis E, Korosoglou G, Katus HA, Steen H. T1 mapping in dilated cardiomyopathy with cardiac magnetic resonance: quantification of diffuse myocardial fibrosis and comparison with endomyocardial biopsy. Eur Heart J Cardiovasc Imaging. 2014;16:210–216. [DOI] [PubMed] [Google Scholar]
- 36. Sado DM, Flett AS, Banypersad SM, White SK, Maestrini V, Quarta G, Lachmann RH, Murphy E, Mehta A, Hughes DA, McKenna WJ, Taylor AM, Hausenloy DJ, Hawkins PN, Elliott PM, Moon JC. Cardiovascular magnetic resonance measurement of myocardial extracellular volume in health and disease. Heart. 2012;98:1436–1441. [DOI] [PubMed] [Google Scholar]
- 37. Pencina MJ, D'Agostino RB Sr, Steyerberg EW. Extensions of net reclassification improvement calculations to measure usefulness of new biomarkers. Stat Med. 2011;30:11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kennedy KF, Pencina MJ. A SAS® macro to compute added predictive ability of new markers predicting a dichotomous outcome. 2010. Available at http://analytics.ncsu.edu/sesug/2010/SDA07.Kennedy.pdf. Accessed June 1, 2011.
- 39. Shah SJ, Heitner JF, Sweitzer NK, Anand IS, Kim HY, Harty B, Boineau R, Clausell N, Desai AS, Diaz R, Fleg JL, Gordeev I, Lewis EF, Markov V, O'Meara E, Kobulia B, Shaburishvili T, Solomon SD, Pitt B, Pfeffer MA, Li R. Baseline characteristics of patients in the treatment of preserved cardiac function heart failure with an aldosterone antagonist trial. Circ Heart Fail. 2013;6:184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA‐21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. [DOI] [PubMed] [Google Scholar]
- 41. Lauer MS, Blackstone EH, Young JB, Topol EJ. Cause of death in clinical research: time for a reassessment? J Am Coll Cardiol. 1999;34:618–620. [DOI] [PubMed] [Google Scholar]
- 42. Allison PD. Survival Analysis Using SAS: A Practical Guide. Cary, NC: SAS Institute; 1995. [Google Scholar]
- 43. Latouche A, Allignol A, Beyersmann J, Labopin M, Fine JP. A competing risks analysis should report results on all cause‐specific hazards and cumulative incidence functions. J Clin Epidemiol. 2013;66:648–653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. In Multivariable Models, Extracellular Volume Fraction in Noninfarcted Myocardium Remained Associated With Hospitalization for Heart Failure (HHF), Death, or the Combined End Point of HHF and Death, Even When Adding the Presence of Nonischemic Late Gadolinium Enhancement to the Models in Table 3
Table S2. In Multivariable Models Extracellular Volume Fraction in Noninfarcted Myocardium Remained Associated With Hospitalization for Heart Failure (HHF), Death, or the Combined End Point of HHF and Death, Even When Adding the Extent of Nonischemic Late Gadolinium Enhancement (Expressed as a Continuous Variable) to the Models in Table 3
Table S3. For Comparison to Extracellular Volume Fraction (ECV), the Presence of Nonischemic Late Gadolinium Enhancement Without ECV in the Multivariable Model was Also Associated With Hospitalization for Heart Failure (HHF), Death, or the Combined End Point of HHF and Death But Less Consistently and to a Lesser Extent Than the Models With ECV Based on the χ2 Values, Net Reclassification Improvement, and Integrated Discrimination Improvement Data in Table 3, Which Used ECV Exclusively to Quantify Myocardial Fibrosis
Table S4. For Comparison to Extracellular Volume Fraction (ECV), the Percentage of Left Ventricular Mass Exhibiting Nonischemic Late Gadolinium Enhancement (With the Hazard Ratio Reflecting a 5% Increase) Without ECV in the Multivariable Model was Also Associated With Outcomes But Less Consistently and to a Lesser Extent Than the Models With ECV Based on the χ2 Values, Net Reclassification Improvement, and Integrated Discrimination Improvement Data in Table 3, Which Used ECV Exclusively to Quantify Myocardial Fibrosis