

Abstract
Background
Ectopic vascular calcification is a common condition associated with aging, atherosclerosis, diabetes, and/or chronic kidney disease. Smooth muscle cells are the best characterized source of osteogenic progenitors in the vasculature; however, recent studies suggest that cells of endothelial origin can also promote calcification. To test this, we sought to increase the osteogenic potential of endothelial cells by overexpressing tissue‐nonspecific alkaline phosphatase (TNAP), a key enzyme that regulates biomineralization, and to determine the pathophysiological effect of endothelial TNAP on vascular calcification and cardiovascular function.
Methods and Results
We demonstrated previously that mice transgenic for ALPL (gene encoding human TNAP) develop severe arterial medial calcification and reduced viability when TNAP is overexpressed in smooth muscle cells. In this study, we expressed the ALPL transgene in endothelial cells following endothelial‐specific Tie2‐Cre recombination. Mice with endothelial TNAP overexpression survived well into adulthood and displayed generalized arterial calcification. Genes associated with osteochondrogenesis (Runx2, Bglap, Spp1, Opg, and Col2a1) were upregulated in the aortas of endothelial TNAP animals compared with controls. Lesions in coronary arteries of endothelial TNAP mice showed immunoreactivity to Runx2, osteocalcin, osteopontin, and collagen II as well as increased deposition of sialoproteins revealed by lectin staining. By 23 weeks of age, endothelial TNAP mice developed elevated blood pressure and compensatory left ventricular hypertrophy with preserved ejection fraction.
Conclusions
This study presented a novel genetic model demonstrating the osteogenic potential of TNAP‐positive endothelial cells in promoting pathophysiological vascular calcification.
Keywords: arteriosclerosis, cardiovascular diseases, endothelium, enzymes, lesion
Subject Categories: Animal Models of Human Disease, Endothelium/Vascular Type/Nitric Oxide, Basic Science Research, High Blood Pressure, Genetically Altered and Transgenic Models
Introduction
Vascular calcification is a pathological hallmark of advanced age, atherosclerosis, diabetes, and chronic kidney disease.1 Coronary artery calcification is an independent predictor of cardiovascular events,2, 3, 4, 5, 6, 7, 8, 9 whereas calcification of peripheral arteries is associated with all‐cause mortality.10 Consequently, calcification of vascular structures represents a significant medical problem.
Mechanisms of vascular calcification are complex; they typically require the relaxation of calcification inhibitors with concomitant increase in activators of biomineralization, both of which are tightly regulated at the cellular level.11 Transdifferentiation of vascular smooth muscle cells (VSMCs) is a well‐characterized phenomenon that shares common molecular pathways with skeletal mineralization.12 During this process, VSMCs upregulate expression of osteogenic transcription factors and begin secreting matrix vesicles, membrane microparticles that initiate hydroxyapatite crystallization.13, 14
Endothelial cells (ECs) could also promote vascular calcification, although evidence of this is still limited. Epidemiological studies suggest that circulating endothelial progenitor cells that express osteogenic markers (osteocalcin and/or alkaline phosphatase [AP]) correlate with vascular calcification in patients with atherosclerosis.15, 16, 17, 18 Moreover, the greater numbers of such cells are detected in patients with diabetes.19 Lineage‐tracing studies show that ECs contribute to the osteoprogenitor pool in aortic lesions in mice with experimental vascular calcification associated with matrix γ‐carboxyglutamic acid protein deficiency (Mgp knockout mice) and in the model of genetically determined diabetes (Akita mice).20 In a model of mitral valve calcification, ECs positioned along mechanically stretched valves express the osteoblastic marker osteocalcin, indicating their transdifferentiation.21 Outside of arterial and valvular examples, ECs isolated from solid tumors of the prostate, in which ectopic calcification colocalizes with endothelial marker CD31, clearly show ex vivo differentiation plasticity either toward chondrocytes expressing cartilage‐specific Col2a1 and Sox9 or osteoblasts expressing osteocalcin and osteopontin.22
Tissue‐nonspecific AP (TNAP) is the key enzyme that regulates availability of extracellular pyrophosphate (PPi), an inhibitor of biomineralization.23 Reduction in extracellular PPi promotes vascular calcification in animals,24, 25, 26 and the decreased levels of circulating plasma PPi have been associated with human genetic or metabolic conditions that predispose affected persons to vascular calcification.25, 27 Conversely, TNAP upregulation has been observed in calcified human arteries.28, 29, 30 Our recent study of transgenic mice overexpressing TNAP in VSMCs31 provided strong genetic evidence that TNAP is sufficient to induce severe vascular calcification and consequent cardiovascular complications—hypertension and heart failure—that drastically reduce the life span of affected animals. In this study, we used a similar strategy to manipulate expression of TNAP specifically in ECs to examine whether endothelial TNAP (eTNAP) might play a critical role in vascular calcification.
Materials and Methods
Animals
Transgenic mice expressing Cre recombinase driven by Tie2 gene promoter (Tie2‐Cre) were obtained from The Jackson Laboratory (Bar Harbor, ME; stock 8863). Characterization of this mouse strain has shown panendothelial expression of the Cre transgene.32 Hprt ALPL knock‐in mice were recently described.31 This mouse strain has a site‐specific transgenic insertion of the ALPL gene into the Hprt locus on the X chromosome. The expression construct contains a ubiquitous CAG promoter, a floxed “stop” cassette, and ALPL cDNA encoding human TNAP. Breeding of homozygous Hprt ALP/ALPL female mice with heterozygous Tie2‐Cre male mice results in the excision of the stop cassette in 50% of male offspring and activation of constitutive transgene expression in the ECs. Using this breeding scheme, male mice were produced and genotyped by polymerase chain reaction with Cre‐specific primers (forward: GCG GTC TGG CAG TAA AAA CTA TC; reverse: GTG AAA CAG CAT TGC TGT CAC TT) and an internal positive control primers (forward: CTA GGC CAC AGA ATT GAA AGA TCT; reverse: GTA GGT GGA AAT TCT AGC ATC ATC C). All animal studies were approved by the Sanford Research institutional animal care and use committee and complied with the National Institutes of Health guidelines for humane treatment of laboratory animals.
Whole‐Body X‐Ray Imaging
Whole body x‐ray images were obtained using small animal imager Carestream In‐Vivo Xtreme (Carestream Health). Mice were anesthetized by inhalation of 1.5% isoflurane in oxygen.
Blood Chemistry
Sera or heparin plasma were prepared from venous blood collected from right ventricles of anesthetized mice. All mice were fasted at least 5 hours before blood collection. AP activity, calcium, phosphorus, and creatinine were determined using ADVIA 1800 Clinical Chemistry analyzer (Siemens) with specific reagents: ALPAMP, CA_2, IP, and CREA. Inorganic PPi was measured in lithium heparin plasma according to the method described previously.33
Histology
AP activity was detected in fresh tissues embedded in optimal cutting temperature compound and cryosectioned at 5 μm. Sections were incubated in 5‐bromo‐4‐chloro‐3‐indolyl‐phosphate/nitro blue tetrazolium phosphatase substrate solution (KPL Inc.) for 5 minutes, rinsed in water, and fixed in 10% neutral buffered formalin. ECs were detected with FITC‐labeled isolectin B4 (IB4) from Bandeiraea simplicifolia (item FL1201; Vector Laboratories). Osteocalcin was detected with rabbit polyclonal antibody (item M173; Tanaka Bio Inc) followed by Alexa 488–labeled anti‐rabbit IgG secondary antibody (item 711‐546‐152; Jackson ImmunoResearch). Runx2 was detected with rat monoclonal antibody (item MAB2006; R&D Systems) followed by Alexa 546–labeled anti‐rat IgG antibody (item 712‐586‐153; Jackson ImmunoResearch). Collagen II was detected with sheep polyclonal antibody (item AF3615; R&D Systems) followed by Alexa 594–labeled anti‐sheep IgG antibody (item 713‐586‐147; Jackson ImmunoResearch). Corresponding secondary antibodies alone served as negative controls for osteocalcin, Runx2, and collagen II staining. Osteopontin was detected with goat polyclonal antibody directly labeled with phycoerythrin (item IC808P; R&D Systems); phycoerythrin‐labeled goat IgG (item IC108P; R&D Systems) served as a negative control. Sialic acid was detected with FITC‐labeled Sambucus nigra lectin (item FL1301; Vector Laboratories). For general histology, mice were perfused with neutral buffered formalin; tissues were dissected and stored in the same fixative (Formal‐Fixx; Thermo Scientific). Tissues were embedded in paraffin, sectioned, and stained with hematoxylin and eosin, Alizarin Red, Masson's trichrome, and von Kossa stains in the Sanford Research Molecular Pathology Core, following standard protocols.
Real‐Time Polymerase Chain Reaction
RNA was isolated from the aortas using a Maxwell 16LEV simplyRNA Tissue kit (item AS1280; Promega). RNA quantity and quality were determined using a NanoDrop 1000 (Thermo Scientific). Equal amounts of RNA from each sample were converted to cDNA using GoScript reverse transcriptase (item A501C; Promega) with random hexamer primers. Gene expression was evaluated using primers designed by Integrated DNA Technologies (Gapdh Mm.PT.39a.1; Alpl Mm.PT.56a.8794492; Sp7 Mm.PT.58.10898265; Bmp2 Mm.PT.58.10419414; Runx2 Mm.PT.58.41866893; Sox9 Mm.PT.58.42739087; Bglap Mm.PT.58.9119501.g; Spp1 Mm.PT.56.43709208; Mgp Mm.PT.58.32674923; Acan Mm.PT.58.10174685; Col2a1 Mm.PT.58.41487458; Opg Mm.PT.56a.414946; Tnfsf11 Mm.PT.55a.29202697) and SYBR green polymerase master mix reagent from Qiagen. Gene expression was normalized using the housekeeping gene Gapdh and expressed in arbitrary units, calculated as follows: · 105, where CtGene is the threshold cycle for target gene amplification, and CtGapdh is the threshold cycle for a reference gene (Gapdh).
Physiology
Echocardiography was performed using a Vevo2100 imaging system (FujiFilm Visual Sonics) equipped with an ultra–high‐frequency linear array solid state transducer (MS550D 22 to 55 MHz). Mice were anesthetized with 0.8% to 1% isoflurane, and a parasternal short‐axis view was obtained in B‐mode and recorded in M‐mode. M‐mode echocardiograms were analyzed by tracing myocardial wall movement over 3 to 5 cardiac cycles to measure heart rate and left ventricular (LV) diameter in systole and in diastole and to calculate ejection fraction, cardiac output, and LV mass.
Blood pressure was measured directly with a pressure transducer (SPR‐671; Millar Instruments). Right carotid artery catheterization was performed on mice anesthetized with 2% isoflurane, and data were recorded over 10 minutes at the maintenance level of 1.25% isoflurane in oxygen. On completion of data collection, mice were deeply anesthetized by 5% isoflurane inhalation prior to blood collection and euthanasia by exsanguination.
Statistics
Data were expressed as mean±SD or mean±SEM. GraphPad Prism version 5.0 (GraphPad Software) statistical software was used to analyze data. Two groups were compared by Student t test or nonparametric Mann–Whitney test when unequal variances were observed. One‐way ANOVA models were calculated to compare multiple groups, followed by Bonferroni‐adjusted multiple comparisons. The Kruskal–Wallis test was used to compare multiple groups when unequal variances were observed. This was followed by Dunn's multiple comparisons test. The significance was accepted at P<0.05.
Results
Panendothelial Expression of TNAP
To test the hypothesis that upregulation of TNAP in ECs would be sufficient to cause vascular calcification, we used a conditional knock‐in mouse model that overexpresses TNAP in a Tie2‐Cre recombinase–dependent manner. Male offspring mice were used in all experiments to avoid random X chromosome inactivation that affects levels of X‐linked ALPL transgene expression in female mice. The eTNAP mice were born at an expected Mendelian ratio (36 of 73 male pups) and survived into adulthood. To confirm TNAP overexpression, we examined the AP activity in eTNAP and wild‐type (WT) mice aged 8 weeks by incubating freshly prepared cryosections of various tissues in chromogenic AP substrate followed by formalin fixation. Using this method, increased AP activity was detected in all tissues of eTNAP compared with WT mice (Figure 1A). AP activity in eTNAP mice was localized to the luminal side of the aorta and vascular networks of heart, lung, kidney, liver, small intestine, and pancreas. A diffuse pattern of AP activity was observed in the parenchyma of lymphoid organs (spleen, thymus, and lymph nodes) of eTNAP mice but was absent in WT mice. In the heart, AP activity, both endogenous (detected in WT mice) and overexpressed (present in eTNAP mice), mirrored the characteristic pattern of capillary network identified by IB4 staining34 (Figure 1B). To rule out possible side effects of eTNAP overexpression on skeletal development, whole‐body x‐ray imaging was performed at 8 weeks of age and showed the lack of detectable skeletal abnormalities in eTNAP mice (Figure 1C).
Figure 1.
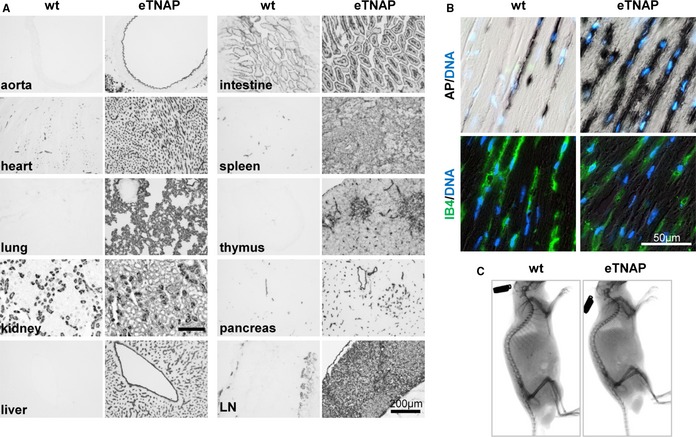
Expression of eTNAP. A, AP activity staining of the aorta, heart, lung, kidney, liver, small intestine, spleen, thymus, pancreas, and mesenteric LN from WT and eTNAP mice aged 8 weeks. B, Colocalization of AP activity with endothelial‐specific lectin (IB4) staining in the consecutive sections of myocardium counterstained with DAPI (DNA). C, Whole‐body x‐ray images of WT and eTNAP mice aged 8 weeks. AP indicates alkaline phosphatase; eTNAP, endothelial tissue‐nonspecific alkaline phosphatase; IB4, isolectin B4; LN, lymph node; WT, wild type.
Serum AP activity was also greatly upregulated in eTNAP compared with WT mice in all age groups examined (aged 8, 13, and 23 weeks), whereas serum calcium, phosphorus, and creatinine were comparable within each age group (Table 1). Plasma PPi concentrations were not significantly different between eTNAP and WT mice aged 13 weeks (Table 1). In this study, plasma samples were not collected from mice aged 8 and 23 weeks, thus we could not perform the PPi assay in those age groups due to artificial increase of PPi, released from platelets during blood coagulation.
Table 1.
Blood Chemistry
| Age | 8 Weeks | 13 Weeks | 23 Weeks | |||
|---|---|---|---|---|---|---|
| Parameter | WT (n=7) | eTNAP (n=7) | WT (n=8) | eTNAP (n=6) | WT (n=10) | eTNAP (n=7) |
| AP, U/L | 124±13 | 2912±11a | 75±15 | 2480±630† | 72±13 | 2425±325† |
| Ca, mg/dL | 8.9±0.5 | 9.0±0.6 | 9.1±0.7 | 9.0±0.3 | 8.8±0.5 | 9.0±0.4 |
| Pi, mg/dL | 6.5±1.5 | 6.8±1.1 | 6.1±0.9 | 5.4±0.6 | 7.1±1.3 | 6.2±0.6 |
| Creatinine, mg/dL | 0.17±0.07 | 0.14±0.05 | 0.13±0.04 | 0.11±0.03 | 0.11±0.04 | 0.11±0.02 |
| PPi, μmol/L | 1.7±1.0‡ | 1.7±0.8§ | ||||
Data are shown as mean±SD. AP indicates alkaline phosphatase; Ca, calcium; eTNAP, endothelial tissue‐nonspecific alkaline phosphatase; Pi, phosphorus; PPi, pyrophosphate; WT, wild type.
P<0.05.
† P<0.01 vs WT, Kruskal–Wallis test followed by Dunn's post hoc pairwise comparisons within each age group. ‡n=4. §n=7.
Histological Phenotype of eTNAP Mice
Histological examination (Alizarin Red staining) revealed the presence of calcified lesions in all tissues in eTNAP mice aged 13 weeks; no lesions were detected in WT mice (Figure 2). Calcified lesions in eTNAP mice were located in the arteries of the heart, kidney, mesentery, pancreas, and spleen as well as in the splenic capsule and lung parenchyma. Only minor lesions were detected in the aortas of eTNAP (distal thoracic aortas were sampled). No extraskeletal calcification was detected in preweaning‐aged eTNAP mice (1 week) by Alizarin Red staining; a panel of tissues included heart, liver, kidney, pancreas, spleen, and bone and served as a positive control (Figure 3).
Figure 2.
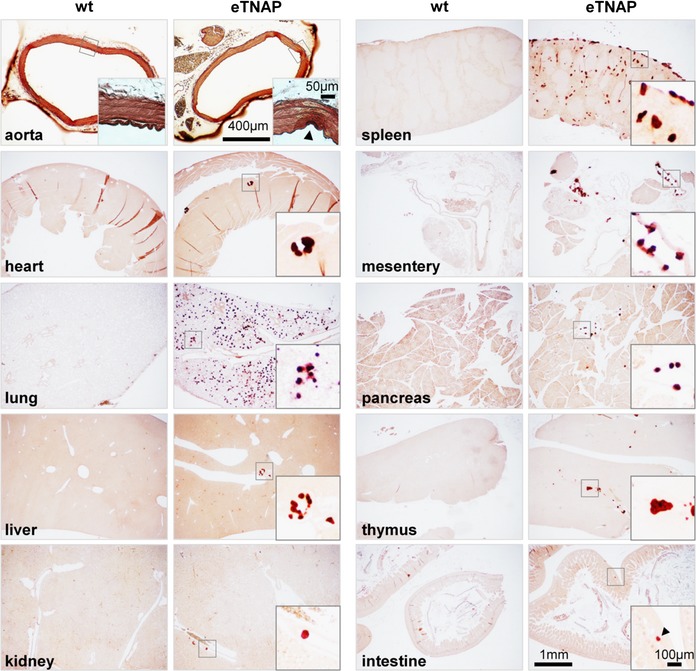
Calcification in eTNAP mice aged 13 weeks. Alizarin Red staining of the aorta, heart, lung, liver, kidney, spleen, mesentery, pancreas, thymus, and intestine from WT and eTNAP mice. eTNAP indicates endothelial tissue‐nonspecific alkaline phosphatase; WT, wild type.
Figure 3.
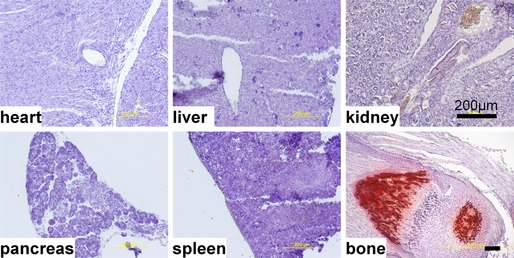
Lack of extraskeletal calcification in endothelial tissue‐nonspecific alkaline phosphatase mice aged 1 week. Heart, liver, kidney, spleen, pancreas, and bone were stained with Alizarin Red and counterstained with hematoxylin.
The distribution and vascular localization of the lesions in different tissues of adult eTNAP mice were examined by hematoxylin and eosin staining. The 5 oldest (aged 24 weeks) eTNAP mice and 6 WT controls were perfusion fixed with formalin, and tissues were dissected after fixation. Three paraffin blocks were prepared from each mouse. The first block contained pancreas, mesentery, and spleen; the second block contained liver (left lateral lobe) and left kidney; and the third block contained mid–cross‐section of the heart, left lung, and distal thoracic aorta. The presence of the lesions in different vascular beds and tissues in eTNAP mice was detected as basophilic inclusions in hematoxylin and eosin sections and confirmed by Alizarin Red staining. Consistent with observations in mice aged 13 weeks, calcification was absent in WT mice (not shown) and present in multiple tissues from eTNAP mice (histological findings are summarized in Table 2 and illustrated in Figure 4). In this sample, no lesions were detected in the aortas (n=3; aortas were not present in the plane of section from 2 other mice). Arterial lesions were present in the mesenteric sections from all 5 mice, and calcification affected arteries of all sizes in every animal (Figure 4A through 4C). In addition, lesions were detected in the large veins of the mesentery (Figure 4D). In the pancreas, lesions were present in the arteries (Figure 4E and 4F) and were not detected in the veins of the same size (Figure 4F). Similarly, multiple lesions were detected in the arteries of the spleens of eTNAP mice (Figure 4G and 4H) and were not detected in the veins draining the spleen. Splenic stromal elements and capsules were also extensively calcified (Figure 4I and 4J). Coronary arteries were affected in every mouse (Figure 4K and 4L); no lesions were detected in the coronary veins. In the lung, neither pulmonary arteries nor veins were involved; however, multiple small round calcification nodules were present throughout the whole parenchyma (Figure 4M). In this histological sample of the kidneys, multiple small lesions were present; however, the sectioning plane did not capture any larger arteries or veins. The small size of the calcified lesions in the kidney did not allow for their clear vascular definition (Figure 4N and 4O). Based on our histological survey, we concluded that calcification primarily affected peripheral and coronary arteries of eTNAP mice.
Table 2.
Tissues Affected by Calcification in eTNAP Mice Aged 24 Weeks (n=5)
| Tissue | Arteries (Size) | Panela | Veins (Size) | Panela | Other Sites | Panela |
|---|---|---|---|---|---|---|
| Aortab | n.d. | |||||
| Mesentery | Large, medium, small | A, B, C | Large | D | n.d. | |
| Pancreas | Medium, small | E, F | n.d. | n.d. | ||
| Spleen | Medium, small | G, H | n.d. | Stroma, capsule | I, J | |
| Heart | Medium, small | K, L | n.d. | n.d. | ||
| Lung | n.d. | n.d. | Parenchyma | M | ||
| Kidney | Smallc | N | Smallc | O | n.d. |
eTNAP indicates endothelial tissue‐nonspecific alkaline phosphatase; n.d., not detected.
shown in Figure 4.
n=3.
Unclear morphological definition.
Figure 4.
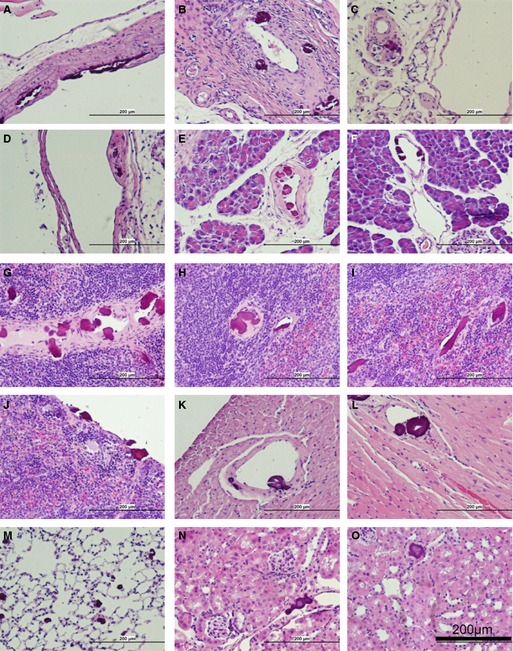
Histopathology of endothelial tissue‐nonspecific alkaline phosphatase mice aged 24 weeks (hematoxylin and eosin staining). Arteries of the mesentery: large (A), medium (B), and small (C) diameter. D, Large vein in the mesentery. Arteries of the pancreas: medium (E) and small (F). Arteries of the spleen: medium (G) and small(H). Nonvascular lesions in the splenic stroma (I) and capsule (J). Coronary arteries: medium (K) and small (L). M, Parenchyma of the lung. N and O, Lesions in the kidney.
In younger adult mice, we found that although small foci of calcification can be detected in the en face preparations of the aorta of eTNAP mice as early as 8 weeks of age (Figure 5A), coronary arteries were more extensively calcified compared with the aorta, as shown by Alizarin Red staining of the base of the heart at the level of the aortic sinus (Figure 5B). To investigate whether arterial lesions in eTNAP mice originate in direct contact with ECs, we examined the consecutive tissue sections of coronary arteries from mice aged 13 weeks by AP and IB4 staining (Figure 5C). Lesions in coronary arteries appeared as protrusions into the arterial lumen and were associated with AP‐ and IB4‐positive cells. The presence of vascular calcification in eTNAP mice aged 13 weeks was confirmed by von Kossa staining, which showed a spectrum of phenotypes. The earliest lesions appeared to originate in the subendothelial space and were negative for von Kossa staining (Figure 5D, left panel); a granular pattern of von Kossa staining was detected in the matrix of intermediate lesions (Figure 5D, middle panel). Advanced lesions contained a von Kossa–positive core and, in some cases, were accompanied by calcification of the elastic lamina and medial layer (Figure 5D, right panel). In hematoxylin and eosin–stained sections, basophilic inclusions were detected in all arterial lesions, including early subendothelial and late neointima‐encapsulated lesions (Figure 5E). Masson's trichrome staining highlighted the matrix core of the early lesions (light‐blue staining) surrounded by cells of endothelial morphology (Figure 5F, top panels), whereas late lesions featured more complex morphology with foci of irregular matrix inclusions and collagen deposits (Figure 5F, bottom panels). In many histological sections at high magnification, lesions displayed lamellar characteristics of their core region (Figure 5B, 5D and 5F).
Figure 5.
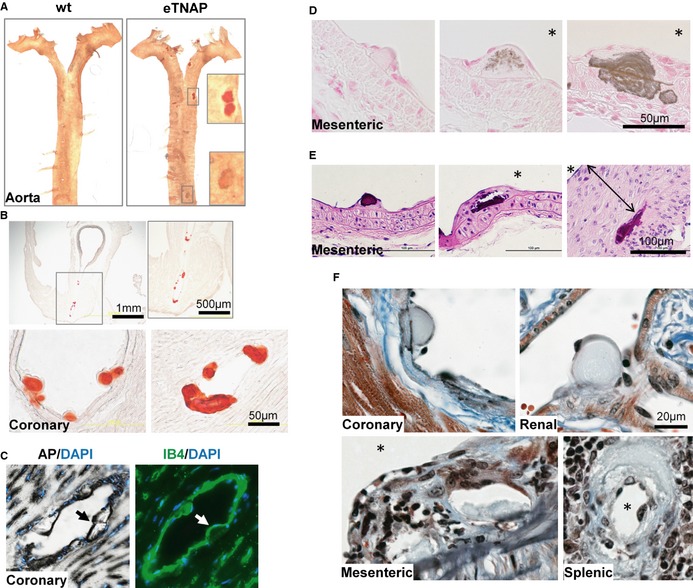
Structural characteristics of arterial lesions in eTNAP mice. A, Detection of Alizarin Red–positive lesions in flat‐mounted preparations of the aortas from eTNAP mice aged 8 weeks. B, Calcification in coronary artery sinus (top) and coronary arteries (bottom) in eTNAP mice aged 8 weeks visualized by Alizarin Red staining. C, Subendothelial localization of coronary lesions (highlighted by arrows) in eTNAP mice aged 13 weeks detected by AP and endothelial‐specific (IB4) staining in consecutive sections counterstained with DAPI (DNA). D, von Kossa–positive calcium deposits in mesenteric arteries of eTNAP mice aged 13 weeks. *, lumen. E, Mesenteric arteries from eTNAP mice aged 13 weeks, with hematoxylin and eosin staining; (double arrow) neointima. *, lumen. F, Masson's trichrome staining of coronary, renal, mesenteric, and spenic arteries from eTNAP mice aged 13 weeks. *, lumen. AP indicates alkaline phosphatase; eTNAP, endothelial tissue‐nonspecific alkaline phosphatase; IB4, isolectin B4; WT, wild type.
Composition of Arterial Lesions in eTNAP Mice
The composition of the matrix core was further examined for the presence of lipids and immunoreactivity for osteocalcin, a specific bone matrix protein.35 To detect lipid, cryosections of the hearts from WT and eTNAP mice aged 8 weeks were developed with lipophilic Oil Red O stain. No lipid inclusions were detected in the normal coronary arteries of WT or in association with the lesions in the arteries of eTNAP mice (Figure 6A). Immunofluorescent staining for osteocalcin specifically detected the presence this protein in the coronary lesions (both small and large) of eTNAP mice aged 8 weeks, whereas osteocalcin expression was undetectable in the normal coronary arteries of WT animals (Figure 6B).
Figure 6.
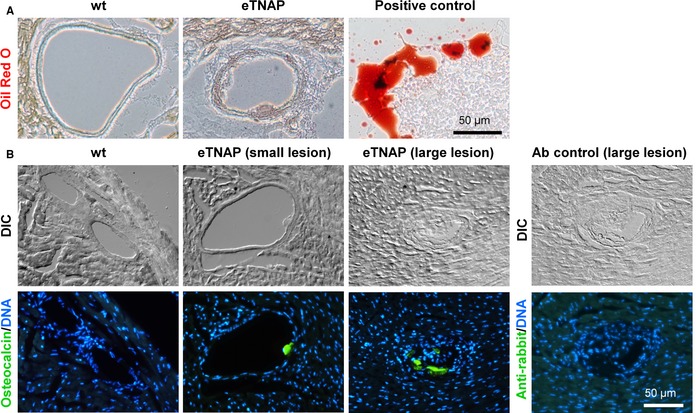
Matrix components of coronary lesions in eTNAP mice aged 8 weeks. A, Coronary arteries stained with lipophilic stain Oil Red O; artificial lipid pool detected in the same sections served as a positive control. B, Detection of osteocalcin by immunohistochemistry; secondary antibody alone served as a negative control; sections were counterstained with DAPI (DNA); DIC imaging. Ab, antibody; DIC indicates differential interference contrast; eTNAP, endothelial tissue‐nonspecific alkaline phosphatase; WT, wild type.
To examine the activation of an osteochondrogenic transdifferentiation program in the arteries of eTNAP mice that might potentially explain their histological phenotype, we isolated RNA from the aortas of TNAP and WT mice aged 13 weeks and analyzed the expression of a set of specific osteochondrogenic markers (Table 3). Several markers of osteogenesis—Runx2, osteocalcin, osteopontin, and osteoprotegerin—were significantly upregulated at mRNA level in the aortas of eTNAP mice compared with WT mice. In addition, the expression of the Col2a1 gene, encoding cartilage‐specific type II collagen, was also increased in eTNAP mice compared with controls. To confirm these observations at the protein level, we performed immunohistochemical analysis of coronary lesions from eTNAP mice using antibodies specific to Runx2, collagen II, and osteopontin. Runx2 staining was detected primarily in the arterial sections devoted to the nuclear DAPI stain, a region probably representing calcified VSMCs, and in the core of the lesions (Figure 7A). Collagen II was consistently detected in the core of coronary lesions (Figure 7B). Osteopontin staining appeared to localize to the periphery of coronary inclusions identified by differential interference contrast imaging (Figure 7C). Because osteopontin belongs to a group of sialylated matrix proteins (modified by carbohydrates, containing sialic acid36), we used a specific reagent, fluorescently labeled S nigra lectin, to test for the presence of sialic acid in the arterial lesions. Coronary lesions detected by differential interference contrast imaging were reactive with S nigra lectin, whereas unaffected arteries and surrounding normal cardiac tissue were negative for S nigra lectin staining (Figure 7D). Similar to osteopontin staining, S nigra lectin staining was brighter at the periphery of the coronary inclusions.
Table 3.
Gene Expression in the Aortas of Mice Aged 13 Weeks
| Gene | WT (n=5) | eTNAP (n=8) | Fold Changea |
|---|---|---|---|
| Alpl | 1484±256 | 2042±364 | 1.4 |
| Runx2 | 321±110 | 610±51 | 1.9† |
| Osterix | 11±4 | 23±8 | 2.1 |
| Sox9 | 473±145 | 966±204 | 2.0 |
| Bmp2 | 832±211 | 1366±191 | 1.6 |
| Osteocalcin | 15±5 | 52±11 | 3.5† |
| Osteopontin | 78±16 | 963±212 | 12.3‡ |
| Mgp | 113 436±33 570 | 200 105±28 472 | 1.8 |
| Aggrecan | 1772±600 | 2846±357 | 1.6 |
| Col2a1 | 5±1 | 40±15 | 8.0§ |
| Opg | 2668±846 | 6242±1060 | 2.3† |
| Rankl | 0.06±0.03 | 0.13±0.03 | 2.2 |
Data are shown as arbitrary units, mean±SEM. eTNAP indicates endothelial tissue‐nonspecific alkaline phosphatase; WT, wild type.
eTNAP relative to WT.
† P<0.05, Student t test, ‡ P<0.01. § P<0.05, Mann–Whitney test.
Figure 7.
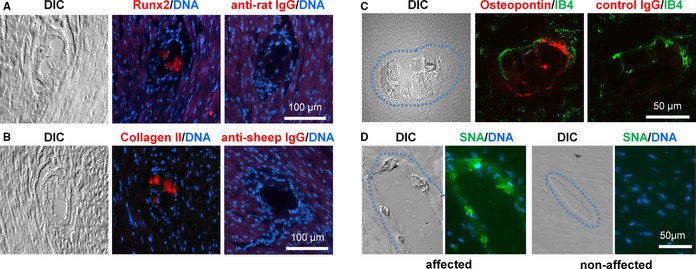
Expression of Runx2, collagen II, osteopontin, and sialic acid in coronary lesions of eTNAP mice. A through D, coronary lesions were identified by DIC imaging. A, Consecutive sections of the hearts from eTNAP mice aged 8 weeks were stained with Runx2 antibody or secondary antibody only, DNA binding dye DAPI was used as counterstain. B, Lesions from eTNAP mice aged 8 weeks were stained with collagen II antibody or secondary antibody alone and counterstained with DAPI. C, Osteopontin expression was detected in the lesions from eTNAP mice aged 13 weeks by direct immunofluorescent staining; IB4 was used as counterstain. D, The presence of sialic acid was detected in the coronary lesions from eTNAP mice aged 13 weeks with fluorescein‐labeled SNA lectin; DAPI was used as nuclear counterstain. Affected (left panels) and unaffected (right panels) coronary arteries from the same tissue section are shown. DIC indicates differential interference contrast; eTNAP, endothelial tissue‐nonspecific alkaline phosphatase; IB4, isolectin B4; SNA, Sambucus nigra.
Cardiovascular Phenotype of eTNAP Mice
Hemodynamic assessment was performed by catheterization of right carotid artery in eTNAP mice and control littermates aged 8, 13, and 23 weeks. Blood pressure was within the normal range and similar between groups at baseline (aged 8 weeks) and at an intermediate time point (aged 13 weeks) despite the presence of disseminated arterial calcification in eTNAP mice. At age 23 weeks, however, blood pressure became significantly elevated in eTNAP mice compared with controls (systolic, 126 versus 109 mm Hg; diastolic, 90 versus 79 mm Hg; P<0.01 for both) (Table 4). Analysis of cardiac function in eTNAP and WT animals was performed by M‐mode echocardiography (Table 4). The calculated LV mass showed LV hypertrophy in an older group (aged 23 weeks) of eTNAP mice. No differences in end‐diastolic or end‐systolic LV diameters were observed between groups, and the ejection fraction normalized to body weight was not significantly different between eTNAP and WT mice, indicating that LV hypertrophy in eTNAP mice aged 23 weeks was compensatory in nature and in line with elevated blood pressure (Table 4).
Table 4.
Cardiovascular Physiology
| Age | 8 Weeks | 13 Weeks | 23 Weeks | |||
|---|---|---|---|---|---|---|
| Parameter | WT (n=29) | eTNAP (n=31) | WT (n=18) | eTNAP (n=15) | WT (n=10) | eTNAP (n=8) |
| BW, g | 30.0±2.0 | 28.2±2.0 | 34.9±3.5 | 32.5±2.8 | 38.5±5.4 | 36.1±7.3 |
| HR, bpm | 451±25 | 434±34 | 439±31 | 427±28 | 505±66 | 524±52 |
| SBP, mm Hga | 101±4 | 99±7 | 102±7 | 107±11 | 109±6 | 126±8b |
| DBP, mm Hga | 75±3 | 73±5 | 75±5 | 81±10 | 79±5 | 90±7b |
| LV Diam(s), mm | 2.82±0.25 | 2.95±0.33 | 2.98±0.29 | 2.92±0.25 | 2.89±0.35 | 2.91±0.21 |
| LV Diam(d), mm | 4.11±0.19 | 4.14±0.25 | 4.28±0.20 | 4.16±0.18 | 4.21±0.31 | 4.11±0.16 |
| EF, % | 59.4±5.7 | 55.7±6.7 | 58.0±6.1 | 57.4±5.2 | 59.7±6.3 | 56.2±6.1 |
| CO/BW, mL/min per gram | 0.67±0.07 | 0.65±0.08 | 0.60±0.11 | 0.58±0.06 | 0.62±0.11 | 0.62±0.12 |
| LV/BW, mg/g | 3.23±0.33 | 3.25±0.42 | 3.12±0.40 | 3.23±0.54 | 3.01±0.49 | 3.79±1.19b |
Data are shown as mean±SD. bmp indicates beats per minute; BW, body weight; CO, cardiac output; DBP, diastolic blood pressure; EF, ejection fraction; eTNAP, endothelial tissue‐nonspecific alkaline phosphatase; HR, heart rate; LV Diam(d), diameter of left ventricle in diastole; LV Diam(s), diameter of left ventricle in systole; LV/BW, left ventricular mass/body weight ratio; SBP, systolic blood pressure; WT, wild type.
n=7 to 9.
P<0.01, vs same age WT, 1‐way ANOVA followed by Bonferroni‐adjusted multiple comparisons.
Discussion
In this study, we tested the hypothesis that elevated TNAP activity in ECs might shift the balance toward calcification of vascular structures; however, we did not anticipate that calcification would preferentially affect arteries and be largely absent in veins and microvasculature. This observation, combined with the fact that endogenous TNAP is normally expressed in a subset of microvascular ECs,37 points to a restricted role of osteogenic (TNAP‐positive) ECs in calcification of elastic arteries. Outside the arteries, calcification was observed in the lungs and splenic stroma and capsule, both tissues with high elastic content. Interestingly, haploinsufficiency of elastin was shown to reduce vascular calcification in one of the most severe models of vascular calcification, Mgp knockout mice,38 pointing to the importance of the mineral scaffolding function of elastic structures when other procalcifying conditions are equal.39 We can speculate that in our model of panendothelial TNAP‐driven arterial calcification, the “wear and tear” of extracellular matrix, proximal to the endothelial layer, might play a role in the initiation of osteochondrogenic transdifferentiation and early vascular lesions. To this end, a recent study demonstrated that activation of a group of endothelial elastases and kallikreins is sufficient to induce endothelial–mesenchymal transitions of ECs and contributes to vascular calcification in Mgp knockout mice.40
The eTNAP mice display massive upregulation of circulating TNAP activity, which raises a question about whether circulating TNAP should reduce the levels of plasma PPi, a known inhibitor of vascular calcification,24, 25, 26 and promote calcification independent of ECs. Previously, we reported findings from 2 nonvascular models of tissue‐specific TNAP overexpression (under the control of Col1a1 or apoE promoters41), in which we observed up to 20‐fold higher average TNAP activity in plasma in the absence of vascular calcification. Moreover, when TNAP was expressed in VSMCs31 or in ECs (this study), different patterns of vascular calcification were observed despite equally elevated circulating TNAP activity. Consequently, it appears that tissue distribution of TNAP (as opposed to its activity in the blood) is responsible for vascular calcification. Conversely, no significant reduction in plasma PPi was detected in any of these studies,31, 41 including the data presented in this report. TNAP is likely to be partially inhibited in whole plasma by the physiological concentrations of inorganic phosphate, whereas dilution of plasma in the conditions of clinical assay relieves this inhibition42 (but not the fold change). We can also speculate that the reduction in circulating PPi levels could be partially compensated by upregulation of PPi production, thus sustaining elevated TNAP activity. In our previous study, we detected higher expression of the PPi‐producing enzyme ectonucleotide pyrophosphatase/phosphodiesterase 1 (Enpp1), in TNAP transgenic mice compared with WT mice.31 The lack of systemic PPi reduction in this model and others suggests that local tissue concentrations of PPi (and the activity of TNAP) are more critical in regulating mineralization compared with their systemic levels.
Our initial assumption was that overexpression of TNAP and consequent reduction of tissue PPi would be sufficient to promote endothelial‐driven vascular calcification. Unexpectedly, we observed that TNAP overexpression induced the formation of arterial lesions that were apparently initiated by subendothelial accumulation of nonmineralized material. This matrix served as a scaffold for calcification at the later stages of lesion progression; therefore, we could not exclude that transdifferentiation of ECs into a chondogenic or osteogenic type is also required to promote vascular calcification, at least in this model of TNAP overexpression.
Despite TNAP overexpression in essentially every EC, the foci of calcification were rather infrequent, indicating that additional stimuli are required for ECs to initiate arterial lesions, even in the presence of high AP activity. A possible explanation is that ECs have to retain a certain level of pluripotency to progress toward transdifferentiation into the synthetic type capable of mounting an appropriate gene expression and secretion of matrix vesicles containing large amounts of substrates for mineral deposition. This theory would be in line with the recent epidemiological studies pointing out the important role of osteogenic endothelial progenitor cells in vascular calcification.15, 16, 17, 18
In this study, we showed that a selective rise of TNAP expression in the ECs is sufficient to trigger arterial calcification. The phenotype of eTNAP mice, however is substantially different from mice bearing TNAP in VSMCs.31 VSMC‐driven TNAP expression results in extensive calcification of the aorta, which leads to early lethality,31 whereas panendothelial TNAP manifests primarily and systemically as disseminated lesions in the smaller diameter arteries. Lesions nucleate in the subendothelial space of medium‐sized arteries and induce neointimal hyperplasia. Arterial remodeling, secondary to calcification, likely resulted in increased vessel wall stiffness, which can explain elevated blood pressure in older eTNAP mice. In this scenario, LV hypertrophy in eTNAP mice aged 23 weeks can be interpreted as a compensatory physiological response counteracting increased systemic vascular resistance. Decreased coronary perfusion due to partial occlusion of coronary arteries, however, can also play a role at this stage of cardiac remodeling.
Healthy absence of calcification in vasculature is maintained by a fine balance between activators and inhibitors of mineralization.11 These include factors promoting osteochondrogenic differentiation of cells residing in the vessel wall, enzymes and transporters maintaining in situ phosphorus and PPi levels, and extracellular matrix components that regulate calcium phosphate nucleation and crystal growth. We showed that ECs can induce vascular calcification and that upregulation of a single inducer of calcification, TNAP, is sufficient to promote the osteogenic potential of ECs.
Sources of Funding
This work was supported by in part by grants NIH DE12889 (Millán), Juvenile Diabetes Research Foundation 47‐2013‐522 (Savinov), the subcontract from NIH UC4 DK104194 grant (Savinov), and by Sanford Research Program Funds (Savinova). Sanford Research Molecular Pathology (histology), Imaging (microscopy), Tumor Biology (small animal imaging), and Molecular Genetics (qPCR) Cores were supported by the NIH grants P20 GM103548 and P20 GM103620.
Disclosures
None.
Acknowledgments
We thank Jacob Ellefson and Kelly Graber for technical assistance.
(J Am Heart Assoc. 2015;4:e002499 doi: 10.1161/JAHA.115.002499)
References
- 1. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. 2010;7:528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Polonsky TS, McClelland RL, Jorgensen NW, Bild DE, Burke GL, Guerci AD, Greenland P. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA. 2010;303:1610–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jain A, McClelland RL, Polak JF, Shea S, Burke GL, Bild DE, Watson KE, Budoff MJ, Liu K, Post WS, Folsom AR, Lima JA, Bluemke DA. Cardiovascular imaging for assessing cardiovascular risk in asymptomatic men versus women: the Multi‐Ethnic Study of Atherosclerosis (MESA). Circ Cardiovasc Imaging. 2011;4:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Folsom AR, Kronmal RA, Detrano RC, O'Leary DH, Bild DE, Bluemke DA, Budoff MJ, Liu K, Shea S, Szklo M, Tracy RP, Watson KE, Burke GL. Coronary artery calcification compared with carotid intima‐media thickness in the prediction of cardiovascular disease incidence: the Multi‐Ethnic Study of Atherosclerosis (MESA). Arch Intern Med. 2008;168:1333–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Taylor AJ, Bindeman J, Feuerstein I, Cao F, Brazaitis M, O'Malley PG. Coronary calcium independently predicts incident premature coronary heart disease over measured cardiovascular risk factors: mean three‐year outcomes in the Prospective Army Coronary Calcium (PACC) project. J Am Coll Cardiol. 2005;46:807–814. [DOI] [PubMed] [Google Scholar]
- 6. Arad Y, Goodman KJ, Roth M, Newstein D, Guerci AD. Coronary calcification, coronary disease risk factors, C‐reactive protein, and atherosclerotic cardiovascular disease events: the St. Francis Heart Study. J Am Coll Cardiol. 2005;46:158–165. [DOI] [PubMed] [Google Scholar]
- 7. Greenland P, LaBree L, Azen SP, Doherty TM, Detrano RC. Coronary artery calcium score combined with Framingham score for risk prediction in asymptomatic individuals. JAMA. 2004;291:210–215. [DOI] [PubMed] [Google Scholar]
- 8. Kondos GT, Hoff JA, Sevrukov A, Daviglus ML, Garside DB, Devries SS, Chomka EV, Liu K. Electron‐beam tomography coronary artery calcium and cardiac events: a 37‐month follow‐up of 5635 initially asymptomatic low‐ to intermediate‐risk adults. Circulation. 2003;107:2571–2576. [DOI] [PubMed] [Google Scholar]
- 9. Vliegenthart R, Oudkerk M, Hofman A, Oei HH, van Dijck W, van Rooij FJ, Witteman JC. Coronary calcification improves cardiovascular risk prediction in the elderly. Circulation. 2005;112:572–577. [DOI] [PubMed] [Google Scholar]
- 10. Allison MA, Hsi S, Wassel CL, Morgan C, Ix JH, Wright CM, Criqui MH. Calcified atherosclerosis in different vascular beds and the risk of mortality. Arterioscler Thromb Vasc Biol. 2012;32:140–146. [DOI] [PubMed] [Google Scholar]
- 11. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34:715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. [DOI] [PubMed] [Google Scholar]
- 13. Kapustin AN, Davies JD, Reynolds JL, McNair R, Jones GT, Sidibe A, Schurgers LJ, Skepper JN, Proudfoot D, Mayr M, Shanahan CM. Calcium regulates key components of vascular smooth muscle cell‐derived matrix vesicles to enhance mineralization. Circ Res. 2011;109:e1–e12. [DOI] [PubMed] [Google Scholar]
- 14. Chen NX, O'Neill KD, Chen X, Moe SM. Annexin‐mediated matrix vesicle calcification in vascular smooth muscle cells. J Bone Miner Res. 2008;23:1798–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang H, Wang LJ, Si DL, Wang C, Yang JC, Jiang P, Du C, Wang JJ. Correlation between osteocalcin‐positive endothelial progenitor cells and spotty calcification in patients with coronary artery disease. Clin Exp Pharmacol Physiol. 2015;42:734–739. [DOI] [PubMed] [Google Scholar]
- 16. Gossl M, Khosla S, Zhang X, Higano N, Jordan KL, Loeffler D, Enriquez‐Sarano M, Lennon RJ, McGregor U, Lerman LO, Lerman A. Role of circulating osteogenic progenitor cells in calcific aortic stenosis. J Am Coll Cardiol. 2012;60:1945–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Flammer AJ, Gossl M, Widmer RJ, Reriani M, Lennon R, Loeffler D, Shonyo S, Simari RD, Lerman LO, Khosla S, Lerman A. Osteocalcin positive CD133+/CD34‐/KDR+ progenitor cells as an independent marker for unstable atherosclerosis. Eur Heart J. 2012;33:2963–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gossl M, Modder UI, Atkinson EJ, Lerman A, Khosla S. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J Am Coll Cardiol. 2008;52:1314–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Flammer AJ, Gossl M, Li J, Matsuo Y, Reriani M, Loeffler D, Simari RD, Lerman LO, Khosla S, Lerman A. Patients with an HbA1c in the prediabetic and diabetic range have higher numbers of circulating cells with osteogenic and endothelial progenitor cell markers. J Clin Endocrinol Metab. 2012;97:4761–4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yao Y, Jumabay M, Ly A, Radparvar M, Cubberly MR, Bostrom KI. A role for the endothelium in vascular calcification. Circ Res. 2013;113:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wylie‐Sears J, Aikawa E, Levine RA, Yang JH, Bischoff J. Mitral valve endothelial cells with osteogenic differentiation potential. Arterioscler Thromb Vasc Biol. 2011;31:598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dudley AC, Khan ZA, Shih SC, Kang SY, Zwaans BM, Bischoff J, Klagsbrun M. Calcification of multipotent prostate tumor endothelium. Cancer Cell. 2008;14:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Millan JL. Mammalian Alkaline Phosphatases: From Biology to Applications in Medicine and Biotechnology. Weinheim, Germany: Wiley‐VCH Verlag GmbH & Co; 2006. [Google Scholar]
- 24. Villa‐Bellosta R, Rivera‐Torres J, Osorio FG, Acin‐Perez R, Enriquez JA, Lopez‐Otin C, Andres V. Defective extracellular pyrophosphate metabolism promotes vascular calcification in a mouse model of Hutchinson‐Gilford progeria syndrome that is ameliorated on pyrophosphate treatment. Circulation. 2013;127:2442–2451. [DOI] [PubMed] [Google Scholar]
- 25. Jansen RS, Duijst S, Mahakena S, Sommer D, Szeri F, Varadi A, Plomp A, Bergen AA, Oude Elferink RP, Borst P, van de Wetering K. ABCC6‐mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation‐brief report. Arterioscler Thromb Vasc Biol. 2014;34:1985–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lomashvili KA, Narisawa S, Millan JL, O'Neill WC. Vascular calcification is dependent on plasma levels of pyrophosphate. Kidney Int. 2014;85:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lomashvili KA, Khawandi W, O'Neill WC. Reduced plasma pyrophosphate levels in hemodialysis patients. J Am Soc Nephrol. 2005;16:2495–2500. [DOI] [PubMed] [Google Scholar]
- 28. Moe SM, Duan D, Doehle BP, O'Neill KD, Chen NX. Uremia induces the osteoblast differentiation factor Cbfa1 in human blood vessels. Kidney Int. 2003;63:1003–1011. [DOI] [PubMed] [Google Scholar]
- 29. Shroff RC, McNair R, Figg N, Skepper JN, Schurgers L, Gupta A, Hiorns M, Donald AE, Deanfield J, Rees L, Shanahan CM. Dialysis accelerates medial vascular calcification in part by triggering smooth muscle cell apoptosis. Circulation. 2008;118:1748–1757. [DOI] [PubMed] [Google Scholar]
- 30. Shanahan CM, Cary NR, Salisbury JR, Proudfoot D, Weissberg PL, Edmonds ME. Medial localization of mineralization‐regulating proteins in association with Monckeberg's sclerosis: evidence for smooth muscle cell‐mediated vascular calcification. Circulation. 1999;100:2168–2176. [DOI] [PubMed] [Google Scholar]
- 31. Sheen CR, Kuss P, Narisawa S, Yadav MC, Nigro J, Wang W, Chhea TN, Sergienko EA, Kapoor K, Jackson MR, Hoylaerts MF, Pinkerton AB, O'Neill WC, Millan JL. Pathophysiological role of vascular smooth muscle alkaline phosphatase in medial artery calcification. J Bone Miner Res. 2015;30:824–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2‐Cre transgenic mice: a new model for endothelial cell‐lineage analysis in vivo. Dev Biol. 2001;230:230–242. [DOI] [PubMed] [Google Scholar]
- 33. Yadav MC, Huesa C, Narisawa S, Hoylaerts MF, Moreau A, Farquharson C, Millan JL. Ablation of osteopontin improves the skeletal phenotype of phospho1(‐/‐) mice. J Bone Miner Res. 2014;29:2369–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Savinova OV, Liu Y, Aasen GA, Mao K, Weltman NY, Nedich BL, Liang Q, Gerdes AM. Thyroid hormone promotes remodeling of coronary resistance vessels. PLoS One. 2011;6:e25054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Price PA. Gla‐containing proteins of bone. Connect Tissue Res. 1989;21:51–57; discussion 57‐60. [DOI] [PubMed] [Google Scholar]
- 36. Butler WT. The nature and significance of osteopontin. Connect Tissue Res. 1989;23:123–136. [DOI] [PubMed] [Google Scholar]
- 37. Zoellner HF, Hunter N. Histochemical identification of the vascular endothelial isoenzyme of alkaline phosphatase. J Histochem Cytochem. 1989;37:1893–1898. [DOI] [PubMed] [Google Scholar]
- 38. Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. [DOI] [PubMed] [Google Scholar]
- 39. Khavandgar Z, Roman H, Li J, Lee S, Vali H, Brinckmann J, Davis EC, Murshed M. Elastin haploinsufficiency impedes the progression of arterial calcification in MGP‐deficient mice. J Bone Miner Res. 2014;29:327–337. [DOI] [PubMed] [Google Scholar]
- 40. Yao J, Guihard P, Blazquez‐Medela AM, Guo Y, Moon JH, Jumabay M, Bostrom KI, Yao Y. Serine protease activation essential for endothelial‐mesenchymal transition in vascular calcification. Circ Res. 2015;117:758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Narisawa S, Yadav MC, Millan JL. In vivo overexpression of tissue‐nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J Bone Miner Res. 2013;28:1587–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coburn SP, Mahuren JD, Jain M, Zubovic Y, Wortsman J. Alkaline phosphatase (EC 3.1.3.1) in serum is inhibited by physiological concentrations of inorganic phosphate. J Clin Endocrinol Metab. 1998;83:3951–3957. [DOI] [PubMed] [Google Scholar]