

Abstract
Background
Preeclampsia (PE) is a life‐threatening hypertensive disorder of pregnancy associated with autoantibodies, termed AT 1‐AA, that activate the AT 1 angiotensin receptor. Although the pathogenic nature of these autoantibodies has been extensively studied, little is known about the molecular cause of their generation.
Methods and Results
Here we show that tissue transglutaminase (TG2), an enzyme that conducts posttranslational modification of target proteins, directly modified the 7‐amino acid (7‐aa) epitope peptide that localizes to the second extracellular loop of the AT 1 receptor. These findings led us to further discover that plasma transglutaminase activity was induced and contributed to the production of AT 1‐AA and disease development in an experimental model of PE induced by injection of LIGHT, a tumor necrosis factor superfamily member. Key features of PE were regenerated by adoptive transfer of purified IgG from LIGHT‐injected pregnant mice and blocked by the 7‐amino acid epitope peptide. Translating our mouse research to humans, we found that plasma transglutaminase activity was significantly elevated in PE patients and was positively correlated with AT 1‐AA levels and PE features.
Conclusions
Overall, we provide compelling mouse and human evidence that elevated transglutaminase underlies AT 1‐AA production in PE and highlight novel pathogenic biomarkers and innovative therapeutic possibilities for the disease.
Keywords: AT1‐AA, autoimmunity, preeclampsia, transglutaminase
Subject Categories: Preeclampsia, High Blood Pressure, Hypertension
Introduction
Preeclampsia (PE) is a life‐threatening disorder of pregnancy with high morbidity and mortality for both mothers and babies, with symptoms of hypertension, renal dysfunction, abnormal fetal growth, and circulating antiangiogenic factors.1, 2, 3, 4 PE5, 6, 7, 8, 9, 10, 11 and other hypertensive conditions12, 13 are characterized by the presence of autoantibodies, termed AT1‐AAs, that activate the AT1 angiotensin receptor (AT1R). These autoantibodies are found in the maternal circulation of 70% to 90% of PE patients5, 11, 14 and are likely to contribute to disease by activation of AT1Rs on various cell types.5, 6, 8, 9 These autoantibodies cause the defining clinical features of PE when introduced into pregnant mice and are likely to contribute to these features in the majority of women with PE who harbor these autoantibodies.10 Available evidence suggests that the antibody titers correlate to the severity of the disease.11 Because AT1‐AAs are significant contributors to the pathophysiology of PE, it is important to define the molecular basis initiating their production. These findings will provide a better understanding of the pathogenesis of PE and will likely identify new therapeutic options to interfere with AT1‐AA production and disease progression.
AT1‐AAs recognize a specific seven amino acid (7‐aa) epitope (AFHYESQ) found on the second extracellular loop of AT1Rs.5 The immunological basis for the epitope specificity is not understood. Although previous studies have shown that infusion of inflammatory cytokines into pregnant rats results in the production of AT1‐AA and many other PE features,15, 16, 17 the mechanisms by which these cytokines cause the production of AT1‐AAs that recognize the 7‐aa epitope peptide of AT1Rs remain a mystery. Molecular mimicry with a homologous sequence on human parvovirus was originally considered as the molecular basis for the immunological origin of AT1‐AA.18, 19 However, this possibility was not supported by epidemiological data that failed to show a correlation between PE and prior parvovirus infection.20 A commonly considered mechanism for an autoimmune response is posttranslational modification (PTM), resulting in the creation of a neoantigen that is recognized as nonself by the immune system.21, 22, 23, 24, 25, 26 A large percentage of proteins in the body are targets of PTM, and it is now clear that some of these modifications create new antigens that stimulate an autoimmune response.21, 22, 23, 24, 25, 26 One of the best‐studied autoimmune diseases associated with PTM is celiac disease, a condition affecting ≈1% of the population of developed countries.27 Celiac disease is a chronic small bowel disorder caused by an abnormal immune response to a tissue transglutaminase (TG2)‐modified dietary protein, termed gliadin (a component of glutens that are prominent in wheat).28, 29, 30 The TG2 modification of glutamine‐rich gliadin creates a modified neoantigen peptide that stimulates an autoimmune response.28, 29, 30 TG2 is the most ubiquitous member of the transglutaminase (TGase) family of enzymes that catalyze the PTM of glutamine residues on target proteins.31 TGase‐catalyzed posttranslational modifications include glutamine deamination, and isopeptide bond formation between a peptidyl glutamine residue and a peptidyl lysine residue or a primary amine.31 PE is characterized by elevated pro‐inflammatory cytokines32, 33, 34, 35, 36, 37, 38 that promote TG2 gene expression.39 We have recently reported that activated TG2 posttranslationally modifies AT1Rs with isopeptide bond formation in placentas of women with PE and that circulating TGase activity is significantly elevated.40 Moreover, in preeclampsia the elevated inflammatory condition favoring the activation and expression of TG239 has been demonstrated to give rise to the production of AT1‐AA.15, 16, 17 Thus, here we hypothesize that TGase activation is required for the production of AT1‐AA induced by the pro‐inflammatory environment of PE.
Materials and Methods
Human Subjects
Patients admitted to the Memorial Hermann Hospital were identified by the obstetrics faculty of the University of Texas Medical School at Houston. Preeclamptic patients were diagnosed with severe disease on the basis of the definition set forth by the National High Blood Pressure Education Working Group Report. Inclusion criteria, including no previous history of hypertension, have been reported previously and include blood pressure ≥160/110 mm Hg and urinary protein ≥300 mg in a 24‐hour period or a dipstick value of ≥1. (Twenty‐four‐hour urinary protein values were available for some of the PE patients. The rest of them had dipstick values.) Other criteria include the presence of persistent headache, visual disturbances, epigastric pain, or hemolysis, elevated liver enzymes, and low platelets syndrome in women with blood pressure ≥140/90 mm Hg. Control pregnant women were selected on the basis of having an uncomplicated, normotensive pregnancy with a normal term delivery. The research protocol was approved by the Institutional Committee for the Protection of Human Subjects. All participants gave informed consent. Clinical features of human subjects are presented in Table.
Table 1.
Clinical Features of Human Subjects
| NT | PE | |
|---|---|---|
| N | 24 | 55 |
| Age, y | 26.35±5.95 | 27.04±8.46 |
| Race, % | ||
| African American | 50 | 51 |
| White | 35 | 33 |
| Hispanic | 15 | 16 |
| Gravity | 2.25±1.74 | 2.25±0 |
| BMI | 31.60±5.23 | 35.96±15.40a |
| Weeks gestational age | 38.70±0.94 | 34.28±5.59a |
| Systolic BP, mm Hg | 119.45±13.32 | 153.95±12.00a |
| Diastolic BP, mm Hg | 70.15±9.97 | 90.81±0.68a |
| Proteinuria, mg/24 h | N/A | 846.62±101.02 |
This table demonstrates that the blood pressure and proteinuria are significantly elevated in PE women vs NT pregnant women at term. BMI values are prepregnancy estimates. The value in each category is indicated as mean±SD. Clinical characteristic features of human subjects used in bacterial peptide display assay (Figure 2D) were reported before.48 BMI indicates body mass index; BP, blood pressure; N/A, nonapplicable; NT, normotensive; PE, preeclampsia.
P<0.001 vs normotensive pregnant women.
Animal Protocols
Pregnant C57Bl/6J mice (Harlan) were anesthetized with isofluorane, and 0.15 mL sterile PBS dissolved with or without recombinant mouse LIGHT (2 ng, R&D Systems) was introduced by retro‐orbital sinus injection on gestation day (GD) 13.5 and 14.5. Some mice were also injected with 2.25 mg cystamine dihydrochloride (Sigma‐Aldrich, MO) dissolved in the 0.15 mL sterile PBS to inhibit TGase activity. Since the retro‐orbital injections of cystamine dihydrochloride were only performed on GD 13.5 and 14.5 together with LIGHT molecules, to ensure a constant and effective cystamine treatment afterwards, these pregnant mice were also administered drinking water containing 0.9 g/L cystamine dihydrochloride throughout the rest of their pregnancy. Both treatment routes were reported and validated in earlier publications.40, 41, 42, 43, 44, 45 All protocols involving animal research were reviewed and approved by the Institutional Animal Welfare committee of the University of Texas Health Science Center at Houston. All animal procedures were in accordance with institutional guidelines.
In Vitro TG2 Function Assay, Western Blot, and Immunoprecipitation Followed by Mass Spectral Analysis
Approximately 100 μg TG2 purified from guinea pig liver (Sigma) was incubated with 0.1 mmol/L AT1‐AA epitope peptide AFHYESQ (7‐aa) for 30 minutes at 37°C in 100 μL reaction buffer containing 50 mmol/L Tris‐HCl, pH 7.4 and 5 mmol/L CaCl2. Under the same conditions, a negative control assay was set up without the addition of the 7‐aa epitope peptide. Following an incubation, a portion of the reaction mixture was examined by Western blot analysis using antibody raised against the 7‐aa epitope peptide. A similar assay was reported before46 to test whether TG2 reacts with the synthesized gliadin peptide in celiac disease. After the reaction, the assay mix was directly examined by Western blot using antibody directed against the 7‐aa epitope peptide to assess the cross‐linking efficiency.
To purify the cross‐linking product for mass spectrometry analysis, the assay mix was also subject to immunoprecipitation using the 7‐aa epitope antibody‐bound to Protein G Sepharose High Performance beads (GE Healthcare Life Sciences) following manufacturer's instructions. The immunoprecipitated product was resolved by SDS‐PAGE and subsequently stained with SimplyBlue™ SafeStain (Life Sciences). A band appearing near 80 kDa was excised and examined by mass spectrometry at the proteomics core facility of the MD Anderson Cancer Center.
Blood Pressure Measurement and Quantification of Proteinuria
The systolic blood pressure was noninvasively measured by a volume pressure recording sensor and an occlusion tail‐cuff (CODA System, Kent Scientific). This method shows good agreement with radiotelemetry measurements of blood pressure.47 Blood pressure was measured at the same time daily (±1 hour) while the mice were kept warm using a warming pad. Twenty‐four‐hour urine was collected for analysis using metabolic cages (Nalgene). Mice were trained in metabolic cages for 2 days prior to urine collection. On GD18.5, the mean arterial pressure was determined by cannulating the right carotid artery with a mouse jugular catheter connected to a pressure transducer and an amplifier unit, just prior to euthanasia. The amplifier was connected to a data acquisition module and mean arterial pressure was recorded on a personal computer by Chart 5 Software (AD Instruments, Inc). All of the mice were euthanized on GD18.5 before delivery when their serum and organs were collected. We quantified urinary albumin by ELISA (Exocell) and measured urinary creatinine by a picric acid colorimetric assay (Exocell). We used the ratio of urinary albumin to urinary creatinine as an index of urinary protein.
Measurement of AT1‐AA
ELISA
AT1‐AA was measured using a commercially available sandwich ELISA (CellTrend GmbH, Luckenwalde, Germany, catalog #12000), which is now marketed by One Lambda (catalog number EIA‐AT2RX). The human plasma samples were diluted 10‐fold rather than 100‐fold as suggested by the manufacturer's instructions. The kit uses straightforward ELISA‐based procedures on 96‐well plates for the quantitative determination of AT1‐AA in human plasma or serum. The kit was developed using membrane preparations from mammalian cells that overproduce human AT1R as the bait. The kit has intra‐assay and interassay coefficients of variation of ≈7% and 12% according to the manufacturer. For measurement of murine AT1‐AA, the HRP‐linked anti‐mouse IgG secondary antibody (Jackson ImmunoResearch Labs, Catalog#715‐035‐150) was employed. Other steps in this measurement were carried out according to vendor instructions.
Peptide display
Plasma samples from humans and mice were assayed for binding to the 7‐aa epitope peptide using an assay in which the epitope sequence AFHYESQ is displayed in a high‐avidity format on the bacterial cell surface as reported before.48 In this assay, AT1‐AA detection can be performed with the 7‐mer and does not require the entire second extracellular loop. This peptide display assay is important for showing the epitope specificity of the AT1‐AA produced in the LIGHT‐injected mice.
Plasma TG Activity
TGase activity in human and mouse samples were determined using in vitro TGase assay kits (Sigma‐Aldrich, MO) following the manufacturer's instructions.
Purification of Total Mouse IgG and Adoptive Transfer Method
Affinity purification of IgG from pregnant mice injected with saline or LIGHT
Total IgG was purified by GammaBind G Sepharose chromatography according to the manufacturer's instructions from sera of mice injected either with saline or LIGHT (Saline‐IgG or LIGHT‐IgG). Briefly, 400 μL serum from mice with either saline or LIGHT injection were loaded on the columns and incubated for 3 hours. The flow‐through was collected by centrifugation at 1000g for 1 minute. The eluted bound IgG was further collected by centrifugation at 1000g for 1 minute after incubation with 100 μL 100 mmol/L glycine (pH 2.7) for 10 minutes. The low pH of the eluted fraction was neutralized by adding 5 μL of 1 mol/L Tris, pH 9.0. We performed all steps at 4°C. The levels of IgG in the elution fraction (affinity‐purified total IgG) were quantified by ELISA (Jackson ImmunoResearch Labs, Catalog#715‐035‐150).
Introduction of antibody into mice
Nonpregnant C57BL/6J mice were used (18–22 g; Harlan) in our study. Mice were anesthetized with isofluorane. Purified total IgG (≈800 μg) from 200 μL sera were concentrated by lyophilization and resuspended in ≈200 μL saline and then introduced into nonpregnant mice by osmotic minipump. Some mice also received a 7‐aa peptide corresponding to an epitope on the second extracellular loop of the AT1 receptor (1.5 mg/mice). This peptide effectively neutralizes AT1‐AA. Mouse systolic blood pressure, mean arterial pressure, and proteinuria were measured as described above. Plasma AT1‐AA concentrations on day 18.5 were quantified by ELISA (CellTrend GmbH, catalog #12000).
Statistical Analysis
Data were expressed as the mean±SEM. GraphPad Prism software was employed to run the statistical programs. Measurements of blood pressure, urinary protein concentration, and plasma concentrations of autoantibodies and enzymes typically approximated a normal distribution. Student t tests (paired or unpaired as appropriate) were applied in 2‐group analysis. The means of multiple groups were compared by the 1‐way ANOVA, followed by a Tukey multiple comparisons test. Data presented in Figure 2A were analyzed using a repeated‐measures ANOVA followed by a Tukey multiple comparisons test. For the bacterial epitope peptide display assay, statistical significance was determined by 1‐ or 2‐tail Mann–Whitney U test. A value of P<0.05 was chosen as the threshold of statistical significance. Pearson product‐moment correlation coefficient r was employed to determine the correlation between the 2 variables in Figure 4.
Figure 2.
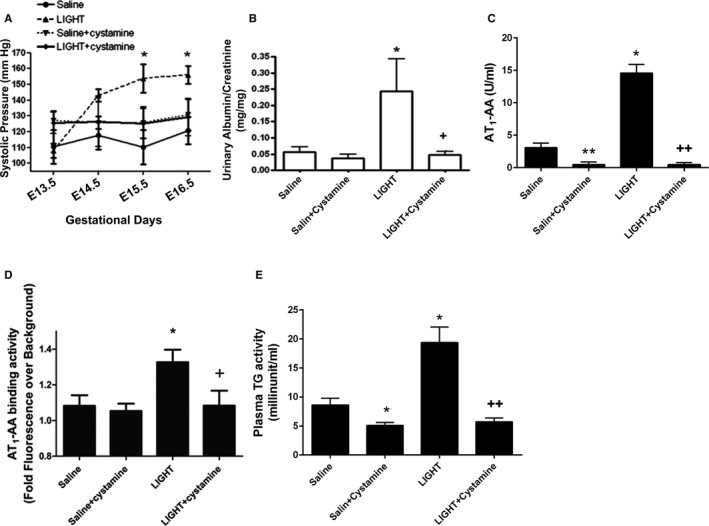
Elevated TGase is required for LIGHT‐induced production of AT 1‐AA and PE features in pregnant mice. Pregnant mice were injected with LIGHT on GD 13.5 and 14.5 in the presence or absence of cystamine, a competitive inhibitor of TGase. Cystamine treated animals continued to receive the TGase inhibitor in the drinking water. Animals were sacrificed on GD18.5. A and B, Blood pressure and proteinuria were induced in the pregnant mice with LIGHT injection. C, Plasma AT 1‐AA levels—Results shown represent specific binding to AT 1Rs in a cell‐based ELISA assay. D, Plasma AT 1‐AA levels—Results shown represent binding to the 7‐aa epitope peptide presented in a bacterial peptide display format. E, Plasma TGase activity. *P<0.05, **P<0.01 vs saline injected mice; + P<0.05, ++ P<0.01 vs LIGHT injected mice. Saline (n=8), Saline + cystamine (n=3), LIGHT (n=13), and LIGHT + cystamine (n=4). AT1‐AA indicates angiotensin II receptor 1 agonistic autoantibody; GD, gestation days; PE, preeclampsia; TGase, transglutaminase.
Figure 4.
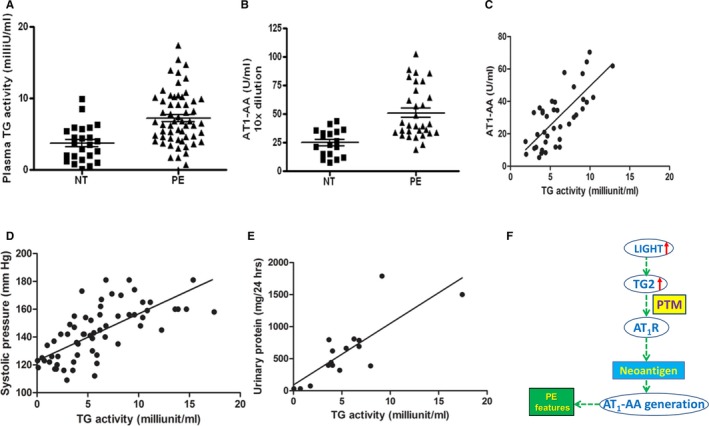
Increased plasma TGase activity in PE patients is positively correlated with AT 1‐AA titer, systolic blood pressure, and urinary protein. A, Plasma TGase activity in normotensive pregnant women (NT, n=24) and PE patients (PE, n=55) (P<0.05). B, Cell‐based whole receptor ELISA assay of circulating AT 1‐AA (NT, n=18; PE, n=31, P<0.05). C through E, The correlation of plasma TGase activity to AT 1‐AA (ELISA) (correlation coefficient r>0.5, P<0.01, n=39), systolic blood pressure (correlation coefficient r>0.5, P<0.01, n=61), and urinary protein (correlation coefficient r>0.5, P<0.01, n=16). F, Working model‐essential role of TGase in the production of AT 1‐AA in PE. Our data support a model in which inflammatory cytokines such as LIGHT induce TGase, which modifies AT 1 receptors. TGase modified AT 1 receptors serve as neoantigens that stimulate the autoimmune production of AT 1‐AA. Thus, our findings identify novel mechanisms for autoantibody production and reveal innovative therapeutic possibilities for PE. AT1‐AA indicates angiotensin II receptor 1 agonistic autoantibody; AT1R, AT1 angiotensin receptor; NT, normotensive; PE, preeclampsia; PTM, posttranslational modification; TGase, transglutaminase.
Results
The 7‐aa Epitope Peptide of the AT1R is Modified by TG2 In Vitro
TG2 catalyzes the PTM of glutamine residues on target proteins by deamination or by isopeptide bond formation with a lysine residue or a primary amine. Of the 359 amino acids that constitute the AT1R, only 5 are glutamine residues (marked red in Figure 1A), and represent potential sites of TGase modification. One glutamine (Q187) resides at the end of the 7‐aa epitope sequence (AFHYESQ) in the second extracellular loop that is uniformly recognized by AT1‐AA in women with PE.5 Thus, TG2‐mediated PTM of Q187 may generate a neoantigen that is recognized by AT1‐AA. Previously, we demonstrated that AT1Rs are modified by TGase in PE placental trophoblasts, but due to the low cellular abundance of AT1R as a membrane bound GPCR, we were unable to purify enough receptor from PE placentas or trophoblasts to thoroughly determine the endogenous isopeptide modification sites and the chemical nature of their amine donor. Thus, to overcome this obstacle, we conducted in vitro TGase assays to determine whether TG2 can directly modify the 7‐aa epitope peptide of AT1R (illustrated in Figure 1B). This was done by incubating purified TG2 with or without the specific 7‐aa epitope peptide. If the epitope peptide is recognized as a substrate by TG2, then it should become covalently attached to the enzyme, either at the active site as a covalent intermediate, or crosslinked to available lysine residues on other TG2 enzymes in the reaction mix.31, 49 To determine whether the 7‐aa epitope peptide was covalently attached to TG2 enzymes, we examined the reaction products39 by denaturing gel electrophoresis and Western blot analysis using an antibody generated specifically to the 7‐aa peptide of the AT1R.32 We also used this antibody in pull‐down experiments followed by mass spectral analysis, to determine whether the epitope peptide became covalently associated with TG2 during the in vitro reaction (illustrated in Figure 1C).
Figure 1.
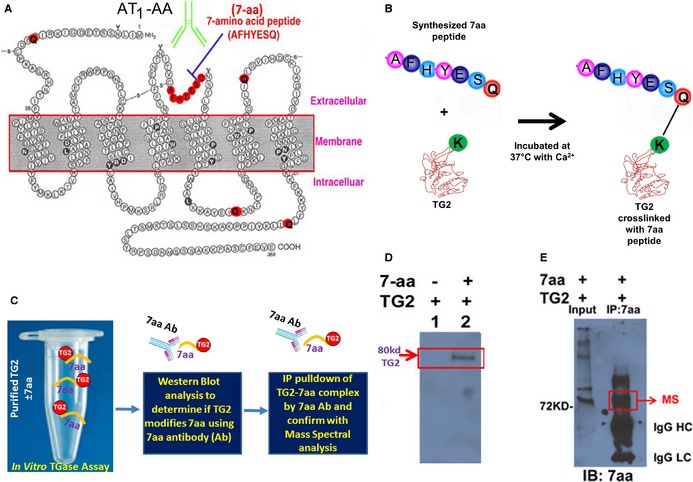
AT 1 receptor seven amino acid (7‐aa) epitope peptide is modified by TG2 in vitro. A, Two dimensional illustration of AT 1 receptor showing location of the 7‐aa epitope peptide and 5 glutamines (Q). B and C, Illustration of experimental strategy to determine whether TG2 modifies the 7‐aa epitope peptide in vitro. D, 7‐aa epitope peptide was covalently cross‐linked to TG2 in the in vitro assay. TG2 was incubated in the presence or absence of the 7‐aa epitope peptide (AFHYESQ). The reaction mix was subsequently fractionated by denaturing gel electrophoresis and transferred to a blot. Antibody specific for the 7‐aa epitope peptide recognized an 80‐kDa protein, the size expected for TG2, indicating that the 7‐aa peptide was covalently cross‐linked to TG2. E, Mass spectral characterization of TG2 cross‐linked with the 7‐aa epitope peptide. After the in vitro reaction, proteins covalently linked with 7‐aa epitope peptide in the reaction mix were immunoprecipitated with the antibody directed against this peptide and resolved in the denaturing gel electrophoresis. The 80‐kDa protein in the immunoprecipitation products was characterized as TG2 in mass spectral analysis. The presence of 7‐aa epitope in the immunoprecipitation products was also confirmed in Western blot using the antibody. AT receptor indicates angiotensin receptor 1; IgG HC, immunoglobulin G heavy chain; IgG LC, immunoglobulin G light chain; TG2, transglutaminase 2; MS, mass spectral analysis.
Western blot analysis showed that antibody to the 7‐aa epitope peptide recognized an 80‐kDa protein the size expected for TG2 (Figure 1D). No cross‐reactivity was observed in control reactions that lacked the 7‐aa epitope peptide (Figure 1D). Mass spectral analysis confirmed that the 80‐kDa protein immunoprecipitated by the 7‐aa epitope peptide antibody was TG2 (Figure 1E). Thus, Western blot analysis following denaturing gel electrophoresis and mass spectral analysis of co‐immunoprecipitated protein indicated that the 7‐aa epitope peptide was modified and covalently cross‐linked to TG2.
Plasma Transglutaminase Activity and AT1‐AA Specifically Recognizing the 7‐aa Epitope Peptide Are Induced in an Experimental Model of PE Based on LIGHT Injection
Although we demonstrated the direct modification of the 7‐aa epitope peptide by TG2 in vitro, the importance of TG activation in AT1‐AA production in vivo is undetermined. To address this question, we used a mouse model of PE based on injection of the inflammatory cytokine LIGHT (tumor necrosis factor [TNF] superfamily member 14) into pregnant mice on GD 13.5 and 14.5.32 We have previously shown that the injection of LIGHT into pregnant mice induced key features of PE including hypertension, proteinuria, reduced fetal and placental weight, elevated sFlt‐1, and elevated endothelin‐1, thus making this a convenient and relevant experimental model of PE.44 We confirmed previously published results showing that LIGHT infusion induced hypertension and proteinuria (Figure 2A and 2B). To determine whether LIGHT also induced AT1‐AA production, we used 2 assays to quantify AT1‐AA in the circulation of the pregnant mice with or without LIGHT injection. Using a cell‐based ELISA to quantify the presence of AT1‐AA, we found that autoantibody titers in LIGHT‐injected pregnant mice were confidently identified and significantly higher than the saline controls (Figure 2C). The cell‐based ELISA measures antibodies that recognize native AT1Rs, and is not specific for binding to the 7‐aa epitope peptide of the AT1R. Thus, to determine whether the AT1‐AA produced in LIGHT injected pregnant mice recognized the same 7‐aa epitope that is recognized by AT1‐AA produced in women with PE, we used a recently developed flow cytometry–based bacterial peptide display assay48 to measure binding to the epitope peptide (AFHYESQ). Similar to the cell‐based ELISA, we found that LIGHT injection induced specific autoantibody directed to the 7‐aa epitope peptide of AT1Rs. Thus, 2 independent assays confirmed that LIGHT infusion into pregnant mice leads to the generation of autoantibody directed against the AT1R (Figure 2D).
Essential Role of Elevated TGase Activity in LIGHT‐Induced PE Features and AT1‐AA Production in Pregnant Mice
In addition to inducing the production of AT1‐AA, we found that LIGHT injection significantly increased plasma TGase activity in pregnant mice (Figure 2E). To determine the functional role of elevated plasma TGase in LIGHT‐induced maternal PE features and AT1‐AA production, we co‐injected LIGHT with PBS or cystamine, a competitive inhibitor of TGase,41, 50 into pregnant mice on GD 13.5 and 14.5. To continuously inhibit TGase activity, cystamine‐injected mice were also provided drinking water containing 0.9 g/L cystamine. First, we confirmed that cystamine treatment significantly inhibited the increase in plasma TGase activity in LIGHT‐injected pregnant mice (Figure 2E). Subsequently, we found that cystamine treatment significantly attenuated LIGHT‐induced PE features including hypertension and proteinuria (Figure 2A and 2B). Finally, we used both the cell‐based ELISA and the bacterial peptide display assay to quantify the effect of cystamine treatment on LIGHT‐induced AT1‐AA production in pregnant mice. We found that cystamine treatment significantly reduced AT1‐AA production in the LIGHT‐injected pregnant mice based on the cell‐based ELISA (Figure 2C) and the bacterial peptide display assay (Figure 2D). Thus, 2 independent assays provide in vivo evidence that elevated TGase activity is required for LIGHT‐induced AT1‐AA production in pregnant mice and that the LIGHT‐induced autoantibodies recognized the same 7‐aa epitope as AT1‐AA present in women with PE. Altogether, we established the importance of elevated TGase activity in the pathophysiology of PE and AT1‐AA production in vivo.
IgG from LIGHT‐Injected Pregnant Mice Causes Hypertension and Proteinuria Following Transfer into Recipient Mice
To determine whether the AT1‐AAs produced in LIGHT‐injected pregnant mice have the ability to contribute to the pathophysiology of PE, we conducted adoptive transfer experiments as illustrated in Figure 3A. For this purpose we isolated total IgG from the saline or LIGHT‐injected pregnant mice described above and tested the isolated IgG for the presence of AT1‐AA before and after introduction into recipient mice. We found that IgG from LIGHT‐injected pregnant mice (ie, LIGHT‐IgG), in contrast to IgG from saline‐injected mice (saline‐IgG), displayed significant titers of AT1‐AA (Figure 3B). Next, LIGHT‐IgG or saline‐IgG was infused into receipt mice for 7 days by osmotic minipump, systolic blood pressure was monitored by tail cuff plethysmography, and urine was collected in metabolic cages on the seventh day for determination of urinary protein. To validate successful antibody transfer, we analyzed AT1‐AA titers in the sera from infused mice at the end of experiments. We found that AT1‐AA was readily detected in the animals infused with LIGHT‐IgG compared to mice infused with saline‐IgG (Figure 3C). A pronounced increase in systolic blood pressure was observed following infusion of LIGHT‐IgG and no significant change in blood pressure was observed in the animals infused with saline‐IgG (Figure 3D). To validate the tail cuff measurements of blood pressure, the intracarotid mean arterial blood pressure was measured in the mice under anesthesia on the final day of saline‐ or LIGHT‐IgG infusion. As shown in Figure 3E, the mean arterial pressure was significantly elevated in the LIGHT‐IgG‐injected mice in contrast to that of the saline‐IgG‐injected mice. A significant increase in urinary protein was also observed in mice infused with LIGHT‐IgG in contrast to mice infused with saline‐IgG (Figure 3F). Thus, the pathologic properties of LIGHT‐induced AT1‐AA were clearly demonstrated by introduction into recipient mice followed by the appearance of hypertension and proteinuria.
Figure 3.
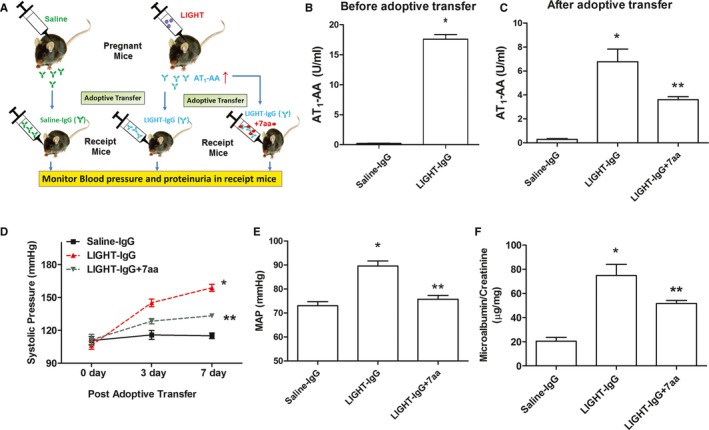
Adoptive transfer experiments show that IgG from LIGHT‐injected mice causes hypertension and proteinuria following introduction into recipient mice. A, Illustration of adoptive transfer experiments. B, Prior to infusion AT 1‐AA levels in IgG from saline (n=6) or LIGHT injected mice (n=8) were determined by ELISA. These IgG preparations, termed saline‐IgG or LIGHT‐IgG (± pre‐incubation with 7‐aa epitope peptide) were subsequently infused into mice for 7 days. Following IgG infusion the recipient mice were analyzed for (C) plasma AT 1‐AA levels on day 7, (D) systolic blood pressure measured by tail cuff plethysmography on different days, (E) intracarotid mean arterial blood pressure was measured on day 7, and (F) urinary protein on day 7. *P<0.01 vs saline‐IgG injected mice, **P<0.05 vs LIGHT‐IgG injected mice. Saline‐IgG (n=5), LIGHT‐IgG (n=6), LIGHT‐IgG+7‐aa (n=4); AT1‐AA indicates angiotensin II receptor 1 agonistic autoantibody; MAP, mean arterial pressure.
Finally, to determine whether LIGHT‐IgG induced hypertension and proteinuria via interaction with the 7‐aa epitope peptide of AT1 receptor, the LIGHT‐IgG was pre‐incubated with the 7‐aa epitope peptide prior to introduction into recipient mice. ELISA analysis of IgG from injected mice showed that the titer of LIGHT‐IgG pre‐incubated with the 7‐aa epitope peptide was significantly lower than that of LIGHT‐IgG that was not pre‐incubated with the 7‐aa epitope peptide (Figure 3C). Moreover, the stimulatory effects of LIGHT‐IgG on blood pressure and urinary protein were significantly reduced when the LIGHT‐IgG was incubated with the 7‐aa epitope peptide prior to injection (Figure 3D through 3F). Overall, these data provide functional evidence that AT1‐AAs produced in LIGHT‐injected pregnant mice specifically recognize the 7‐aa epitope peptide of AT1Rs and adoptively transfer hypertension and proteinuria to recipient mice.
Increased Plasma Transglutaminase Activity in PE Patients is Positively Correlated with AT1‐AA Titer, Systolic Blood Pressure and Urinary Protein
The results presented above indicate that increased plasma TGase is required for AT1‐AA production in pregnant mice. To translate our mouse findings to humans, we measured TGase activity and AT1‐AA titer in plasma samples from normotensive and PE patients. Similar to our mouse results, we found that plasma TGase activity was significantly elevated in PE patients compared to normotensive pregnant women (Figure 4A). Additionally, AT1‐AA levels were independently determined by the cell‐based whole receptor ELISA (Figure 4B). Thus, we provide evidence that plasma TGase activity and AT1‐AA titer were significantly elevated in PE patients compared to normotensive individuals. However, because these values were determined at term, and because women with PE usually deliver early, we cannot rule out the possibility that the observed differences are related to differences in gestational age.
To assess the potential significance of plasma TGase activity in the pathophysiology of PE, we examined the relationship of circulating TGase activity with AT1‐AA titer, systolic blood pressure, and urinary protein in women with PE. We found that plasma TGase activity was positively and significantly correlated with AT1‐AA titer blood pressure, and urinary protein (Figure 4C through 4E). Altogether, our translational studies with human samples are consistent with our mouse findings and suggest a pathological role for TGase activity in AT1‐AA production and subsequent disease development.
Discussion
The presence of AT1‐AAs in the circulation of PE patients was first reported in 1999.5 Since then, a large and growing body of evidence has demonstrated that AT1‐AAs stimulate the Gq‐coupled AT1 receptor5, 8, 9 and contribute to the pathophysiology of PE.6, 7, 10, 11 Although the pathogenic role of these autoantibodies has been extensively studied, the molecular basis triggering their generation remains a mystery. Research reported here revealed that TGase is required for the production of AT1‐AA against 7‐aa epitope peptide of AT1R in a cytokine‐induced model of PE in pregnant mice. Our findings are strongly supported by multiple lines of evidence: (1) In vitro studies demonstrated that TG2 directly modifies the 7‐aa epitope peptide of the AT1R; (2) In vivo data revealed that elevated TGase activity is essential for the production of AT1‐AA with specificity against the 7‐aa epitope peptide and for disease development in PE; and (3) Human translational studies showed that plasma TGase activity is elevated in PE patients and positively correlated with AT1‐AA levels, hypertension, and proteinuria. Overall, in vitro direct evidence of TG2‐mediated modification of 7‐aa epitope peptide of AT1R and in vivo mouse findings coupled with human translational studies have added significant new insight to the pathogenesis of PE and open up innovative diagnostic and therapeutic possibilities.
Our results show that most women with PE have elevated AT1‐AA titers and increased plasma TGase activity compared to those with normotensive pregnancies. The fact that not all women with PE harbor AT1‐AA or have elevated TGase activity is consistent with the mounting evidence that PE is a heterogeneous disease. It is also noteworthy that a small portion of pregnant women who were not diagnosed with PE had AT1‐AA titers and plasma TGase activities that overlapped with the lower end of the distribution for women diagnosed with PE. We have previously reported similar observations using a functional bioassay for AT1‐AA.11 Because our studies only determined AT1‐AA titers and plasma TGase activities at term, we do not know how long these values were elevated. Thus, it is possible that these values were only recently elevated in these women, in comparison to those observed in women with PE, and had not been elevated for a sufficient length time to cause features of PE. This possibility underscores the need for a prospective clinical study measuring these parameters throughout pregnancy to determine when they first appear and what temporal relationship this has with the onset of PE symptoms.
One of the commonly accepted causes for autoantibody production is PTM of proteins to generate neoantigens.21, 22, 23, 24, 25, 26 Celiac disease is the most common autoimmune disease caused by TG2‐mediated PTM. Specifically, in celiac disease TG2 mediates PTM of glutamine‐rich gluten peptides, resulting in the generation of neoantigens that subsequently trigger autoantibody production.28, 29, 30 We have recently found that TGase activity is increased in the placentas and plasma of PE patients.40 Moreover, TGase mediates PTM of the AT1R in the placentas from PE patients.40 However, whether TGase‐mediated modification of AT1Rs contributes to the production of autoantibodies in PE has not been determined. Our mutational analysis of exogenous human AT1Rs produced in Chinese Hamster Ovary (CHO) cells determined that intracellular TG2 modifies AT1Rs on the cytoplasmic tail glutamine residue (Q315) and thereby increases receptor stability by inhibiting ubiquitin‐mediated degradation.40 We found that 1 of the 5 glutamine residues (Q187) present in AT1Rs is present at the end of the 7‐aa epitope peptide (AFHYESQ) in the second extracellular loop of the receptor. This finding raises a possibility that TG2 functions extracellularly to mediate PTM of glutamine residue Q187 of the 7‐aa epitope peptide on the second extracellular loop of AT1R. Indeed, TG2 is also known to function extracellularly in the microparticles shed from the cell membrane.51, 52 In contrast to celiac disease where TG2‐mediated PTM of a short dietary peptide (gliadin) is easily detected,28 it has been challenging to purify sufficient quantities of AT1R from placentas of women with PE to determine whether glutamine Q187 is modified endogenously. However, here we overcame this hurdle by conducting in vitro functional assays to determine whether TG2 can specifically modify the 7‐aa epitope peptide of the AT1R. With the antibody specifically developed against the 7‐aa epitope peptide,32 Western blot analysis following denaturing gel electrophoresis and mass spectral analysis of the protein immunoprecipitated with the antibody show that the 7‐aa epitope peptide was modified and covalently cross‐linked to TG2 (Figure 1). These in vitro findings support the possibility that TG2‐mediated PTM of AT1Rs may contribute to autoantigen origination of AT1‐AA and account for its epitope specificity.
In vitro experiments are unable to determine whether TGase modification of AT1Rs promotes AT1‐AA production. Thus, we extended our in vitro studies to in vivo to address the importance of TGase in AT1‐AA production in PE. PE is associated with an increased maternal inflammatory response.33 A pathogenic role for the inflammatory response is provided by data showing that infusion of inflammatory cytokines (TNF‐α, IL‐6, and IL‐17)15, 16, 17 results in key features of PE in pregnant rats. Supporting this finding, our studies presented here demonstrated that infusion of LIGHT,32 a TNF superfamily member,53, 54 into pregnant. mice results in PE features and AT1‐AA production. Using an adoptive transfer experimental strategy, we demonstrated for the first time that AT1‐AAs produced in LIGHT‐infused pregnant mice are sufficient to regenerate PE features including hypertension and proteinuria in recipient mice, indicating the pathogenic role of LIGHT‐induced autoantibody. More importantly, we further demonstrated that pre‐incubation of autoantibody purified from LIGHT‐infused pregnant mice with the 7‐aa epitope peptide significantly attenuated PE features in receipt mice. Thus, our adoptive transfer experiments provide compelling evidence that LIGHT‐induced AT1‐AA has a pathogenic role and is directed to the same 7‐aa epitope as the AT1‐AAs present in women with PE.
Successful establishment of an animal model with production of AT1‐AA specific for the 7‐aa epitope peptide of AT1Rs provides us with an important investigative tool to test the importance of TGase in AT1‐AA production in vivo. Consistent with AT1‐AA production triggered in LIGHT‐infused pregnant mice, we discovered that plasma TGase activity was also induced by LIGHT. To test the importance of elevated TGase in AT1‐AA production and disease development, we treated LIGHT‐infused pregnant mice with cystamine, a well‐accepted and commonly used TGase inhibitor.32, 33, 34, 35, 36, 37, 38, 45 We found that inhibition of TGase activity significantly reduced LIGHT‐induced AT1‐AA production and maternal disease development in pregnant mice. While it is clear that cystamine inhibits TGase activity, the possibility of off‐target effects cannot be ruled out at this time. Nevertheless, the fact that cystamine blocks LIGHT‐induced features of PE, including AT1‐AA production, is a very important finding with significant mechanistic and therapeutic implications. Taken together, our findings suggest that elevated TGase activity is essential for LIGHT‐induced pathophysiology of PE including hypertension, proteinuria, and AT1‐AA production.
Our preclinical studies with mouse models suggest that elevated TGase may play an important role in the pathophysiology of PE and the production of AT1‐AA. To extend these findings to humans, we conducted translational studies and found that plasma TGase activity was significantly elevated in patients with PE. A cell‐based ELISA was used to determine the presence of antibody that binds to intact AT1 receptors55. We observed a strong positive correlation between plasma TGase activity and circulating AT1‐AA titers, blood pressure, and urinary protein in women with PE. This strong correlation is consistent with a crucial role for TGase in autoantibody production, a possibility supported by the ability of cystamine to block LIGHT‐induced AT1‐AA production in pregnant mice.
PE is a complicated pregnancy disorder featuring an increase in maternal inflammatory cytokines.3, 32, 33, 34, 35, 36, 37, 38 Rodent models of PE based on infusion of inflammatory cytokines (TNF‐α, IL‐6, IL‐1715, 16, 17) are characterized by the presence of AT1‐AA. It is of special note that the promoter region of the TG2 gene contains gene regulatory elements for the binding of transcription factor NF‐κB,39 which functions in response to inflammatory cytokines such as TNF, or members of the TNF superfamily (e.g., LIGHT), as well as regulatory elements that respond to IL‐6.39 These cytokines (TNF, LIGHT, and IL‐6) are increased in most, but not all, women with PE and are likely to contribute to the increased TG2 production39, 56, 57 observed in most PE pregnancies.40 Consistent with this expectation, we have shown here that circulating levels of TGase activity are significantly increased in women with PE and in LIGHT‐injected pregnant mice. We have previously shown that increased TG2 modifies AT1Rs by ubiquitination‐preventing isopeptide modification in PE placental syncytiotrophoblasts,40 where the expression of TG2 and AT1Rs is the highest in the placenta. We believe that the TG2‐mediated AT1 receptor modification and stabilization antagonizes AT1R downregulation in PE and may thereby contribute to disease development by providing increased abundance of AT1Rs for activation and downstream signaling and for presentation to the immune system, resulting in autoimmunity and autoantibody production.
TG2 is also known to be present in microparticles shed from cell membranes.51, 52 We show here that TG2 is able to modify and cross‐link the 7‐aa epitope peptide in vitro. Thus, similar to celiac disease,28, 29, 30 where TG2 modifies a dietary peptide gliadin, it is possible that in PE the circulating autoantigen of AT1‐AA is created by elevated TG2 modification of the AT1‐AA epitope from the second extracellular loop of the receptor in the microparticles shed from placental syncytiotrophoblasts.58 In this way, the TG2‐generated autoantigen may be presented to the immune system in the circulation where we identified an increased level of TGase activity in PE. From this perspective, our results are in good agreement with earlier results15, 16, 17 showing that TNF, IL‐6, and IL‐17 can stimulate AT1‐AA production in pregnant rats. An especially important finding reported in our study is that cytokine‐mediated induction of AT1‐AA production is inhibited by cystamine, an inhibitor of TGase. Our findings implicate TGase‐mediated modification of AT1 receptors as a key factor contributing to cytokine‐induced autoantibody production. The increased AT1‐AA will in turn activate AT1 receptors, leading to increased TG2 production and AT1 receptor modification.32 Taken together, we believe that AT1‐AA, AT1 receptor activation, and TG2‐mediated receptor modification form a vicious cycle driving disease development.
In conclusion, nothing was known about the role of TGase in AT1‐AA production and disease development prior to our studies. Therefore, our findings about elevated TGase activity in AT1‐AA production are novel. First, a strong correlation of plasma TGase activity with AT1‐AA titers and disease features in humans is highly significant for diagnostic purpose. Our data also support a new working model (Figure 4F) in which a heightened inflammatory response characteristic of PE contributes to the induction of TGase, which modifies AT1Rs. The TGase‐modified AT1Rs serve as neoantigens that stimulate the autoimmune production of AT1‐AA. These studies suggest that PE may be an autoimmune condition in which cytokine‐ and/or placental hypoxia–induced production of TG2 results in increased AT1 receptor modification leading to AT1‐AA production and thereby contributing to other disease features. The novel but compelling concept of PE as an autoimmune condition offers new insight regarding the origin and management of PE.59 Finally, our findings that elevated TGase underlies autoantigen origination is likely not limited to PE and may also apply to the generation of agonistic autoantibodies in other autoimmune‐related cardiovascular diseases. Examples include several forms of hypertension associated with autoantibodies that activate α1‐adrenergic receptors,60, 61, 62 idiopathic dilated cardiomyopathy associated with β1‐adrenergic receptor activating autoantibodies,63, 64 idiopathic dilated cardiomyopathy associated with autoantibodies that activate the muscarinic M2 receptor,65, 66 and scleroderma associated with autoantibodies that activate the endothelin type A receptor.67 Thus, our discovery of the contribution of TGase in autoantibody production in PE may provide a clue about why self‐reacting antibodies are generated in other autoimmune‐related cardiac diseases that are also associated with hypoxia and increased inflammatory cytokines. Taken together, our current studies highlight TG2 as an attractive therapeutic target in PE and potentially other cardiovascular and hypertensive disorders.
Sources of Funding
This work was supported by National Institutes of Health Grants HL19549 (to Xia), RC4HD067977 and HD34130 (to Xia and Kellems), and by China National Science Foundation 81228004 (to Xia).
Disclosures
None.
(J Am Heart Assoc. 2015;4:e002323 doi: 10.1161/JAHA.115.002323)
References
- 1. Roberts JM, Cooper DW. Pathogenesis and genetics of pre‐eclampsia. Lancet. 2001;357:53–56. [DOI] [PubMed] [Google Scholar]
- 2. Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. [DOI] [PubMed] [Google Scholar]
- 3. Granger JP, Alexander BT, Llinas MT, Bennett WA, Khalil RA. Pathophysiology of preeclampsia: linking placental ischemia/hypoxia with microvascular dysfunction. Microcirculation. 2002;9:147–160. [DOI] [PubMed] [Google Scholar]
- 4. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms‐like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest. 1999;103:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dechend R, Homuth V, Wallukat G, Kreuzer J, Park JK, Theuer J, Juepner A, Gulba DC, Mackman N, Haller H, Luft FC. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000;101:2382–2387. [DOI] [PubMed] [Google Scholar]
- 7. Dechend R, Viedt C, Muller DN, Ugele B, Brandes RP, Wallukat G, Park JK, Janke J, Barta P, Theuer J, Fiebeler A, Homuth V, Dietz R, Haller H, Kreuzer J, Luft FC. AT1 receptor agonistic antibodies from preeclamptic patients stimulate NADPH oxidase. Circulation. 2003;107:1632–1639. [DOI] [PubMed] [Google Scholar]
- 8. Thway TM, Shlykov SG, Day MC, Sanborn BM, Gilstrap LC III, Xia Y, Kellems RE. Antibodies from preeclamptic patients stimulate increased intracellular Ca2+ mobilization through angiotensin receptor activation. Circulation. 2004;110:1612–1619. [DOI] [PubMed] [Google Scholar]
- 9. Bobst SM, Day MC, Gilstrap LC III, Xia Y, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human mesangial cells and induce interleukin‐6 and plasminogen activator inhibitor‐1 secretion. Am J Hypertens. 2005;18:330–336. [DOI] [PubMed] [Google Scholar]
- 10. Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre‐eclampsia in pregnant mice. Nat Med. 2008;14:855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: correlation with disease severity. Hypertension. 2010;55:386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fu ML, Herlitz H, Schulze W, Wallukat G, Micke P, Eftekhari P, Sjogren KG, Hjalmarson A, Muller‐Esterl W, Hoebeke J. Autoantibodies against the angiotensin receptor (AT1) in patients with hypertension. J Hypertens. 2000;18:945–953. [DOI] [PubMed] [Google Scholar]
- 13. Liao YH, Wei YM, Wang M, Wang ZH, Yuan HT, Cheng LX. Autoantibodies against AT1‐receptor and alpha1‐adrenergic receptor in patients with hypertension. Hypertens Res. 2002;25:641–646. [DOI] [PubMed] [Google Scholar]
- 14. Herse F, Verlohren S, Wenzel K, Pape J, Muller DN, Modrow S, Wallukat G, Luft FC, Redman CW, Dechend R. Prevalence of agonistic autoantibodies against the angiotensin II type 1 receptor and soluble fms‐like tyrosine kinase 1 in a gestational age‐matched case study. Hypertension. 2009;53:393–398. [DOI] [PubMed] [Google Scholar]
- 15. LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension. 2008;52:1168–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gadonski G, LaMarca BB, Sullivan E, Bennett W, Chandler D, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of interleukin 6. Hypertension. 2006;48:711–716. [DOI] [PubMed] [Google Scholar]
- 17. Dhillion P, Wallace K, Herse F, Scott J, Wallukat G, Heath J, Mosely J, Martin JN Jr, Dechend R, LaMarca B. IL‐17‐mediated oxidative stress is an important stimulator of AT1‐AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol. 2012;303:R353–R358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zaki M, Greenwood C, Redman CW. The spontaneous reversal of pre‐eclampsia associated with parvovirus‐induced hydrops and the placental theory of pre‐eclampsia: a case report. BJOG. 2003;110:1125–1126. [PubMed] [Google Scholar]
- 19. Yeh SP, Chiu CF, Lee CC, Peng CT, Kuan CY, Chow KC. Evidence of parvovirus B19 infection in patients of pre‐eclampsia and eclampsia with dyserythropoietic anaemia. Br J Haematol. 2004;126:428–433. [DOI] [PubMed] [Google Scholar]
- 20. Stepan H, Wallukat G, Schultheiss HP, Faber R, Walther T. Is parvovirus B19 the cause for autoimmunity against the angiotensin II type receptor? J Reprod Immunol. 2007;73:130–134. [DOI] [PubMed] [Google Scholar]
- 21. Cloos PA, Christgau S. Post‐translational modifications of proteins: implications for aging, antigen recognition, and autoimmunity. Biogerontology. 2004;5:139–158. [DOI] [PubMed] [Google Scholar]
- 22. Anderton SM. Post‐translational modifications of self antigens: implications for autoimmunity. Curr Opin Immunol. 2004;16:753–758. [DOI] [PubMed] [Google Scholar]
- 23. Dieker J, Muller S. Post‐translational modifications, subcellular relocation and release in apoptotic microparticles: apoptosis turns nuclear proteins into autoantigens. Folia Histochem Cytobiol. 2009;47:343–348. [DOI] [PubMed] [Google Scholar]
- 24. Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol. 2012;24:112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zavala‐Cerna MG, Martinez‐Garcia EA, Torres‐Bugarin O, Rubio‐Jurado B, Riebeling C, Nava A. The clinical significance of posttranslational modification of autoantigens. Clin Rev Allergy Immunol. 2014;47:73–90. [DOI] [PubMed] [Google Scholar]
- 26. Bhat S, Mary S, Banarjee R, Giri AP, Kulkarni MJ. Immune response to chemically modified proteome. Proteomics Clin Appl. 2014;8:19–34. [DOI] [PubMed] [Google Scholar]
- 27. Fasano A, Catassi C. Celiac disease. N Engl J Med. 2012;367:2419–2426. [DOI] [PubMed] [Google Scholar]
- 28. Molberg O, McAdam SN, Korner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Noren O, Roepstorff P, Lundin KE, Sjostrom H, Sollid LM. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut‐derived T cells in celiac disease. Nat Med. 1998;4:713–717. [DOI] [PubMed] [Google Scholar]
- 29. Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2:647–655. [DOI] [PubMed] [Google Scholar]
- 30. Sollid LM, Jabri B. Celiac disease and transglutaminase 2: a model for posttranslational modification of antigens and HLA association in the pathogenesis of autoimmune disorders. Curr Opin Immunol. 2011;23:732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. [DOI] [PubMed] [Google Scholar]
- 32. Wang W, Parchim NF, Iriyama T, Luo R, Zhao C, Liu C, Irani RA, Zhang W, Ning C, Zhang Y, Blackwell SC, Chen L, Tao L, Hicks MJ, Kellems RE, Xia Y. Excess LIGHT contributes to placental impairment, increased secretion of vasoactive factors, hypertension, and proteinuria in preeclampsia. Hypertension. 2014;63:595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Borzychowski AM, Sargent IL, Redman CW. Inflammation and pre‐eclampsia. Semin Fetal Neonatal Med. 2006;11:309–316. [DOI] [PubMed] [Google Scholar]
- 34. Conrad KP, Miles TM, Benyo DF. Circulating levels of immunoreactive cytokines in women with preeclampsia. Am J Reprod Immunol. 1998;40:102–111. [DOI] [PubMed] [Google Scholar]
- 35. Irani RA, Zhang Y, Zhou CC, Blackwell SC, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Autoantibody‐mediated angiotensin receptor activation contributes to preeclampsia through tumor necrosis factor‐alpha signaling. Hypertension. 2010;55:1246–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Raghupathy R. Cytokines as key players in the pathophysiology of preeclampsia. Med Princ Pract. 2013;22(suppl 1):8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xie C, Yao MZ, Liu JB, Xiong LK. A meta‐analysis of tumor necrosis factor‐alpha, interleukin‐6, and interleukin‐10 in preeclampsia. Cytokine. 2011;56:550–559. [DOI] [PubMed] [Google Scholar]
- 38. Lau SY, Guild SJ, Barrett CJ, Chen Q, McCowan L, Jordan V, Chamley LW. Tumor necrosis factor‐alpha, interleukin‐6, and interleukin‐10 levels are altered in preeclampsia: a systematic review and meta‐analysis. Am J Reprod Immunol. 2013;70:412–427. [DOI] [PubMed] [Google Scholar]
- 39. Gundemir S, Colak G, Tucholski J, Johnson GV. Transglutaminase 2: a molecular Swiss army knife. Biochim Biophys Acta. 2012;1823:406–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu C, Wang W, Parchim N, Irani RA, Blackwell SC, Sibai B, Jin J, Kellems RE, Xia Y. Tissue transglutaminase contributes to the pathogenesis of preeclampsia and stabilizes placental angiotensin receptor type 1 by ubiquitination‐preventing isopeptide modification. Hypertension. 2014;63:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med. 2002;8:143–149. [DOI] [PubMed] [Google Scholar]
- 42. Dedeoglu A, Kubilus JK, Jeitner TM, Matson SA, Bogdanov M, Kowall NW, Matson WR, Cooper AJ, Ratan RR, Beal MF, Hersch SM, Ferrante RJ. Therapeutic effects of cystamine in a murine model of Huntington's disease. J Neurosci. 2002;22:8942–8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fox JH, Barber DS, Singh B, Zucker B, Swindell MK, Norflus F, Buzescu R, Chopra R, Ferrante RJ, Kazantsev A, Hersch SM. Cystamine increases L‐cysteine levels in Huntington's disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem. 2004;91:413–422. [DOI] [PubMed] [Google Scholar]
- 44. Borrell‐Pages M, Canals JM, Cordelieres FP, Parker JA, Pineda JR, Grange G, Bryson EA, Guillermier M, Hirsch E, Hantraye P, Cheetham ME, Neri C, Alberch J, Brouillet E, Saudou F, Humbert S. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006;116:1410–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davies JE, Rose C, Sarkar S, Rubinsztein DC. Cystamine suppresses polyalanine toxicity in a mouse model of oculopharyngeal muscular dystrophy. Sci Transl Med. 2010;2:34ra40. [DOI] [PubMed] [Google Scholar]
- 46. Di Niro R, Mesin L, Zheng NY, Stamnaes J, Morrissey M, Lee JH, Huang M, Iversen R, du Pre MF, Qiao SW, Lundin KE, Wilson PC, Sollid LM. High abundance of plasma cells secreting transglutaminase 2‐specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012;18:441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Feng M, Whitesall S, Zhang Y, Beibel M, D'Alecy L, DiPetrillo K. Validation of volume‐pressure recording tail‐cuff blood pressure measurements. Am J Hypertens. 2008;21:1288–1291. [DOI] [PubMed] [Google Scholar]
- 48. Elliott SE, Parchim NF, Liu C, Xia Y, Kellems RE, Soffici AR, Daugherty PS. Characterization of antibody specificities associated with preeclampsia. Hypertension. 2014;63:1086–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev. 2009;89:991–1023. [DOI] [PubMed] [Google Scholar]
- 50. Siegel M, Khosla C. Transglutaminase 2 inhibitors and their therapeutic role in disease states. Pharmacol Ther. 2007;115:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robinson NJ, Baker PN, Jones CJ, Aplin JD. A role for tissue transglutaminase in stabilization of membrane‐cytoskeletal particles shed from the human placenta. Biol Reprod. 2007;77:648–657. [DOI] [PubMed] [Google Scholar]
- 52. van den Akker J, van Weert A, Afink G, Bakker EN, van der Pol E, Boing AN, Nieuwland R, VanBavel E. Transglutaminase 2 is secreted from smooth muscle cells by transamidation‐dependent microparticle formation. Amino Acids. 2012;42:961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. [DOI] [PubMed] [Google Scholar]
- 54. Zhai Y, Guo R, Hsu TL, Yu GL, Ni J, Kwon BS, Jiang GW, Lu J, Tan J, Ugustus M, Carter K, Rojas L, Zhu F, Lincoln C, Endress G, Xing L, Wang S, Oh KO, Gentz R, Ruben S, Lippman ME, Hsieh SL, Yang D. LIGHT, a novel ligand for lymphotoxin beta receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J Clin Invest. 1998;102:1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Reinsmoen NL, Lai CH, Heidecke H, Haas M, Cao K, Ong G, Naim M, Wang Q, Mirocha J, Kahwaji J, Vo AA, Jordan SC, Dragun D. Anti‐angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation. 2010;90:1473–1477. [DOI] [PubMed] [Google Scholar]
- 56. Kuncio GS, Tsyganskaya M, Zhu J, Liu SL, Nagy L, Thomazy V, Davies PJ, Zern MA. TNF‐alpha modulates expression of the tissue transglutaminase gene in liver cells. Am J Physiol. 1998;274:G240–G245. [DOI] [PubMed] [Google Scholar]
- 57. Suto N, Ikura K, Sasaki R. Expression induced by interleukin‐6 of tissue‐type transglutaminase in human hepatoblastoma HepG2 cells. J Biol Chem. 1993;268:7469–7473. [PubMed] [Google Scholar]
- 58. Chua S, Wilkins T, Sargent I, Redman C. Trophoblast deportation in pre‐eclamptic pregnancy. Br J Obstet Gynaecol. 1991;98:973–979. [DOI] [PubMed] [Google Scholar]
- 59. Xia Y, Kellems RE. Is preeclampsia an autoimmune disease?. Clin Immunol. 2009;133:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Fu ML, Herlitz H, Wallukat G, Hilme E, Hedner T, Hoebeke J, Hjalmarson A. Functional autoimmune epitope on alpha 1‐adrenergic receptors in patients with malignant hypertension. Lancet. 1994;344:1660–1663. [DOI] [PubMed] [Google Scholar]
- 61. Luther HP, Homuth V, Wallukat G. Alpha 1‐adrenergic receptor antibodies in patients with primary hypertension. Hypertension. 1997;29:678–682. [DOI] [PubMed] [Google Scholar]
- 62. Wenzel K, Haase H, Wallukat G, Derer W, Bartel S, Homuth V, Herse F, Hubner N, Schulz H, Janczikowski M, Lindschau C, Schroeder C, Verlohren S, Morano I, Muller DN, Luft FC, Dietz R, Dechend R, Karczewski P. Potential relevance of alpha(1)‐adrenergic receptor autoantibodies in refractory hypertension. PLoS One. 2008;3:e3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wallukat G, Wollenberger A. Effects of the serum gamma globulin fraction of patients with allergic asthma and dilated cardiomyopathy on chronotropic beta adrenoceptor function in cultured neonatal rat heart myocytes. Biomed Biochim Acta. 1987;46:S634–S639. [PubMed] [Google Scholar]
- 64. Limas CJ, Goldenberg IF, Limas C. Autoantibodies against beta‐adrenoceptors in human idiopathic dilated cardiomyopathy. Circ Res. 1989;64:97–103. [DOI] [PubMed] [Google Scholar]
- 65. Fu ML. Anti‐peptide antibodies against an autoimmune epitope on human muscarinic receptor mimic functional autoantibodies against the same epitope in patients with idiopathic dilated cardiomyopathy. Eur Heart J. 1995;16(Suppl O):89–91. [DOI] [PubMed] [Google Scholar]
- 66. Fu ML, Wallukat G, Hjalmarson A, Hoebeke J. Agonist‐like activity of anti‐peptide antibodies directed against an autoimmune epitope on the heart muscarinic acetylcholine receptor. Receptors Channels. 1994;2:121–130. [PubMed] [Google Scholar]
- 67. Riemekasten G, Philippe A, Nather M, Slowinski T, Muller DN, Heidecke H, Matucci‐Cerinic M, Czirjak L, Lukitsch I, Becker M, Kill A, van Laar JM, Catar R, Luft FC, Burmester GR, Hegner B, Dragun D. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis. 2011;70:530–536. [DOI] [PubMed] [Google Scholar]