

Abstract
Background
Duchenne muscular dystrophy is a fatal cardiac and skeletal muscle disease resulting from mutations in the dystrophin gene. We have previously demonstrated that a dystrophin‐associated protein, sarcospan (SSPN), ameliorated Duchenne muscular dystrophy skeletal muscle degeneration by activating compensatory pathways that regulate muscle cell adhesion (laminin‐binding) to the extracellular matrix. Conversely, loss of SSPN destabilized skeletal muscle adhesion, hampered muscle regeneration, and reduced force properties. Given the importance of SSPN to skeletal muscle, we investigated the consequences of SSPN ablation in cardiac muscle and determined whether overexpression of SSPN into mdx mice ameliorates cardiac disease symptoms associated with Duchenne muscular dystrophy cardiomyopathy.
Methods and Results
SSPN‐null mice exhibited cardiac enlargement, exacerbated cardiomyocyte hypertrophy, and increased fibrosis in response to β‐adrenergic challenge (isoproterenol; 0.8 mg/day per 2 weeks). Biochemical analysis of SSPN‐null cardiac muscle revealed reduced sarcolemma localization of many proteins with a known role in cardiomyopathy pathogenesis: dystrophin, the sarcoglycans (α‐, δ‐, and γ‐subunits), and β1D integrin. Transgenic overexpression of SSPN in Duchenne muscular dystrophy mice (mdx TG) improved cardiomyofiber cell adhesion, sarcolemma integrity, cardiac functional parameters, as well as increased expression of compensatory transmembrane proteins that mediate attachment to the extracellular matrix.
Conclusions
SSPN regulates sarcolemmal expression of laminin‐binding complexes that are critical to cardiac muscle function and protects against transient and chronic injury, including inherited cardiomyopathy.
Keywords: cardiac hypertrophy, cell adhesion molecules, Duchenne muscular dystrophy, gene therapy
Subject Categories: Hypertrophy, Echocardiography, Gene Therapy, Genetically Altered and Transgenic Models
Introduction
Duchenne muscular dystrophy (DMD) is the most prevalent, lethal genetic disorder of childhood and the most common of the muscular dystrophies.1 Cardiomyopathy develops by the 2nd and 3rd decade of life in over 90% of cases. Genetic mutations in the dystrophin gene cause DMD, resulting in loss of dystrophin protein and the entire dystrophin‐associated glycoprotein complex (DGC).1, 2 The dystrophin gene encodes a large protein (427 kDa) with spectrin‐like repeats that is localized to the sarcolemma of skeletal and cardiac muscles.1, 2 The DGC is a laminin‐binding adhesion complex that confers structural linkage between the extracellular matrix and the intracellular actin cytoskeleton, thereby providing protection to the sarcolemma from contraction‐induced injury.2, 3 Loss of the DGC reduces attachment of the myofiber to its surrounding extracellular matrix and renders the myofiber susceptible to contraction‐induced damage, eventually leading to muscle degeneration.3, 4, 5, 6
The connection between cardiac disease and DMD has been long recognized. Mutations linked to DMD are thought to first exert their effects as skeletal muscle weakness with a subsequent loss of ambulation, whereas cardiac decline occurs in later decades of life. In contrast, X‐linked cardiomyopathy, caused by mutations in the dystrophin gene, is a distinct form of heart disease identified as a rapidly progressive primary myocardial disorder that manifests in teenage males as congestive heart failure. Female carriers (one copy of the mutated dystrophin gene on the X chromosome) exhibit cardiac disease with later onset (fifth decade of life) and typically slower progression.7 The mechanisms governing differential expression of X‐linked cardiomyopathy mutations in the DMD gene remain unclear. Linkage analysis identified genetic variants in the dystrophin gene (DMD), which segregated with X‐linked dilated cardiomyopathy without identifiable skeletal muscle abnormalities in 2 separate families.8, 9 A characteristic of X‐linked dilated cardiomyopathy is that the mutated dystrophin transcript arises primarily in cardiac tissue, with nearly normal to normal dystrophin transcripts present in other tissue types. A search for distinct regions of the dystrophin gene associated with primary cardiac involvement revealed a number of mutations in the extreme 5′ end of the dystrophin gene that give rise to X‐linked dilated cardiomyopathy.9, 10
Dystrophin is localized to the subsarcolemma, where it associates with a transmembrane glycoprotein, β‐dystroglycan (DG), which in turn associates with the extracellular receptor for laminin, α‐dystroglycan (DG).1, 2 Dystrophin and α/β‐DG form a larger complex by interactions with 4 integral membrane sarcoglycan subunits (α‐, β‐, δ‐ and γ‐SGs). Genetic mutations in the genes encoding α‐, β‐, δ‐ and γ‐SG cause autosomal recessive limb‐girdle muscular dystrophy (AR‐LGMD) type 2D, 2E, 2C, and 2F, respectively.11, 12, 13, 14, 15 Severe cardiac manifestations, including dilated cardiomyopathy, are evident in cases of AR‐LGMD‐type 2E, 2C, and 2F, which are caused by mutations in β‐ δ‐, and γ‐SG genes, respectively. In sarcoglycanopathies, loss of one sarcoglycan leads to loss of all 4 sarcoglycan components from the sarcolemma.16 Cardiomyopathy is rare in AR‐LGMD type 2D, as loss of α‐SG is likely compensated by increased expression of ε‐SG.17, 18, 19 Membrane instability is emerging as a critical initiating event in DMD and sarcoglycan‐deficient AR‐LGMD; therefore, mechanisms promoting restoration of cell surface–extracellular matrix connections have great potential to alleviate muscle damage and restore function. Sarcoglycanopathies are additionally characterized by an infarct pattern of fibrosis attributed to vasospasm.20, 21, 22
The abundance of the utrophin–glycoprotein complex (UGC), a laminin‐binding complex analogous to the DGC, is increased in DMD muscle to partially compensate for the loss of the DGC.23, 24, 25, 26, 27, 28 Transgenic overexpression studies have demonstrated that further increasing the levels of the UGC can fully ameliorate DMD disease, and testing of compounds that increase utrophin transcription activity is currently under way.23 It is unknown whether increased abundance of utrophin ameliorates skeletal and cardiac disease in sarcoglycan‐deficient AR‐LGMD. Additionally, recent studies have revealed that overexpression of α7β1 integrin ameliorates DMD in relevant murine models by promoting muscle cell adhesion in a manner similar to utrophin.29, 30, 31 Similarly, integrin loss contributes to cell membrane instability and is associated with increased fibrosis that compromises cardiac muscle function and increases susceptibility to arrhythmias.32, 33 Under pathological conditions, integrins colocalize to the borders of cardiac fibrosis.34, 35
Sarcospan (SSPN) is a tetraspanin‐like protein that facilitates protein interactions between transmembrane proteins, and associates with the 3 major adhesion complexes in skeletal muscle including the DGC, UGC, and α7β1 integrin.36, 37, 38 This unique biochemical property of SSPN makes it an attractive therapeutic target for the muscular dystrophies. Transgenic overexpression of SSPN increased the levels of dystrophin at the sarcolemma in wild‐type skeletal muscle. In dystrophin‐deficient murine models of DMD (mdx mice), SSPN increased expression of the UGC and α7β1D integrin at the mdx sarcolemma, leading to restoration of laminin‐binding and amelioration of skeletal muscle disease.39, 40 Recent studies have revealed that SSPN also mediates efficient skeletal muscle regeneration after transient injury by regulating intracellular Akt signaling pathways that control these processes.39, 40 Lebakken and colleagues initially reported that SSPN ablation did not alter DGC protein abundance or skeletal muscle function in young mice.41 However, investigation of older SSPN‐null mice revealed an increased susceptibility to eccentric contraction‐induced damage due to reduction of DGC and UGC at the sarcolemma.42 Since SSPN is fundamental to maintenance of cellular integrity of skeletal muscle, we investigated its role in SSPN‐deficient cardiac muscle in the current study and tested the ability of SSPN to prevent DMD‐associated cardiomyopathy.
Methods
Animal Models
All mice used in this study were males and different ages are listed in the figure legends. The SSPN‐deficient mice used in this study were a generous gift from Dr Kevin P. Campbell (University of Iowa Medical School, Iowa City, IA). SSPN‐null mice were maintained using a SSPN‐null×SSPN‐null breeding strategy. Male WT C57BL/6J mice were maintained and age matched with SSPN‐null progeny to generate age‐matched controls. Mice were genotyped at various time points to ensure correct genetics. Generation and characterization of human SSPN‐transgenic mdx TG mice have been described previously by our group. The breeding strategy employed was mdx:human SSPN (Line 3) TG male×mdx female crossings. Age‐matched mdx mice that resulted from this cross were used as controls. Mice were maintained in the Terasaki Life Sciences Vivarium, and all procedures followed guidelines established by the Institutional Animal Care and Use Committee at the University of California, Los Angeles; approval for these studies was granted by the UCLA Animal Welfare Assurance.
Histochemical and Immunofluorescence Assays
H&E staining was used to visualize changes in tissue architecture including myocyte disarray, hypertrophy, and fibrosis. Longitudinal sections of fresh frozen cardiac tissue (7 μm) were acclimated to room temperature for 15 minutes before staining and stained with H&E obtained from Thermo Fisher Scientific and according to previously published methods.43 Masson's Trichrome staining was utilized for collagen detection in cardiac tissue according to described methods.44 Cardiac section images were captured under identical conditions using an Axioplan 2 fluorescent microscope equipped with a Plan Neofluar 40× NI 1.3 oil differential interference contrast objective and the Axiovision Rel 4.5 software (Carl Zeiss, Inc). Average fiber area measurements were performed by manually measuring the area of fiber bundles in transversely cut cardiac sections using Thermo Scientific myImage Analysis v1.1 software. A similar procedure was used to measure fibrotic areas compared relative to total area (%). Immunofluorescence assays were performed and visualized as described.39
Protein Preparations and Immunoblot Analysis
Cardiac muscle was snap‐frozen in liquid nitrogen and stored immediately at −80°C, and tissue samples were prepared as described for immunoblot analysis. Prior to tissue collection, mice were euthanized using isoflurane administered using a nose cone. For the DG pulldown assays, cardiac muscle was extracted in 0.1% digitonin and adhesion complexes were enriched by wheat germ agglutinin (sWGA) and eluted with 0.3 mol/L N‐acetylglucosamine (Sigma‐Aldrich).42 Total protein samples were prepared in ice‐cold radioimmunoprecipitation assay buffer; 89901 (Thermo Scientific) along with phosphatase inibitors. Equal concentrations of samples were resolved by SDS‐PAGE (4–20% PreciseTM Protein Gels) and transferred to nitrocellulose membranes. Immunoblots were performed using 30 μg of sWGA eluates or 60 μg of radioimmunoprecipitation assay lysate. The following primary antibodies were used: dystrophin (MANDYS1, 1:200; Development Studies Hybridoma Bank), Utr (MANCHO3, 1:50; Development Studies Hybridoma Bank), α‐DG IIH6 (sc‐53987, 1:500; Santa Cruz Biotechnology, Inc), β‐DG (MANDAG2, 1:250; Development Studies Hybridoma Bank), α‐SG (VP‐A105, 1:100; Vector Labs), β‐SG (VP‐B206, 1:100; Vector Labs), δ‐SG (VP‐D501, 1:50; Vector Labs), γ‐SG (VP‐G803, 1:100; Vector Labs), β1D integrin (MAB1900, 1:200; Chemicon), mouse SSPN (R18, 1:100; in house), human SSPN (R15; in house), and GAPDH (MAB374, 1:50 000; Chemicon). Coomassie blue staining was used as an additional loading control. Horseradish peroxidase–conjugated anti‐rabbit IgG (GE Healthcare), anti‐mouse IgG (GE Healthcare) were used at 1:2000 dilutions in 5% BLOTTO and incubated at room temperature for 3 hours. Blots were developed using enhanced chemiluminescence (SuperSignal West Pico Chemiluminescent Substrate; Thermo Scientific). Densitometry was performed using Thermo Scientific myImage analysis, and the average integrated density values of the protein of interest were determined and normalized to controls.
Quantitative Reverse Transcription–Polymerase Chain Reaction
Total RNA was extracted from hearts of 12‐month‐old mice using TRIzol® Reagent (Ambion) according to manufacturer's instructions, RNA concentrations were determined using the NanoDrop 1000 (Thermo Scientific), and reverse transcription was performed with 0.5 μg RNA using the iScript cDNA Synthesis kit (Bio‐Rad Laboratories, Inc). Changes in relative gene expression were quantified using the 2∆∆ CT method and normalized to an internal control mL32 (a ribosomal protein that does not vary in conditions tested here). Quantitative reverse transcription‐–polymerase chain reaction was performed in duplicate per sample using the TaqMan ABI PRISM 7900 (Applied Biosystems) according to the instructions provided by in the SsoFastTM EvaGreen® Kit (Bio‐Rad Laboratories, Inc).
Evans Blue Dye Uptake
Sarcolemmal membrane damage was assessed using an Evans blue dye (EBD) tracer analysis. Mice were injected peritoneally with EBD and maintained for 12 hours prior to euthanasia by isoflurane administration. The percentage of EBD‐positive fibers (fluoresce red) against laminin counterstain (green) was obtained by quantitating the number of EBD‐positive myofibers/total number of myofibers. Sarcolemmal membrane damage was assessed using an EBD tracer analysis that was performed by intraperitoneal injection of 1 g EBD/10 g body weight in 12‐ to 14‐month‐old mice, 8 hours prior to dissection.
Laminin Overlay
A nitrocellulose membrane was prepared as described in the immunoblot analysis methods and sWGA enriched eluates were blocked with 5% bovine serum albumin (BSA) in laminin binding buffer (LBB; 10 mmol/L triethanolamine, 140 mmol/L NaCl, 1 mmol/L MgCl2, 1 mmol/L CaCl2, pH 7.6) then incubated with 20 nmol/L mouse ultrapure Engelbreth–Holm–Swarm laminin (354239, BD Biosciences) in laminin binding buffer for 6 hours at 4°C. Subsequently, the membranes were washed in laminin binding buffer‐tween (LBBT) and incubated with primary rabbit anti‐laminin (L9393, 1:5000; Sigma) secondary horseradish peroxidase–conjugated anti‐rabbit IgG (GE Healthcare) and blots were developed using standard techniques.
Creatine Kinase Measurements
The Stanbio CK‐MB Liqui‐UV® kit was used to quantitatively determine levels of CK‐MB (cardiac iso‐enzyme) in the blood serum of wild‐type, mdx and mdx TG mice following procedures outlined by the manufacturer. Blood was collected from mice retroorbitally in red‐cap blood collection tubes (no anticoagulant) and was allowed to clot at room temperature for 30 minutes and spun for 10 minutes at 2000g in a refrigerated centrifuge. The sera were collected and immediately stored at −80°C. A Molecular Devices Spectra Max (ML) plate reader was used and absorbance readings were taken at 340 nm and 1‐cm light path. The following equation was used to determine % CK‐MB Activity=CK‐MB Activity (U/L)×100/Total CK activity (U/L).
Echocardiography
Left ventricular (LV) size, mass, wall thickness, ventricular and valve function, and Doppler blood flows were obtained using methods previously described and animals were anesthesized using inhaled isoflurane.45, 46 Mice were lightly sedated with isoflurane vaporized in oxygen (2.5% induction, 1.0% maintenance), weighed, and their chests were shaved. The mice were positioned in the left lateral decubitus position for ultrasonic imaging with an Acuson Sequoia C256 equipped with a 15L8 15 MHz probe (Siemens Medical Solutions, Malvern, PA). Needle electrodes attached to the extremities allowed simultaneous recording of the ECG to determine timing and heart rates. Also, the long‐axis, short‐axis, and apical views were used to obtain 2‐dimensional, M‐mode, and spectral Doppler images at physiological heart rates (500–600 bpm). Measurements were made using the Acuson and AccessPoint software (Freeland Systems, LLC, Santa Fe, NM).
Chronic β‐Adrenergic Stimulation
Mice were anesthetized with isoflurane and miniosmotic pumps (Alzet, Durect Corp, Cupertino, CA), filled with sterile saline (sham) or isoproterenol, were implanted subcutaneously to challenge the myocardium with isoproterenol (0.8 mg/day) (dosage determined empirically) for 14 days, and mice were monitored by echocardiography.47 The dose and length of experiment were chosen that allowed changes in cardiac function but not high enough to cause lethality or β‐receptor desensensitization. The sham‐ and isoproterenol‐treated mice were echoed prior to pump implantation and at the end of the study to identify any ventricular remodeling and functional changes from baseline.
Statistics
All values in the text and figures are presented as mean±SEM. The statistical significance of the differences between means was assessed using Sigma Plot 13.0 software. One‐way ANOVA with Bonferroni post hoc test was used to determine significance among different treatment groups. Data that failed the assumptions of normality were analyzed using the nonparametric Kruskal–Wallis test. The statistical test utilized for each set of data is indicated in the figure legends. P values of <0.05 were considered significant, and significant and near‐significant P values are reported in the figure legends.
Results
In skeletal muscle, ablation of SSPN significantly reduces skeletal muscle function as determined by lowered specific diaphragmatic force production in SSPN‐null mice. The decreased force between the first and the fifth eccentric contraction observed in SSPN‐null diaphragms was evidence that loss of SSPN weakens the sarcolemma.42 Mechanistic studies revealed that SSPN‐deficiency reduced abundance of laminin‐binding adhesion complexes at the sarcolemma, rendering the myofiber susceptible to contraction‐induced injury.42 Based on these findings, we tested the hypothesis that SSPN deficiency would negatively impact cardiac function.
Cardiac Morphology in Isoproterenol‐Treated SSPN‐Null Mice
Tetraspanin‐like proteins, such as SSPN, often reveal their dysfunction when subjected to conditions of chronic stress.48 This study was founded upon an initial observation that 12‐month‐old SSPN‐null mice exhibited cardiac enlargement compared to wild‐type mice. To further investigate the effects of SSPN deficiency on cardiac morphology, we exposed 12‐month‐old SSPN‐null mice to a 2‐week β‐adrenergic challenge using continual administration of isoproterenol (0.8 mg/day) via osmotic pump. Comparison of body weights revealed that SSPN‐null mice were heavier after isoproterenol‐treatment compared to wild‐type mice (Figure 1A, left panel). Both isoproterenol and saline‐treated SSPN‐null mice exhibited significantly greater heart weight/body weight ratios (HWt/BWt; mg/g) compared to the relevant wild‐type controls (Figure 1A, right panel). After saline administration, HWt/BWt ratios for wild‐type and SSPN‐null mice were 5.32±0.67 and 6.58±0.27 mg/g, respectively, whereas after isoproterenol treatment, they increased to 6.63±0.58 and 7.41±0.64 mg/g for wild‐type and SSPN‐null mice, respectively. We observed that SSPN‐null hearts were larger than wild‐type hearts after isoproterenol treatment, as visualized in wet‐mount samples (Figure 1B).
Figure 1.
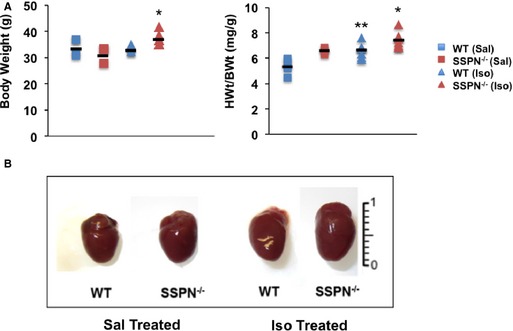
Cardiac morphology is altered in isoproterenol‐treated SSPN‐null mice. Mice in this study were designated as saline‐treated (Sal) or isoproterenol‐treated (Iso) for both SSPN‐null (SSPN −/−) and WT mice. All mice were 12 months of age. A, BWts of WT and SSPN −/− mice after saline‐ or isoproterenol‐treatment and the HWt/BWt ratio (mg/g) were compared for saline‐ and isoproterenol‐treated mice. Data are shown as mean and error expressed as SEM. B, Representative wet mount images of hearts from treated and untreated WT and SSPN −/− mice used in this study. In (A) BWt (g) (left panel) and HWt/BWt (right panel) measurements were plotted for saline (Sal, WT, n=4 and SSPN −/− mice, n=3) and isoproterenol (Iso, both WT and SSPN −/− mice, n=7) treatment for each genotype after completion of the 2‐week study. P‐values <0.05 were considered significant and statistics were calculated using 1‐way ANOVA with Bonferroni post hoc test. For BWt (g) left panel, *P=0.022 SSPN −/− (Sal) vs SSPN −/− (Iso), P=0.115 SSPN −/− (Iso) vs WT (Iso), P=0.408 SSPN −/− (Iso) vs WT (Sal). For HWt/BWt (mg/g) right panel, *P<0.001 SSPN −/− (Iso) vs WT (Sal), **P=0.016 WT (Iso) vs WT (Sal), P=0.363 SSPN −/− (Iso) vs SSPN −/− (Sal), P=0.149 SSPN −/− (Iso) vs WT (Iso). HWt/BWt indicates heart weight/body weight; SSPN, sarcospan; WT, wild‐type.
Cardiac Dysfunction in SSPN‐Null Hearts
Echocardiography was used to evaluate cardiac function of SSPN‐null mice relative to controls. All parameters at baseline and after treatment are summarized in Table 1. Increasing age further exacerbated hypertrophy in SSPN‐null hearts; at 18 months, SSPN‐null hearts (95.89±6.71 mg) exhibited greater left ventricular mass (LV mass) relative to age‐matched wild‐type controls (67.18±2.89 mg) (Figure 2A). The LV mass of wild‐type mice remained unchanged between 12 and 18 months of age (12 months, 67.54±3.76 mg; 18 months, 67.18±2.89 mg). LV mass was significantly increased in 12‐month‐old SSPN‐null mice after isoproterenol treatment (116.1±12.7 mg) compared with saline‐administered controls (82.6±18 mg) as depicted in Figure 2B. However, saline‐administered SSPN‐null and wild‐type mice had similar LV masses at 12 months of age.
Table 1.
Summary of Echocardiographic Data Obtained From β‐Adrenergic Stimulation of WT and SSPN‐null Mice
| Baseline | Saline | Isoproterenol | ||||
|---|---|---|---|---|---|---|
| WT (n=7) | SSPN−/− (n=7) | WT (n=4) | SSPN−/− (n=4) | WT (n=7) | SSPN−/− (n=7) | |
| ECHO parameter | ||||||
| HR, bpm | 508.6±24.6 | 526.3±15.6 | 517.64±21.1 | 535.19±30.8 | 626.98±12.7§ | 598.1±21.7 |
| LVEDD, mm | 4.10±0.11 | 4.22±0.08 | 4.08±0.18 | 4.28±0.16 | 4.23±0.26 | 4.76±0.11†, || |
| LVESD, mm | 2.74±0.10 | 2.95±0.09* | 2.83±0.23 | 2.83±0.12 | 2.37±0.23 | 3.3±0.08† |
| LVEF, % | 68.60±2.04 | 62.78±2.44 | 64.5±4.8 | 68.6±4.25 | 80.6±2.61§ | 64.5±1.4† |
| LV % FS | 33.31±1.44 | 30.00±1.61 | 30.9±3.7 | 34.03±2.5 | 44.4±2.72§ | 30.0±0.91† |
| Vcf, % | 6.41±0.49 | 6.08±0.34 | 6.4±0.85 | 7.1±0.65 | 10.2±0.6§ | 6.5±0.64† |
| MV E, mm/s | 0.71±0.08 | 0.81±0.08 | 0.89±0.1 | 0.92±0.1 | 1.08±0.08 | 0.96±0.04 |
| MV A, mm/s | 0.44±0.04 | 0.50±0.04 | 0.61±0.1 | 0.64±0.1 | 0.58±0.04 | 0.43±0.08 |
| MV E/A | 1.6±0.11 | 1.6±0.08 | 1.4±0.35 | 1.5±0.1 | 1.9±0.26 | 2.3±0.02† |
| PWT, mm | 0.48±0.01 | 0.49±0.02 | 0.53±0.04 | 0.54±0.03 | 0.56±0.02 | 0.63±0.04† |
| IVS, mm | 0.51±0.0 | 0.51±0.02 | 0.54±0.04 | 0.57±0.03 | 0.56±0.01 | 0.63±0.001‡ |
| LVmass, mg | 66.4±3.63 | 70.85±2.31 | 68.5±3.2 | 82.8±9.0 | 82.6±8.73§ | 116.1±4.80#, †, ‡ |
Data are mean±SE. A indicates atrial peak flow velocity; E, early peak flow velocity; FS, fractional shortening; HR, heart rate; IVS, interventricular septum thickness; LVEDD, left ventricular end diastolic dimension; LVEF, left ventricular ejection fraction; LVESD, left ventricular end systolic dimension; MV, mitral valve; PWT, posterior wall thickness; SSPN, sarcospan; SSPN−/−, SSPN‐null; Vcf, velocity of circumferential shortening; WT, wild‐type.
One‐way ANOVA with Bonferroni correction was used to calculate significance, *P<0.05 (comparison between baseline wild‐type and SSPN‐deficient mice), †P<0.05 (comparison between isoproterenol treated wild‐type and SSPN‐null mice), ‡P<0.05 (comparison between saline treated and isoproterenol treated SSPN‐null mice), §P<0.05 (comparison between saline treated and isoproterenol treated WT mice). ||P<0.05 (comparison between saline treated wild‐type and isoproterenol‐treated SSPN‐null mice). #P<0.05 (comparison between saline treated SSPN‐null and isoproterenol‐treated wild‐type mice). Values reported as averages and errors presented as SEM.
Figure 2.
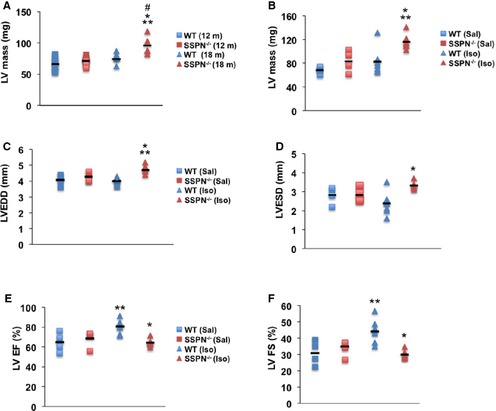
Cardiac dysfunction in SSPN‐null mice in response to isoproterenol treatment. Echocardiographic measurements change in mice as they age, (A) LV mass for 12‐ and 18‐month‐old WT and SSPN −/− mice. All mice used in (B through F) were 12 months of age. B, Echocardiographic measurements of LV mass, mg were obtained after saline‐ and isoproterenol‐administration to WT and SSPN‐null (SSPN −/−) mice. C, Increased left ventricle dimension in millimeters (mm) in SSPN‐null (SSPN −/−) compared to WT mice during diastole (LVEDD) and (D) systole (LVESD) measured by echocardiography after isoproterenol challenge. E, Echocardiographic measurements of LV EF% and (F) LV FS% are shown for WT and SSPN‐null (SSPN −/−) mice after saline‐ and isoproterenol treatment. For (A), 12‐month‐old WT and SSPN‐null (SSPN −/−) mice (each, n=11) and 18‐month‐old WT and SSPN‐null (SSPN −/−) mice (each, n=5) were compared. For (B through F) samples are compared for saline (Sal, n=4) and isoproterenol (Iso, n=6) treatment for each genotype. The data demonstrate the range of values, the black line symbol indicates the mean of each data set. * P‐values <0.05 were considered significant, and statistics were calculated using 1‐way ANOVA with Bonferroni post hoc test unless otherwise noted. For (A) *P<0.001 SSPN −/− (18) vs WT (12), **P<0.001 SSPN −/− (18) vs SSPN −/− (12), # P=0.016 SSPN −/− (18) vs WT (18), P=0.340 WT (18) vs WT (12); (B) *P=0.006 SSPN −/− (Sal) vs SSPN −/− (Iso), the Kruskal–Wallis test was utilized for **P=0.009 WT (Sal) vs SSPN −/− (Iso), P=0.086 SSPN −/− (Iso) vs WT (Iso); (C) *P<0.001 SSPN −/− (Iso) vs WT (Iso), **P=0.006 SSPN −/− (Iso) vs WT (Sal), P=0.076 SSPN −/− (Sal) vs SSPN −/− (Iso); (D) *P=0.006 SSPN −/− (Iso) vs WT (Iso); (E) *P=0.002 SSPN −/− (Iso) vs WT (Iso), **P=0.010 WT (Iso) vs WT (Sal), P=0.077 WT (Iso) vs SSPN −/− (Sal) and (F) *P=0.001 SSPN −/− (Iso) vs WT (Iso), **P=0.008 WT (Iso) vs WT (Sal), P=0.057 SSPN −/− (Sal) vs WT (Iso). LV indicates left ventricular; LVEDD, left ventricular end diastolic dimension; LV EF, left ventricular ejection fraction; LVESD, left ventricular end systolic dimension; LV FS, left ventricular fractional shortening; SSPN, sarcospan; WT, wild‐type.
Echocardiography revealed additional parameters that were negatively impacted in SSPN‐null mice after isoproterenol treatment. Posterior wall thickness and interventricular septum thickness were elevated in treated SSPN‐nulls relative to treated wild‐type controls (Table 1). Additionally, the left ventricular end diastolic dimension of isoproterenol‐treated SSPN‐null hearts was statistically increased relative to saline‐treated SSPN‐nulls, while isoproterenol did not affect left ventricular end diastolic dimension (LVEDD) from wild‐type mice (Figure 2C). Upon isoproterenol treatment, SSPN‐null hearts exhibited greater left ventricular end systolic dimension (LVESD) compared with wild‐type hearts, which responded to isoproterenol treatment in a predictable manner with a decrease in LVESD (Figure 2D). Specifically, treated SSPN‐null hearts displayed left ventricular end systolic dimension values of 3.3±0.2 mm compared with isoproterenol treated wild‐type control values of 2.37±0.6 mm (Table 1). Representative M‐mode images (Figure S1) showed enlargement of the left cardiac chamber in SSPN‐nulls after isoproterenol stimulation. Overall, cardiac function in SSPN‐null mice was significantly altered after isoproterenol treatment, evident as LV hypertrophy and dilatation.
LV function following β‐adrenergic stimulation was assessed in 12‐month‐old SSPN‐null and wild‐type mice using echocardiography. As expected, isoproterenol increased LV contractility, as revealed by ejection fraction values (LVEF) in hearts of wild‐type mice. Specifically, LVEF values for isoproterenol‐treated wild‐type mice was 80.6±6.9% compared with saline‐administered wild‐type controls (64.5±9.6%). Baseline LVEF values were similar for wild‐type and SSPN‐nulls (Table 1). In obvious contrast to the wild‐type response to isoproterenol, SSPN‐null hearts exhibit an attenuated contractile response to isoproterenol, as revealed by LVEF (%) values of 64.5±3.8% (Figure 2E). Echocardiographic analysis also revealed large differences in LV fractional shortening (LVFS) in SSPN‐nulls upon isoproterenol challenge. In response to β‐receptor agonists, wild‐type hearts responded with increased LVFS as evidenced by values after saline treatment (30.9±7.4%) and isoproterenol treatment (44.4±7.2%) (Figure 2F). However, we observed that SSPN‐null mice responded aberrantly to isoproterenol administration, evident by the depressed (LVFS) (Sal, 34.03±5.0%; Iso, 30.0±2.4%). Despite these changes, SSPN‐nulls maintain cardiac function as shown by the preserved ejection fraction and modest decreases in fractional shortening (Figure 2E and 2F). Furthermore, isoproterenol treatment affected diastolic dysfunction, as revealed by E/A ratios (early peak/atrial peak flow velocities) that were significantly increased after isoproterenol stimulation in SSPN‐null hearts compared with wild‐type (Table 1).
Isoproterenol Treatment Accelerates Cardiac Pathology in SSPN‐Null Mice
Analysis of cardiac muscle cryosections from 12‐month‐old SSPN‐null mice revealed significant interstitial and extensive replacement fibrosis after isoproterenol administration (Figure 3A). Fibrosis in isoproterenol‐treated wild‐type hearts was not as extensive (Figure 3A). Masson's trichrome staining further supported widespread collagen deposition in SSPN‐null hearts (Figure 3B). Quantification of fibrosis in cross‐sections from isoproterenol‐treated SSPN‐null hearts demonstrated that 12% of the total area was fibrotic, whereas wild‐type hearts exhibited 4.6% fibrosis (Figure 3C). In saline‐treated SSPN‐nulls, ≈2% of the total area was fibrotic compared to ≈0.5% total area for wild‐type (Figure 3C). Additionally, we found that the cross‐sectional areas of fibers were differentially affected by isoproterenol treatment in SSPN‐nulls (Figure 3D and 3E).
Figure 3.
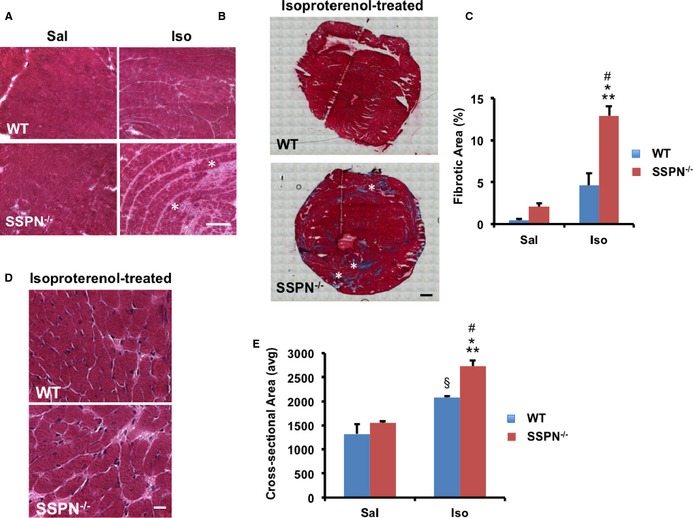
Isoproterenol treatment alters cardiac architecture of SSPN‐null mice. A, Transverse cardiac sections from 12‐month‐old WT and SSPN‐null (SSPN −/−) mice were stained with H&E, revealing myocyte disarray, hypertrophy, and fibrosis in SSPN‐nulls. White asterisks indicate focal fibrotic regions, Bar, 50 μm. B, Masson's trichrome staining identifies regions of collagen deposition (dark blue) in cardiac cross‐sections (axial, short axis). White asterisks indicate focal fibrotic regions. Bar, 900 μm. C, Fibrosis was quantified by measuring the area of collagen staining in H&E‐stained whole heart cross‐sections after saline (Sal) and isoproterenol (Iso) administration. Data are represented as percentage of fibrosis per total area. D, Enlarged H&E cardiac images reveal myofiber hypertrophy in SSPN‐nulls after isoproterenol treatment. Bar, 50 μm. E, Cardiac myofiber cross‐sectional area was quantified after saline and isoproterenol challenge. H&E stained panels highlight fiber size differences in WT and SSPN −/− mice. For (C and E), data are presented as an average from 12‐month‐old, WT mice and SSPN‐null (SSPN −/−) that were saline‐ (Sal) (n=3) and isoproterenol‐treated (Iso) (n=3). Statistical analysis was performed using 1‐way ANOVA with Bonferroni post hoc test and *P‐values <0.05 were considered significant. For (C) *P<0.001 SSPN −/− (Iso) vs WT (Sal), **P<0.001 SSPN −/− (Iso) vs SSPN −/− (Sal), # P=0.002 SSPN −/− (Iso) vs WT (Iso), P=0.093 WT (Iso) vs WT (Sal) and (E) *P<0.001 SSPN −/− (Iso) vs WT (Sal), **P<0.001 SSPN −/− (Iso) vs SSPN −/− (Sal), # P=0.034 SSPN −/− (Iso) vs WT (Iso), § P=0.013 WT (Iso) vs SSPN −/− (Sal), P=0.091 WT (Iso) vs WT (Sal). H&E indicates hematoxylin and eosin; SSPN, sarcospan; WT, wild‐type.
SSPN‐Null Mice Are Responsive to β‐Adrenergic Stimulation
To determine whether the reduction of β‐adrenergic responsiveness to isoproterenol in SSPN‐null mice was caused by β‐adrenergic receptor down regulation, quantitative reverse transcription–polymerase chain reaction analysis was performed in 12‐month‐old untreated and isoproterenol‐treated (wild‐type and SSPN‐null) mice. Unlike wild‐type mice, SSPN‐null mice did not exhibit enhanced cardiac contractility (LVEF and LV fractional shortening, Figure 2E and 2F). We found similar levels of β1, β2, β3 adrenergic receptor expression in isoproterenol‐treated SSPN‐null hearts compared with wild‐type (Figure 4A through 4C). In summary, SSPN‐null hearts appear to respond normally to isoproterenol treatment in their upregulation of β‐adrenergic receptor mRNAs.
Figure 4.
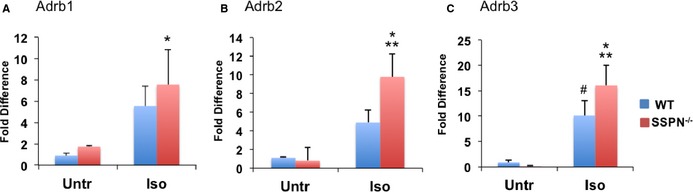
SSPN‐null mice have a blunted response to β‐adrenergic stimulation. Relative changes in gene expression changes of β‐adrenergic receptors (β1, β2, and β3) upon isoproterenol challenge were assessed by qRT‐PCR in cardiac muscle obtained from untreated (Untr) and isoproterenol‐treated (Iso) 12‐month‐old WT and SSPN‐null (SSPN −/−) mice. A, Relative gene expression values of the ADRB1 gene encoding (β‐AR 1). (B) Relative gene expression values for the ADRB2 gene encoding (β‐ΑR 2). (C) Relative gene expression values for the ADRB3 gene encoding (β‐AR 3) receptors in untreated (Untr) and isoproterenol‐treated (Iso) WT and SSPN −/− hearts. Data were normalized to mL32 (a ribosomal protein) as an internal control and are presented as an average. All mice were 12 months of age. Data are presented as an average from WT and SSPN‐null (SSPN −/−) mice, either saline (Sal) (n=6) or isoproterenol‐treated (Iso) (n=6) and error bars represent SEM. The Kruskal–Wallis test was applied since data did not meet assumptions of normality and *P‐values <0.05 were considered significant. For (A) *P=0.044 SSPN −/− (Iso) vs WT (Sal), P=0.081 SSPN −/− (Iso) vs SSPN −/− (Sal), P=0.177 WT (Iso) vs WT (Sal); (B) *P=0.005 SSPN −/− (Iso) vs SSPN −/− (Sal), **P=0.020 SSPN −/− (Iso) vs WT (Sal), P=0.091 WT (Iso) vs WT (Sal) and (C) *P=0.014 SSPN −/− (Iso) vs SSPN −/− (Sal), **P=0.018 SSPN −/− (Iso) vs WT (Sal), # P=0.049 WT (Iso) vs SSPN −/− (Sal), P=0.064 WT (Iso) vs WT (Sal). qRT‐PCR indicates quantitative reverse transcription–polymerase chain reaction; SSPN sarcospan; WT, wild‐type.
SSPN Deficiency Reduces Sarcolemmal Content of Cardiomyopathy‐Associated Proteins
SSPN associates with dystrophin, the dystroglycans, and the sarcoglycans to form the DGC, which serves as a laminin‐binding receptor.36 In skeletal muscle, loss of SSPN reduced expression of known disease genes, including dystrophin and certain sarcoglycans, that encode proteins involved in muscle cell adhesion, to the extracellular matrix.39 To determine whether SSPN ablation in cardiac muscle similarly affects these proteins, we examined DGC protein expression using indirect immunofluorescence of cardiac cryosections obtained from 4‐month‐old wild‐type and SSPN‐null mice. We found that loss of SSPN reduced levels of dystrophin and sarcoglycan subunits (α‐, β‐, δ‐, γ‐ and SG) (Figure 5A), and thereby affects localization of sarcolemmal adhesion complexes, which may contribute to muscle pathology (Figure 5A). Even though dystrophin levels were slightly increased in both wild‐type and SSPN‐null hearts upon isoproterenol treatment, SSPN‐null hearts still exhibited reduced dystrophin relative to wild‐type (data not shown).
Figure 5.
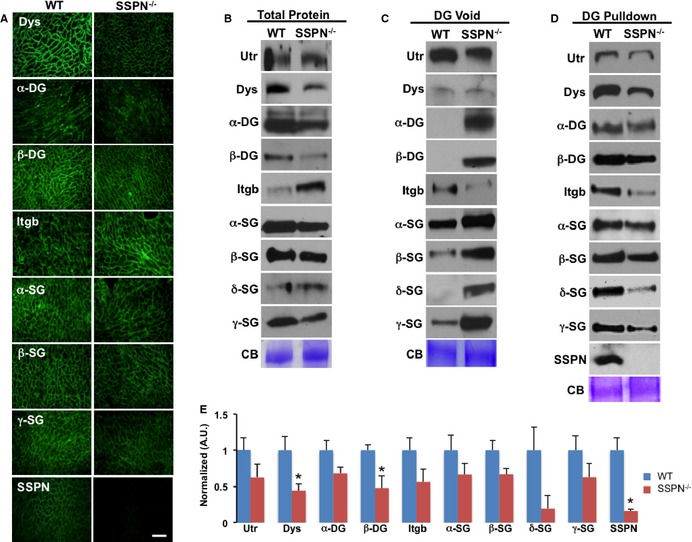
Loss of SSPN influences expression of cardiomyopathy genes. (A) Transverse cardiac cryosections from WT and SSPN‐null mice (SSPN −/−) stained with antibodies against dystrophin (Dys), utrophin (Utr), dystroglycans (α‐ and β‐DG), β1D‐integrin (Itgb) and sarcoglycans (α‐, β‐, δ‐ and γ‐SGs), and SSPN. Bar, 50 μm. B, Total protein lysates obtained from WT and SSPN‐null (SSPN−/−) mice were immunoblotted for DGC and associated proteins. Equal protein samples (60 μg) were resolved by SDS‐PAGE and immunoblotted with the indicated antibodies. DG pulldown assay, whole heart lysates were enriched by sWGA lectin chromatography, which binds DG and allows biochemical determination of associations between members of DGC‐associated complexes with DG, core components, in the presence or absence of SSPN. In (C) immunoblots of DG void samples are shown and in (D) immunoblots of eluates containing DG associated proteins are shown. CB staining is shown as a loading control. Densitometry was used to quantify immunoblots shown in (D) to determine relative changes in DGC and associated proteins in SSPN‐null (SSPN−/−) mice compared to WT and reported as normalized arbitrary units. All mice were 4 months old and similar results were obtained for 12‐month‐old mice (data not shown). Immunoblot data are presented as an average (n=5) for WT and SSPN‐null (SSPN −/−), except for δ‐SG (n=3) for WT and SSPN‐null (SSPN −/−), and error bars represent SEM. * P‐values <0.05 were considered significant, and statistics were calculated using 1‐way ANOVA. For comparisons of protein expression levels in WT vs SSPN −/− mice in (E) *P=0.003 Dys, *P=0.04 β‐DG, *P=0.009 SSPN, P=0.09 Utr, P=0.08 α‐DG, P=0.120 Itgb, P=0.227 α‐SG, P=0.065 β‐SG, P=0.326 δ‐SG, P=0.208 γ‐SG. CB indicates Coomassie blue; DGC, dystrophin‐glycoprotein complex; Itgb, β1D‐integrin; SDS‐PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; SSPN, sarcospan; sWGA, succinylated wheat germ agglutinin; WT, wild‐type.
To determine whether SSPN loss affects expression of adhesion complexes, we performed immunoblots examining total protein levels (Figure 5B). The overall levels of utrophin, dystrophin, α‐/β‐DG, and β‐SG were slightly reduced in SSPN‐null muscle compared to wild‐type (Figure 5B). β1D integrin expression was increased in the SSPN‐null hearts (Figure 5B), similar to that reported in skeletal muscle.42 Overall, total sarcoglycan expression levels were unchanged in SSPN‐null hearts compared to wild‐type.
To quantitate expression of laminin‐binding adhesion complexes in SSPN‐null hearts, we performed a DG pulldown assay. Agarose‐bound sWGA lectin chromatography selectively binds to N‐glycans bound to DG and associated proteins. Using this approach, we can isolate adhesion complexes associated with the DGC.2 Analysis of sWGA void and eluate samples allow detection of unbound and DG bound samples, respectively (Figure 5C and 5D). We found that utrophin and dystrophin binding to DG was reduced in the absence of SSPN. Loss of SSPN also negatively affected binding of sarcoglycans (α‐, β‐, δ‐ and γ‐SG) to DG (Figure 5D). In the absence of SSPN, sarcoglycans bound less tightly to DG (especially δ‐ and γ‐SGs). Examination of sWGA voids by immunoblot showed increased levels of sarcoglycans in SSPN‐nulls, indicating that they require SSPN to effectively associate with the cardiac DGC. Although β1D integrin was increased in the SSPN‐null total protein blots (Figure 5A), its binding to DG was greatly reduced in the absence of SSPN (Figure 5D and 5E). We found that all components of the DGC, along with utrophin and β1D integrin, were present in sWGA eluates isolated from wild‐type hearts (Figure 5D). These data reveal that SSPN mediates attachment of β1D integrin to DG and that loss of SSPN does not alter membrane localization of β1D integrin. However, in SSPN‐null skeletal muscle, β1D integrin levels maintained association with DG,42 revealing discernable differences in cardiac and skeletal muscles in terms of association.
In SSPN‐nulls, DG pulldown revealed significant changes for dystrophin and β‐DG only. Many of these DCG proteins, however, were visibly reduced in SSPN‐null hearts (n=5) (Figure 5E). A schematic diagram (Figure 6) represents the major consequences of SSPN ablation in cardiac tissue: (1) destabilization of and almost complete loss of δ‐ and γ‐SG from the sarcolemma; (2) α‐DG associates less tightly with laminin in the extracellular matrix, reducing cell‐to‐cell adhesion; (3) dystrophin levels are reduced; (4) functional responsiveness to isoproterenol is reduced (cardiac output unchanged) and ventricular dimensions increased; and (5) the hypertrophic response is heightened upon isoproterenol administration and aging.
Figure 6.
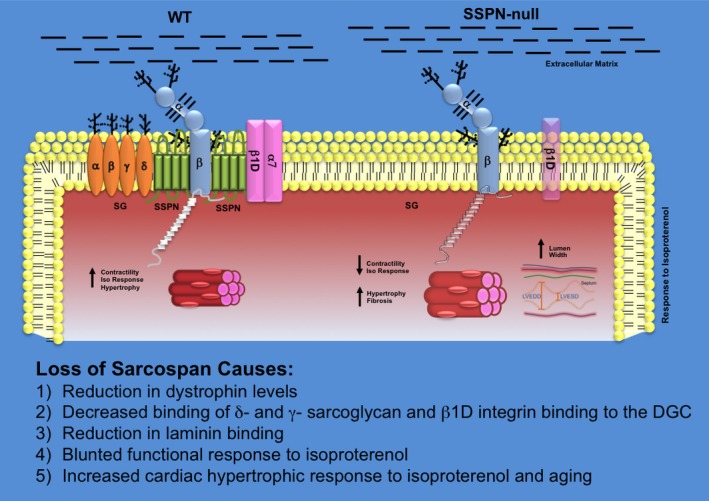
Schematic summarizing effects of SSPN loss in cardiac muscle. A schematic diagram illustrates the changes that occur at the sarcolemma due to alterations in SSPN levels in WT and SSPN‐null (SSPN −/−) mice. Protein levels of the entire DGC as well as β1D‐integrin (light purple) are depicted by changes in color intensity. The sarcoglycans (α‐, β‐, γ‐, δ‐SGs; light orange) are depicted as transmembrane oval shapes. Dystrophin (Dys; white) is shown on the cytoplasmic face of the sarcolemma membrane. Dystroglycan (α‐, β‐ DGs; light blue) levels do not change with SSPN levels. Changes to cardiac muscle fiber cross‐sectional area are shown by increased size. Alterations in left ventricular dimension are demonstrated for SSPN −/− hearts, shown as increased LVEDD and LVESD. Glycosylation is depicted as black branches on both the SGs and DGs. The extracellular matrix containing laminin to which DG associates is depicted as black lines. This schematic summarizes the major consequences of SSPN loss including: (1) reduction in dystrophin levels, (2) almost complete loss of δ‐ and γ‐SG from the sarcolemma, (3) reduction in laminin binding in the extracellular matrix reducing cell‐to‐cell adhesion, (4) blunted functional responsiveness to isoproterenol, and (5) increased hypertrophic response to isoproterenol administration and aging. DGC indicates dystrophin–glycoprotein complex; LVEDD, left ventricular end diastolic dimension; LVESD, left ventricular end systolic dimension; SSPN, sarcospan; WT, wild‐type.
SSPN Attenuates Cardiac Damage in DMD Mice
In skeletal muscle, transgenic overexpression of SSPN ameliorates DMD muscle disease by increasing abundance of compensatory adhesion complexes (UGC and α7β1D integrin) at the sarcolemma that restore connection of the sarcolemma to the extracellular matrix.42 We investigated whether overexpression of human SSPN in cardiac muscle of dystrophin‐deficient mdx mice (mdx TG) would alleviate features of dystrophic cardiac disease. The mdx mice have a mutation in the dystrophin gene and serve as a mouse model of DMD with associated cardiomyopathy. Since mice utilized in this study overexpress human SSPN under control of the α‐actin promoter, we compared relative protein expression levels of the transgene in skeletal muscle and cardiac muscle, shown in Figure S2. Improvements in skeletal muscle pathology appear to positively impact cardiac function,49 however, not in aged mdx mice.50 Therefore, it is unclear whether some of the ameliorative effects in mdx TG hearts compared to mdx could be due to effects of SSPN overexpression in skeletal muscle. Using a well‐documented assay, we injected 13‐ to 15‐month‐old mice with EBD, an in vivo tracer dye, to assess muscle fiber damage. Mdx mice displayed substantial EBD uptake (13.32%) in cardiac tissue, particularly in the septum and right ventricle (Figure 7A through 7C). However, SSPN dramatically reduced membrane damage in mdx cardiac muscle, as revealed by a reduction in EBD incorporation in mdx cardiac myofibers (1.25%). In fact, EBD staining of mdx TG cardiac muscle was restored to wild‐type levels (Figure 7A through 7C). We observed low, but detectable, levels of EBD infiltration (1.02%) in SSPN‐null hearts (Figure 7A through 7C).
Figure 7.
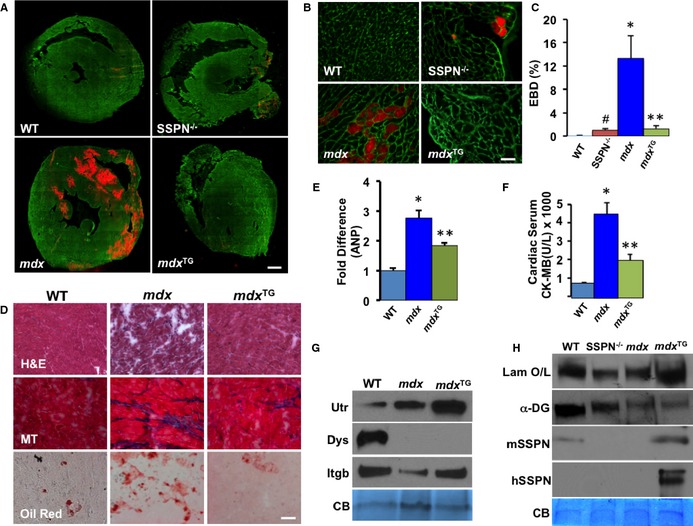
SSPN attenuates membrane damage in DMD hearts. A, Mice were injected with EBD to detect regions of membrane instability (visualized by red fluorescence). Transverse cryosections of heart muscle from 12‐month‐old WT, SSPN‐null (SSPN −/−), DMD model (mdx), and mdx:SSPN‐Tg (mdx TG) mice were stained with laminin antibodies (green) to visualize individual myofibers. Bar, 900 μm. B, Magnified (×40) images of representative sections from (A). Bar, 50 μm. C, Quantification of EBD‐positive fibers (n=3 hearts per genotype). D, Cardiac muscle from 4‐month‐old WT, mdx and mdx TG mice were stained with H&E, MT, and Oil Red (n=2). Bar, 50 μm. E, Relative changes in gene expression changes of ANP were assessed by qRT‐PCR in 4‐month‐old WT, mdx, and mdx TG hearts and number of mice utilized was (WT (n=4), mdx (n=5), mdx TG (n=5). F, Serum CK‐MB levels were detected in WT; mdx and mdx TG groups of mice (WT (n=5), mdx (n=6), mdx TG (n=5). G, Immunoblots of sWGA cardiac eluates show changes in key proteins in WT, mdx and mdx TG hearts (n=3 experiments, samples from 3 different mice of each genotype) whereas, in (H) Immunoblots show changes in laminin binding in a laminin overlay experiment (Laminin O/L) upon overexpression of SSPN in WT, mdx and mdx TG hearts (n=3 experiments, samples from 3 different mice of each genotype). Equal sWGA protein samples (30 μg) were resolved by SDS‐PAGE and immunoblotted with the indicated antibodies. CB staining is shown as a loading control. All data are presented as averages and error bars are expressed as SEM. Statistics were performed by 1‐way ANOVA with Bonferroni correction for individual groups in (C through F) and *P‐values ≤0.05 are indicated. For (C) *P=0.018 mdx vs WT, **P=0.027 mdx vs mdx TG, # P<0.001 SSPN −/− vs WT; (E) *P=0.003 WT vs mdx, **P=0.036 mdx vs mdx TG and (F) * P<0.001 mdx vs WT and **P=0.009 mdx vs mdx TG. ANP indicates atrial natriuretic peptide; CB, Coomassie blue; CK‐MB, cardiac creatine kinase; DMD, Duchenne muscular dystrophy; EBD, Evans blue dye; H&E, hematoxylin & eosin; mdx, Duchenne muscular dystrophy mouse model; mdx TG, mdx mice overexpressing human sarcospan; MT, Masson's trichrome; qRT‐PCR indicates quantitative reverse transcription–polymerase chain reaction; SDS‐PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis; SSPN, sarcospan; sWGA, succinylated wheat germ agglutin; WT, wild‐type.
We analyzed several histological parameters of mdx disease using the following assays: H&E histochemistry, oil red staining (adipose deposition), and Masson's trichrome (collagen deposition). As expected, mdx hearts exhibited extensive fibrosis as well as fat and collagen deposition (Figure 7D). Overexpression of SSPN improved cardiac disease parameters in mdx hearts; however, localized regions of damage still occurred in some areas of the SSPN‐transgenic mdx hearts. We expect that tissue histology can be improved to an even greater extent with higher SSPN expression levels. Improvement of disease at the tissue level was further evidenced by the reduction of ANP (atrial natriuretic peptide) expression in mdx TG hearts compared to mdx controls (Figure 7E). Additional evidence of functional rescue was the significant reduction of CK‐MB (cardiac creatine kinase isoform) in mdx TG mice compared to mdx mice (Figure 7F). Human SSPN overexpression was able to increase levels of key DGC‐ and UGC‐associated proteins utrophin and β1D integrin, but not dystrophin (Figure 7G). Furthermore, the presence of SSPN in mdx TG hearts increased laminin binding in laminin overlay experiments compared to mdx controls (Figure 7H). This indicates that SSPN increased cardiac cell adhesion and was responsible for stabilizing the cardiac cell membrane, leading to decreased EBD uptake in mdx TG hearts and CK‐MB in their bloodstream. Immunoblots showed that the increase in laminin binding appeared dependent on SSPN expression and not upon increasing levels of α‐DG. The levels of mouse and human SSPN are also shown; mouse SSPN is also seen in mdx hearts, however, at a decreased level and not discerned at this exposure. The increase in mouse SSPN in the mdx TG could be due to enhanced SSPN expression; however, likely some cross‐reactivity exists between the mouse SSPN antibody and the human SSPN protein (Figure 7H).
SSPN Improves Cardiac Function in DMD Mice
To assess baseline cardiac parameters and function of mice used in our study, we performed echocardiography on ≈10‐ to 11 month‐old wild‐type, mdx and mdx TG mice. In the mdx mice overexpressing SSPN (mdx TG), the heart weight/body weight (HWt/BWt; mg/g) ratio was significantly increased to 6.17±0.15 in mdx mice compared to wild‐type, whereas in mdx TG mice the HWt/BWt ratio was 5.42±0.25 closer to the wild‐type value 5.33±0.11 (Figure 8A). In regard to function, echocardiographic measurements of cardiac contractility LVEF (%) were significantly higher in mdx TG mice 71.72±4.89 compared to mdx 59.12±4.96 and similar to wild‐type values 69.8±1.52 (Figure 8B). Similarly, LVFS% was significantly improved in mdx TG mice (36.44±3.42) compared to mdx values (27.38±1.27) and slightly higher than wild‐type values (34.06±1.07) (Figure 8C). Echocardiographic measurements of cardiac dimension revealed a trend (although not significant) that mdx (LVmass; mg) values were increased to 81.4±8.79 compared to wild‐type values of 64.51±2.24, whereas hypertrophic growth was reduced in the mdx TG hearts with values of 65.82±7.01 (Figure 8D). Ventricular dimensions during diastole (left ventricular end diastolic dimension; mm) were slightly smaller in mdx TG hearts with values of 3.72±0.29 mm compared to dimensions of 4.14±0.17 mm in mdx and 4.02±0.08 mm in wild‐type hearts, while in systole (left ventricular end systolic dimension; mm) lumenal dimensions were significantly decreased in mdx TG hearts to 2.47±0.20 mm compared to 3.00±0.09 mm in mdx and 2.65±0.07 mm in wild‐type mice (Figure 8E). See Table 2 for additional echocardiography parameters including the values reported here. In Figure 8F, M‐mode echocardiography images are shown for wild‐type, mdx, and mdx TG mice and demonstrate changes in lumenal width and contractility; wild‐type and mdx TG hearts had narrower ventricular lumenal dimensions and heightened wall movement relative to mdx hearts.
Figure 8.
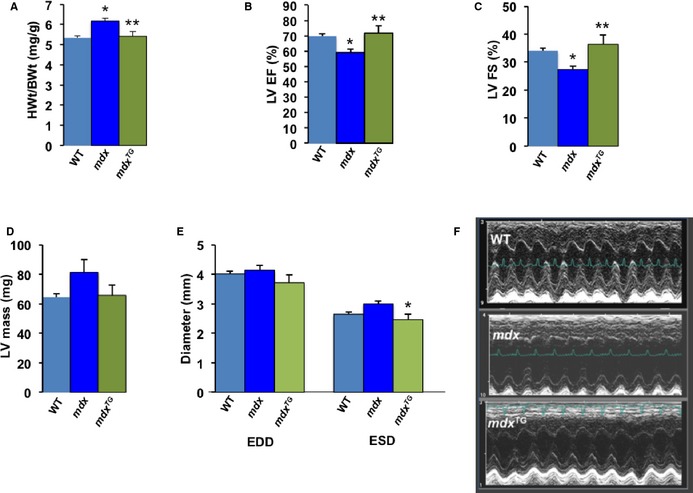
Physiological evaluation of DMD mice overexpressing SSPN. To determine the effects of SSPN on mice exhibiting mdx cardiac pathology we evaluated the following morphological and functional parameters. A, HWt/BWt; mg/g values obtained from 11‐ to 12 month‐old mice: WT (n=8), mdx (n=10), mdx TG (n=6). Echocardiographic data presented was obtained from mice ≈10 to 11 months of age: WT (n=12), mdx (n=5), mdx TG (n=5) Echocardiographic assessment of contractility shows differences in (B) LV EF (%) and (C) LV FS (%) of WT, mdx and mdx TG mice. D, LVmass (mg) measurements obtained by echocardiography indicate differences in ventricular mass in WT, mdx and mdx TG mice. E, Echocardiographic measurements of LVEDD and LVESD in mm of WT, mdx and mdx TG mice. F, M‐mode images obtained by echocardiography for WT, mdx and mdx TG mice. Statistical analysis was performed using 1‐way ANOVA with Bonferroni post hoc test and *P‐values <0.05 are indicated on the plots, all data presented as averages and error bars represent SEM. For (A) *P=0.003 mdx vs WT, **P=0.01 mdx vs mdx TG; (B) *P=0.01 mdx vs WT, **P=0.01 mdx vs mdx TG; (C) *P=0.03 mdx vs WT, **P=0.01 mdx vs mdx TG; (D) P=0.061 mdx vs WT; and (E) ESD *P=0.03 mdx vs mdx TG, P=0.112 mdx vs WT. DMD indicates Duchenne muscular dystrophy; HWt/BWt, heart weight/body weight; LV EF, left ventricular ejection fraction; LV FS, left ventricular fractional shortening; LVEDD, left ventricular end diastolic dimension; LVESD, left ventricular end systolic dimension; mdx, Duchenne muscular dystrophy mouse model; mdx TG, mdx mice overexpressing human sarcospan; SSPN, sarcospan; WT, wild‐type.
Table 2.
Summary of Echocardiographc Data Obtained From DMD Mice
| Baseline | |||
|---|---|---|---|
| WT (n=12) | mdx (n=5) | mdx TG (n=5) | |
| Echo parameter | |||
| HR, bpm | 527.5±19.2 | 536.2±20.0 | 547.4±17.7 |
| LVEDD, mm | 4.02±0.08 | 4.14±0.17 | 3.72±0.29 |
| LVESD, mm | 2.65±0.07 | 3.00±0.09 | 2.47±0.20† |
| LVEF, % | 69.8±1.52 | 59.12±4.96* | 71.72±4.89† |
| LV % FS | 34.06±1.07 | 27.38±1.27* | 36.44±3.42† |
| Vcf, % | 6.75±0.32 | 5.88±0.35 | 8.03±0.74† |
| MV E, mm/s | 0.69±0.04 | 0.84±0.11 | 0.6±0.04 |
| MV A, mm/s | 0.42±0.03 | 0.51±0.06 | 0.30±0.24† |
| MV E/A | 1.66±0.07 | 1.66±0.06 | 2.1±0.11†, ‡ |
| PWT, mm | 0.49±0.01 | 0.56±0.02 | 0.55±0.04 |
| IVS, mm | 0.51±0.01 | 0.58±0.03* | 0.57±0.03 |
| LVmass, mg | 64.51±2.24 | 81.4±8.79 | 65.82±7.01 |
Data are mean±SEM. A indicates atrial peak flow velocity; DMD, Duchenne muscular dystrophy; E, early peak flow velocity; FS, fractional shortening; HR, heart rate; IVS, interventricular septum thickness; LVEDD, left ventricular end diastolic dimension; LVEF, left ventricular ejection fraction; LVESD, left ventricular end systolic dimension; mdx, Duchenne muscular dystrophy mouse model; mdx TG, mdx mice overexpressing human sarcospan; MV, mitral valve; PWT, posterior wall thickness; Vcf, velocity of circumferential shortening; WT, wild‐type.
One‐way ANOVA with Bonferroni post hoc test was utilized to determine differences between groups, *P<0.05 (comparison between baseline WT and mdx mice), † P<0.05 (comparison between mdx and mdxTG mice), ‡ P<0.05 (comparison between WT and mdx TG mice). Values reported as averages and errors presented as SEM. Results for the overall test were: HR (F=0.806), LVEDD (F=0.514), LVESD (F=0.036), LVEF (F=0.009), LVFS (F=0.010), VCF (F=0.024), E (F=0.117), A (F=0.014), E/A (F=0.005) PWT (F=0.047), IVS (F=0.025), LVmass (F=0.061).
Our overall findings are summarized in a schematic that illustrates the influence of SSPN on the integrity of the cardiac sarcolemma and the expression of the DGC and α7β1 integrin. These findings reveal the important role that SSPN plays in their localization at the cell surface. Furthermore, functional consequences of SSPN loss can be attributed to a reduction in important sarcolemmal proteins including SGs and β1D integrin, which have important signaling and structural roles and are known to be involved in cardiomyopathy and vascular dysfunction (Figure 9). Overexpression of SSPN has been shown to improve the localization of utrophin and improve membrane stability by increasing laminin binding (Figure 9). This schematic summarizes our major findings when SSPN is overexpressed including the following: (1) Increased association of utrophin and β1D integrin with α‐DG in the DGC. (2) α‐DG associates more tightly with laminin in the extracellular matrix, increasing cell‐to‐cell adhesion. (3) Increased membrane stability seen by the reduction in EBD uptake and decreased detection of CK‐MB in the blood sera. (4) Tissue histology and cardiac function is improved in DMD mice. (5) Cardiac hypertrophic growth is reduced in DMD mice.
Figure 9.
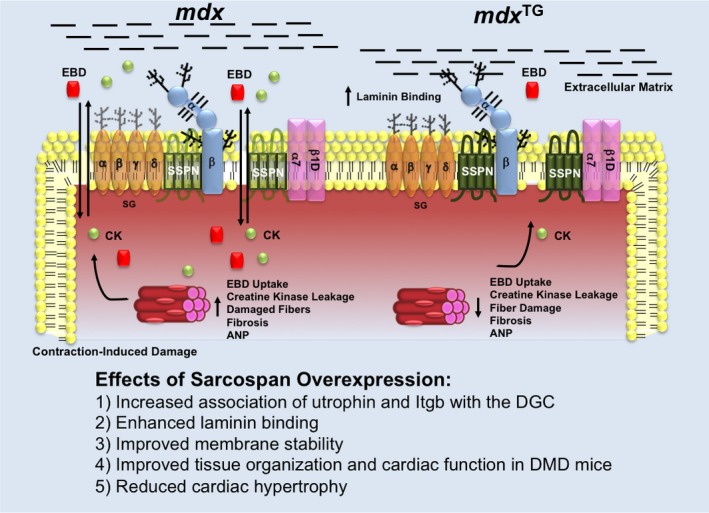
Influence of SSPN on adhesion complexes in cardiac muscle. A schematic diagram illustrates the changes that occur at the sarcolemma due to overexpression of human SSPN in mdx and mdx TG mice. Protein levels of the entire DGC as well as the α7/β1D‐integrin complexes (light purple) are depicted by changes in color intensity. SSPN (green) is represented as a 4‐transmembrane‐spanning protein, where dark green represents its 3‐fold overexpression. The sarcoglycans (α‐, β‐, γ‐, δ‐ SGs; light orange) are depicted as transmembrane oval shapes. Dystrophin (Dys; white) is shown on the cytoplasmic face of the sarcolemma membrane. Dystroglycan (α‐, β‐DGs; light blue) levels do not change with SSPN levels. Glycosylation is depicted as black branches on both the SGs and DGs. CK‐MB is shown as a green sphere and EBD is depicted as a red square. Black lines indicate laminin in the extracellular matrix to which α‐DG and the DGC associate.This schematic summarizes the major findings when SSPN is overexpressed including: (1) increased association of utrophin and Itgb to the DGC, (2) increased laminin binding, (3) improved membrane stability, (4) improved tissue histology and cardiac function in DMD mice, and (5) reduced cardiac hypertrophy. CK‐MB indicates cardiac creatine kinase; DGC, dystrophin–glycoprotein complex; DMD, Duchenne muscular dystrophy; EBD, Evans blue dye; mdx, Duchenne muscular dystrophy mouse model; Itgb, β1D‐integrin; mdx TG, mdx mice overexpressing human sarcospan; SSPN, sarcospan; SSPN, sarcospan.
Discussion
To investigate the role of SSPN in the heart and hypertrophic remodeling processes, we initiated studies exposing SSPN‐null mice to chronic β‐adrenergic stimulation and uncovered a physiological role of SSPN in regulating the cardiac hypertrophic response. In this study, 12‐month‐old SSPN‐null mice demonstrated increased heart weight/body weight after isoproterenol administration, although SSPN‐null mice had increased body weights compared to wild‐type after isoproterenol treatment. Even upon visual inspection, the isoproterenol‐treated hearts were larger than their untreated counterparts; at the tissue level, cross‐sectional area of fibers in SSPN‐null hearts were noticeably larger than controls. Consistent with these findings, echocardiographic measurements indicated increased left ventricle wall thickness and increased lumenal dimension in SSPN‐null mice. This suggests that β‐adrenergic stimulation may have revealed the phenotypic consequence of SSPN‐ablation before the onset of age‐related symptoms.
It was determined that the lack of physiological responsiveness to isoproterenol in the SSPN‐null mice compared to saline‐treated controls was not due to β‐receptor downregulation. These findings suggest that the blunted functional response in SSPN‐null mice may be due to alterations in integrin signaling, which is involved in the hypertrophic response to β‐adrenergic pathways. In this study, we found specifically that integrin interactions with the DGC are dependent on SSPN, although overall levels of β1D‐integrin are upregulated, likely due to sensing a reduction of membrane stability when SSPN is absent. The contribution of β1D‐integrin to membrane stabilization and signaling that is either unassociated or weakly associated with the DGC remains unknown. Specifically, we demonstrate that β1‐integrin association with DG is dependent on SSPN. These effects are expected to reduce integrin signaling, thus diminishing the effects of β‐adrenergic stimulation.51, 52 This in‐depth analysis of SSPN‐null cardiac muscle revealed that loss of SSPN decreased abundance of the DGC at the sarcolemma. We found that SSPN ablation disrupts protein interactions that are crucial for association of β1D‐integrin and SGs with the DGC in the heart, as revealed by sWGA precipitation. The effects on integrin binding to the DGC likely underlie part of the mechanism for cardiac dysfunction in SSPN‐null mice, since it has an important role in stabilizing the sarcolemma. Future studies will focus on investigating effects of SSPN loss on abundance and localization of α and β integrin subunits and the impact of SSPN loss on Akt‐dependent pathways that are altered in skeletal muscle.42
To further test the phenotypic consequences of SSPN loss, 12‐month‐old mice were treated with isoproterenol to model sustained adrenergic stimulation, eliciting compensatory cardiac mechanisms that ultimately culminate in hypertrophy. In response to isoproterenol treatment, the SSPN‐null hearts exhibited enhanced hypertrophy (increased HWt/BWt ratio and LV mass) compared to wild‐type. In this study, parameters of LV function indicated reduced contractility and subsequently diastolic dysfunction, as evidenced by the decreased fractional shortening and increased E/A ratio. The EF (%) remained preserved in SSPN‐null mice, and subsequently cardiac output was unchanged in response to β‐adrenergic stimulation compared to controls. Interestingly, B1KOmdx (dystrophin and β1‐integrin) deficient mice also demonstrate reduced LV function as well as blunted adrenergic responsiveness compared to controls.51 Additionally, remodeling of the hypertrophied myocardium, together with increased vulnerability of cardiomyocytes to hemodynamic stressors, may cause changes in myocyte shape and distorted alignment with the anisotropic laminar muscle, leading to a reduction of ejection fraction and blunted β‐adrenergic response.53, 54 Structural changes such as these in SSPN‐null cardiac tissue may underlie the ventricular remodeling and reduced function that are evident in SSPN‐null mice. Chamber dimension is increased during both contractile phases, indicating a decline in cardiac function. SSPN‐null mice, after β‐adrenergic stimulation, phenocopy functional outcomes in β1KO mice that display replacement fibrosis, LV chamber dilation, increased septal and free wall thicknesses, and depressed ventricular function (fractional shortening and velocity of circumferential fiber shortening) compared to controls.55
Isoproterenol treatment caused a substantial fibrotic response in SSPN‐null hearts and could be contributed by a number of potential sources. It is feasible that decreased membrane stability could increase cardiomyocyte susceptibility to transient ischemia and myocardial damage, resulting in localized fibrotic regions. Isoproterenol induces vasospasms in susceptible individuals, which may contribute to an infarct pattern of fibrosis.56 In the absence of vascular data, we speculate that the large focal fibrotic areas in SSPN‐null hearts may result from increased susceptibility of SSPN‐null mice to vasoconstriction, or alternatively that increased cell membrane fragility makes SSPN‐deficient hearts more susceptible to transient ischemia. A previous study performed in rat hearts described local reductions in dystrophin in areas of myocardial injury post–isoproterenol treatment. The reduction in dystrophin is thought to increase susceptibility to ischemic injury.57 Similarly, the reduction of dystophin in the SSPN‐null hearts relative to wild‐type controls increases susceptibility of SSPN‐nulls to isoproterenol‐induced ischemic injury. Fibrosis could also be attributed to integrin loss from the costameres, since integrins colocalize to cardiomyocyte termini during pathological stress.58 During development of hypertrophic cardiomyopathy, β1‐integrins are shed into the extracellular matrix surrounding the cardiomyocytes, which induces α integrin expression, particularly in the left ventricle.59 Targeted deletion of the β1‐integrin gene compromises myocyte cell membrane integrity and leads to development of postnatal cardiac fibrosis.55 Furthermore, fibrosis can underlie diastolic and systolic dysfunction, and heighten susceptibility to cardiac arrhythmias.60, 61 Disruption of the SG‐SSPN subcomplex in smooth muscle is known to cause vasospasm in relevant murine models, leading to fibrosis and severe cardiomyopathy.21, 62 SSPN loss reduces SGs abundance at the sarcolemma, with a greater reduction in δ‐SG and γ‐SG, suggesting that they form a tighter association with SSPN than other members of the complex. Future studies will be focused on determining whether SSPN loss reduces SGs in the vasculature, thereby contributing to vasospasm.
Overexpression of SSPN ameliorated mdx pathology in dystrophic hearts at the tissue level. EBD uptake was extensively evident in mdx mice; however, the SSPN transgene appears to have substantially restored membrane stability, and mdx TG mice demonstrated EBD uptake levels similar to wild‐type. Reducing sarcolemmal fragility has been well recognized as therapeutically valuable. Studies by Metzger and colleagues have demonstrated the beneficial consequences of in vivo administration of a membrane sealant poloxamer 188 to dystrophic mice.63 The sarcolemmal stabilization provided by the sealant vastly improved ventricular geometry and blocked development of acute cardiac failure in mice during dobutamine‐mediated cardiac stress testing.64 SSPN overexpression in mdx hearts increases utrophin and β1D‐integrin complexes and further promotes utrophin localization around the cardiac sarcolemma, leading to increased laminin binding that promotes cell‐to‐cell adhesion and stabilizes dystrophic cell membranes. SSPN transgenic expression in mdx cardiac tissue reduced levels of collagen deposition and fatty infiltrates, and decreased levels of ANP, a cardiac stress marker. These ameliorative effects by SSPN in mdx cardiac tissue are strongly supported by the significant reduction of serum CK‐MB levels in the mdx TG mice. Serum CK measurements provide a dependable index for membrane stability defects. Examination of skeletal muscle CK levels in DMD patients is routinely performed to assess the level of muscle membrane damage and is an early marker for DMD in undiagnosed patients. Although our assay for cardiac‐specific CK‐MB reliably detects this isoform, we cannot rule out a contribution of CK‐MB from skeletal muscle. Normally skeletal muscle produces low levels of CK‐MB (≈1%); however, regenerative cycles in skeletal muscle commonly seen in muscular dystrophy causes increased CK‐MB expression.65 In our study, the restoration of tissue pathology (H&E and Masson Trichrome) and membrane stabilization (EBD uptake and increased laminin binding) in mdx TG compared to mdx hearts provide evidence of direct effects of SSPN overexpression in the heart. Therefore, it is reasonable to expect that SSPN overexpression in the heart can reduce leakage of CK‐MB into the bloodstream.
Functional improvement was also seen in the hearts of mdx TG mice, which showed significantly enhanced contractile performance compared to mdx hearts, and in fact restored them to WT values. Overall, the hearts of mdx TG mice were less hypertrophic than mdx hearts and exhibited improvements in ventricular geometry. Since mdx mice begin to exhibit cardiac symptoms around 10 months,66 we utilized ≈10‐ to 11‐month‐old mice for this study. Limitations exist; however, since mdx mice do not display severe cardiac disease, the functional improvement by SSPN overexpression was modest, although significant. Increased levels of sarcolemmal proteins including utrophin and β1D integrin are expected to stabilize the cell membrane. This coupled with enhanced cellular adhesion mediated by SSPN may protect against cardiomyocyte loss, improve integrity of cardiac muscle, and subsequently restore function. Future studies will examine the ability of SSPN to rescue severe dystrophic cardiac disease in mdx:utr‐het mice that have reduced expression of utrophin, a compensatory homologue of dystrophin. From these studies it can be determined that SSPN has potential to ameliorate pathology and subsequently improve cardiac performance in DMD mice.
Presently, disease‐associated mutations have not been identified in the SSPN gene.67 The role of SSPN in cardiac and skeletal muscle is complex, and we anticipate that many functions remain undiscovered. Several genome‐wide association studies suggest a role for SSPN in disease, since various polymorphisms that occur in the SSPN gene locus 12p11.2 were found to positively correlate with increased waist circumference and LV mass in several extended and large families.68 It appears that the role of SSPN in the heart is stabilization of sarcolemmal proteins that protect against contraction‐induced damage and participation in signaling, which promotes cardiac function. Furthermore, the close physical association of SSPN with the DGC and integrins suggests its importance in mediating interaction between these complexes, which, when mutated or absent, cause cardiac disease, whereas overexpression of SSPN restores cardiac function and tissue pathology in mdx mice, suggesting that it may hold promise as a future therapeutic option.
Sources of Funding
Funding support was generously provided by the National Research Service Award GM07104, Edith Hyde Fellowship, Eureka Pre‐doctoral Training Fellowship, and the Ruth L. Kirschstein National Research Service Award T32AR059033 from National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) to Marshall; Center for Duchenne Muscular Dystrophy (CDMD)‐CureDuchenne Postdoctoral Fellowship to Parvatiyar; Whitcome Fellowship (UCLA) for Nguyen; William Townsend Porter Predoctoral Fellowship (American Physiological Society) for Richardson. We also acknowledge NIH AR048179, CTSI/CDMD Team Science UL1TR000124/NIH 5P30AR057230, “Restoration of Muscle Cell Adhesion to Treat Cardiomyopathy in Muscular Dystrophy” and Muscular Dystrophy Association MDA274143, National Center for Advancing Translational Sciences UCLA CTSI Grant UL1TR000124 to Crosbie‐Watson for support during this project.
Disclosures
None.
Supporting information
Figure S1. M‐mode images of isoproterenol‐treated SSPN‐null hearts. The representative M‐mode images show WT (top) and SSPN−/− (bottom) mice after β‐agonist treatment.
Figure S2. Expression levels of the SSPN transgene in mdx mice. Total protein samples obtained from total skeletal and cardiac muscle were immunoblotted to determine expression levels of the human SSPN transgene in both tissue types of mdx and mdxTG mice. GAPDH was included as a loading control.
Acknowledgments
We thank Andrew D. Watson, MD, PhD, Elizabeth M. Gibbs, PhD, and Kristen M. Stearns‐Reider, PT, PhD for their critical discussions and reading of the manuscript.
(J Am Heart Assoc. 2015;4:e002481 doi: 10.1161/JAHA.115.002481)
Accompanying Figures S1 and S2 are available at http://jaha.ahajournals.org/content/4/12/e002481/suppl/DC1
References
- 1. Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. [DOI] [PubMed] [Google Scholar]
- 2. Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. [DOI] [PubMed] [Google Scholar]
- 3. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. 1993;90:3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ervasti JM, Campbell KP. A role for the dystrophin‐glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. [DOI] [PubMed] [Google Scholar]
- 6. Weller B, Karpati G, Carpenter S. Dystrophin‐deficient mdx muscle fibers are preferentially vulnerable to necrosis induced by experimental lengthening contractions. J Neurol Sci. 1990;100:9–13. [DOI] [PubMed] [Google Scholar]
- 7. Berko BA, Swift M. X‐linked dilated cardiomyopathy. N Engl J Med. 1987;316:1186–1191. [DOI] [PubMed] [Google Scholar]
- 8. Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X‐linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87:1854–1865. [DOI] [PubMed] [Google Scholar]
- 9. Muntoni F, Melis MA, Ganau A, Dubowitz V. Transcription of the dystrophin gene in normal tissues and in skeletal muscle of a family with X‐linked dilated cardiomyopathy. Am J Hum Genet. 1995;56:151–157. [PMC free article] [PubMed] [Google Scholar]
- 10. Yoshida K, Nakamura A, Yazaki M, Ikeda S, Takeda S. Insertional mutation by transposable element, L1, in the DMD gene results in X‐linked dilated cardiomyopathy. Hum Mol Genet. 1998;7:1129–1132. [DOI] [PubMed] [Google Scholar]
- 11. Bonnemann CG, Modi R, Noguchi S, Mizuno Y, Yoshida M, Gussoni E, McNally EM, Duggan DJ, Angelini C, Hoffman EP. Beta‐sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex [published erratum appears in Nat Genet. 1996:110]. Nat Genet. 1995;11:266–273. [DOI] [PubMed] [Google Scholar]
- 12. Roberds SL, Letrucq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tomé FMS, Romero NB, Fardeau M, Beckmann JS, Kaplan J‐C, Campbell KP. Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell. 1994;78:625–633. [DOI] [PubMed] [Google Scholar]
- 13. Lim LE, Duclos F, Broux O, Bourg N, Sunada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, Tomé FMS, Fardeau M, Jackson CE, Beckmann JS, Campbell KP. Beta‐sarcoglycan: characterization and role in limb‐girdle muscular dystrophy linked to 4q12. Nat Genet. 1995;11:257–265. [DOI] [PubMed] [Google Scholar]
- 14. Noguchi S, McNally EM, Ben Othmane K, Hagiwara Y, Mizuno Y, Yoshida M, Yamamoto H, Bonnemann CG, Gussoni E, Denton PH, Kyriakides T, Middleton L, Hentati F, Ben Hamida M, Nonaka I, Vance JM, Kunkel LM, Ozawa E. Mutations in the dystrophin‐associated protein gamma‐sarcoglycan in chromosome 13 muscular dystrophy. Science. 1995;270:819–822. [DOI] [PubMed] [Google Scholar]
- 15. Nigro V, de Sa Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passos‐Bueno MR, Zatz M. Autosomal recessive limb‐girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta‐sarcoglycan gene. Nat Genet. 1996;14:195–198. [DOI] [PubMed] [Google Scholar]
- 16. Kirschner J, Lochmuller H. Sarcoglycanopathies. Handb Clin Neurol. 2011;101:41–46. [DOI] [PubMed] [Google Scholar]
- 17. Ettinger AJ, Feng G, Sanes JR. Epsilon‐sarcoglycan, a broadly expressed homologue of the gene mutated in limb‐girdle muscular dystrophy 2D. J Biol Chem. 1997;272:32534–32538. [DOI] [PubMed] [Google Scholar]
- 18. Straub V, Ettinger AJ, Durbeej M, Venzke DP, Cutshall S, Sanes JR, Campbell KP. epsilon‐sarcoglycan replaces alpha‐sarcoglycan in smooth muscle to form a unique dystrophin‐glycoprotein complex. J Biol Chem. 1999;274:27989–27996. [DOI] [PubMed] [Google Scholar]
- 19. Lancioni A, Rotundo IL, Kobayashi YM, D'Orsi L, Aurino S, Nigro G, Piluso G, Acampora D, Cacciottolo M, Campbell KP, Nigro V. Combined deficiency of alpha and epsilon sarcoglycan disrupts the cardiac dystrophin complex. Hum Mol Genet. 2011;20:4644–4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Factor SM, Minase T, Cho S, Dominitz R, Sonnenblick EH. Microvascular spasm in the cardiomyopathic Syrian hamster: a preventable cause of focal myocardial necrosis. Circulation. 1982;66:342–354. [DOI] [PubMed] [Google Scholar]
- 21. Cohn RD, Durbeej M, Moore SA, Coral‐Vazquez R, Prouty S, Campbell KP. Prevention of cardiomyopathy in mouse models lacking the smooth muscle sarcoglycan‐sarcospan complex. J Clin Invest. 2001;107:R1–R7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Townsend D, Yasuda S, McNally E, Metzger JM. Distinct pathophysiological mechanisms of cardiomyopathy in hearts lacking dystrophin or the sarcoglycan complex. FASEB J. 2011;25:3106–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM, Davies K. Expression of full‐length utrophin prevents muscular dystrophy in mdx mice. Nat Med. 1998;4:1441–1444. [DOI] [PubMed] [Google Scholar]
- 24. Pons F, Robert A, Marini JF, Leger JJ. Does utrophin expression in muscles of mdx mice during postnatal development functionally compensate for dystrophin deficiency? J Neurol Sci. 1994;122:162–170. [DOI] [PubMed] [Google Scholar]
- 25. Tinsley JM, Blake DJ, Roche A, Fairbrother U, Riss J, Byth BC, Knight AE, Kendrick‐Jones J, Suthers GK, Love DR, Edwards YH, Davies KE. Primary structure of dystrophin‐related protein. Nature. 1992;360:591–593. [DOI] [PubMed] [Google Scholar]
- 26. Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature. 1996;384:349–353. [DOI] [PubMed] [Google Scholar]
- 27. Wakefield PM, Tinsley JM, Wood MJ, Gilbert R, Karpati G, Davies KE. Prevention of the dystrophic phenotype in dystrophin/utrophin‐deficient muscle following adenovirus‐mediated transfer of a utrophin minigene. Gene Ther. 2000;7:201–204. [DOI] [PubMed] [Google Scholar]
- 28. Squire S, Raymackers JM, Vandebrouck C, Potter A, Tinsley J, Fisher R, Gillis JM, Davies KE. Prevention of pathology in mdx mice by expression of utrophin: analysis using an inducible transgenic expression system. Hum Mol Genet. 2002;11:3333–3344. [DOI] [PubMed] [Google Scholar]
- 29. Liu J, Milner DJ, Boppart MD, Ross RS, Kaufman SJ. beta1D chain increases alpha7beta1 integrin and laminin and protects against sarcolemmal damage in mdx mice. Hum Mol Genet. 2012;21:1592–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Burkin DJ, Wallace GQ, Milner DJ, Chaney EJ, Mulligan JA, Kaufman SJ. Transgenic expression of {alpha}7{beta}1 integrin maintains muscle integrity, increases regenerative capacity, promotes hypertrophy, and reduces cardiomyopathy in dystrophic mice. Am J Pathol. 2005;166:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burkin DJ, Wallace GQ, Nicol KJ, Kaufman DJ, Kaufman SJ. Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J Cell Biol. 2001;152:1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minetti C, Cordone G, Beltrame F, Bado M, Bonilla E. Disorganization of dystrophin costameric lattice in Becker muscular dystrophy. Muscle Nerve. 1998;21:211–216. [DOI] [PubMed] [Google Scholar]
- 33. Porter GA, Dmytrenko GM, Winkelmann JC, Bloch RJ. Dystrophin colocalizes with beta‐spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J Cell Biol. 1992;117:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88:1112–1119. [DOI] [PubMed] [Google Scholar]
- 35. Matsushita T, Oyamada M, Fujimoto K, Yasuda Y, Masuda S, Wada Y, Oka T, Takamatsu T. Remodeling of cell‐cell and cell‐extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ Res. 1999;85:1046–1055. [DOI] [PubMed] [Google Scholar]
- 36. Marshall JL, Kwok Y, McMorran BJ, Baum LG, Crosbie‐Watson RH. The potential of sarcospan in adhesion complex replacement therapeutics for the treatment of muscular dystrophy. FEBS J. 2013;280:4210–4229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marshall JL, Crosbie‐Watson RH. Sarcospan: a small protein with large potential for Duchenne muscular dystrophy. Skelet Muscle. 2013;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Crosbie RH, Lebakken CS, Holt KH, Venzke DP, Straub V, Lee JC, Grady RM, Chamberlain JS, Sanes JR, Campbell KP. Membrane targeting and stabilization of sarcospan is mediated by the sarcoglycan subcomplex. J Cell Biol. 1999;145:153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marshall JL, Holmberg J, Chou E, Ocampo AC, Oh J, Lee J, Peter AK, Martin PT, Crosbie‐Watson RH. Sarcospan‐dependent Akt activation is required for utrophin expression and muscle regeneration. J Cell Biol. 2012;197:1009–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peter AK, Ko CY, Kim MH, Hsu N, Ouchi N, Rhie S, Izumiya Y, Zeng L, Walsh K, Crosbie RH. Myogenic Akt signaling upregulates the utrophin‐glycoprotein complex and promotes sarcolemma stability in muscular dystrophy. Hum Mol Genet. 2009;18:318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lebakken CS, Venzke DP, Hrstka RF, Consolino CM, Faulkner JA, Williamson RA, Campbell KP. Sarcospan‐deficient mice maintain normal muscle function. Mol Cell Biol. 2000;20:1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Marshall JL, Chou E, Oh J, Kwok A, Burkin DJ, Crosbie‐Watson RH. Dystrophin and utrophin expression require sarcospan: loss of alpha7 integrin exacerbates a newly discovered muscle phenotype in sarcospan‐null mice. Hum Mol Genet. 2012;21:4378–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peter AK, Crosbie RH. Hypertrophic response of Duchenne and limb‐girdle muscular dystrophies is associated with activation of Akt pathway. Exp Cell Res. 2006;312:2580–2591. [DOI] [PubMed] [Google Scholar]
- 44. Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer‐Crosbie M, Hara K, Rosenzweig A, Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. Am J Physiol Cell Physiol. 2010;299:C1256–C1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jordan A, Lyne J, Wong T. Unusual scarring patterns on cardiac magnetic resonance imaging: a potentially treatable etiology not to be missed. Can J Cardiol. 2010;26:149–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roos KP, Jordan MC, Fishbein MC, Ritter MR, Friedlander M, Chang HC, Rahgozar P, Han T, Garcia AJ, Maclellan WR, Ross RS, Philipson KD. Hypertrophy and heart failure in mice overexpressing the cardiac sodium‐calcium exchanger. J Card Fail. 2007;13:318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jordan MC, Henderson SA, Han T, Fishbein MC, Philipson KD, Roos KP. Myocardial function with reduced expression of the sodium‐calcium exchanger. J Card Fail. 2010;16:786–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Levy S, Shoham T. Protein‐protein interactions in the tetraspanin web. Physiology (Bethesda). 2005;20:218–224. [DOI] [PubMed] [Google Scholar]
- 49. Rafael JA, Tinsley JM, Potter AC, Deconinck AE, Davies KE. Skeletal muscle‐specific expression of a utrophin transgene rescues utrophin‐dystrophin deficient mice. Nat Genet. 1998;19:79–82. [DOI] [PubMed] [Google Scholar]
- 50. Wasala NB, Bostick B, Yue Y, Duan D. Exclusive skeletal muscle correction does not modulate dystrophic heart disease in the aged mdx model of Duchenne cardiomyopathy. Hum Mol Genet. 2013;22:2634–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Elsherif L, Huang MS, Shai SY, Yang Y, Li RY, Chun J, Mekany MA, Chu AL, Kaufman SJ, Ross RS. Combined deficiency of dystrophin and beta1 integrin in the cardiac myocyte causes myocardial dysfunction, fibrosis and calcification. Circ Res. 2008;102:1109–1117. [DOI] [PubMed] [Google Scholar]
- 52. Li R, Wu Y, Manso AM, Gu Y, Liao P, Israeli S, Yajima T, Nguyen U, Huang MS, Dalton ND, Peterson KL, Ross RS. beta1 integrin gene excision in the adult murine cardiac myocyte causes defective mechanical and signaling responses. Am J Pathol. 2012;180:952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maron BJ, Roberts WC. Hypertrophic cardiomyopathy and cardiac muscle cell disorganization revisited: relation between the two and significance. Am Heart J. 1981;102:95–110. [DOI] [PubMed] [Google Scholar]
- 54. Maron BJ, Gottdiener JS, Perry LW. Specificity of systolic anterior motion of anterior mitral leaflet for hypertrophic cardiomyopathy. Prevalence in large population of patients with other cardiac diseases. Br Heart J. 1981;45:206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shai SY, Harpf AE, Babbitt CJ, Jordan MC, Fishbein MC, Chen J, Omura M, Leil TA, Becker KD, Jiang M, Smith DJ, Cherry SR, Loftus JC, Ross RS. Cardiac myocyte‐specific excision of the beta1 integrin gene results in myocardial fibrosis and cardiac failure. Circ Res. 2002;90:458–464. [DOI] [PubMed] [Google Scholar]
- 56. Horimoto M, Wakisaka A, Takenaka T, Igarashi K, Inoue H, Yoshimura H, Miyata S. Familial evidence of vasospastic angina and possible involvement of HLA‐DR2 in susceptibility to coronary spasm. Jpn Circ J. 1998;62:284–288. [DOI] [PubMed] [Google Scholar]
- 57. Campos EC, Romano MM, Prado CM, Rossi MA. Isoproterenol induces primary loss of dystrophin in rat hearts: correlation with myocardial injury. Int J Exp Pathol. 2008;89:367–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dabiri BE, Lee H, Parker KK. A potential role for integrin signaling in mechanoelectrical feedback. Prog Biophys Mol Biol. 2012;110:196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shapiro LM, McKenna WJ. Distribution of left ventricular hypertrophy in hypertrophic cardiomyopathy: a two‐dimensional echocardiographic study. J Am Coll Cardiol. 1983;2:437–444. [DOI] [PubMed] [Google Scholar]
- 60. Janicki JS, Brower GL. The role of myocardial fibrillar collagen in ventricular remodeling and function. J Card Fail. 2002;8:S319–S325. [DOI] [PubMed] [Google Scholar]
- 61. Sonnenblick EH, Fein F, Capasso JM, Factor SM. Microvascular spasm as a cause of cardiomyopathies and the calcium‐blocking agent verapamil as potential primary therapy. Am J Cardiol. 1985;55:179B–184B. [DOI] [PubMed] [Google Scholar]
- 62. Coral‐Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP. Disruption of the sarcoglycan‐sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98:465–474. [DOI] [PubMed] [Google Scholar]
- 63. Townsend D, Turner I, Yasuda S, Martindale J, Davis J, Shillingford M, Kornegay JN, Metzger JM. Chronic administration of membrane sealant prevents severe cardiac injury and ventricular dilatation in dystrophic dogs. J Clin Invest. 2010;120:1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. [DOI] [PubMed] [Google Scholar]
- 65. Jaffe AS, Vasile VC, Milone M, Saenger AK, Olson KN, Apple FS. Diseased skeletal muscle: a noncardiac source of increased circulating concentrations of cardiac troponin T. J Am Coll Cardiol. 2011;58:1819–1824. [DOI] [PubMed] [Google Scholar]
- 66. Fayssoil A, Renault G, Guerchet N, Marchiol‐Fournigault C, Fougerousse F, Richard I. Cardiac characterization of mdx mice using high‐resolution Doppler echocardiography. J Ultrasound Med. 2013;32:757–761. [DOI] [PubMed] [Google Scholar]
- 67. Crosbie RH, Lim LE, Moore SA, Hirano M, Hays AP, Maybaum SW, Collin H, Dovico SA, Stolle CA, Fardeau M, Tome FM, Campbell KP. Molecular and genetic characterization of sarcospan: insights into sarcoglycan‐sarcospan interactions. Hum Mol Genet. 2000;9:2019–2027. [DOI] [PubMed] [Google Scholar]
- 68. Wang L, Beecham A, Di Tullio MR, Slifer S, Blanton SH, Rundek T, Sacco RL. Novel quantitative trait locus is mapped to chromosome 12p11 for left ventricular mass in Dominican families: the Family Study of Stroke Risk and Carotid Atherosclerosis. BMC Med Genet. 2009;10:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. M‐mode images of isoproterenol‐treated SSPN‐null hearts. The representative M‐mode images show WT (top) and SSPN−/− (bottom) mice after β‐agonist treatment.
Figure S2. Expression levels of the SSPN transgene in mdx mice. Total protein samples obtained from total skeletal and cardiac muscle were immunoblotted to determine expression levels of the human SSPN transgene in both tissue types of mdx and mdxTG mice. GAPDH was included as a loading control.