

Abstract
Background
Idiopathic dilated cardiomyopathy (DCM) is typically diagnosed in adulthood, yet familial cases exhibit variable age‐dependent penetrance and a subset of patients develop sporadic DCM in childhood. We sought to discover the molecular basis of sporadic DCM in an 11‐year‐old female with severe heart failure necessitating cardiac transplantation.
Methods and Results
Parental echocardiograms excluded asymptomatic DCM. Whole exome sequencing was performed on the family trio and filtered for rare, deleterious, recessive, and de novo variants. Of the 8 candidate genes identified, only 2 had a role in cardiac physiology. A de novo missense mutation in TNNT2 was identified, previously reported and functionally validated in familial DCM with markedly variable penetrance. Additionally, recessive compound heterozygous truncating mutations were identified in XIRP2, a member of the ancient Xin gene family, which governs intercalated disc (ICD) maturation. Histomorphological analysis of explanted heart tissue revealed misregistration, mislocalization, and shortening of ICDs, findings similar to Xirp2 −/− mice.
Conclusions
The synergistic effects of TNNT2 and XIRP2 mutations, resulting in perturbed sarcomeric force generation and transmission, respectively, would account for an early‐onset heart failure phenotype. Whereas the importance of Xin proteins in cardiac development has been well established in animal models, this study implicates XIRP2 as a novel modifier gene in the pathogenesis of DCM.
Keywords: dilated cardiomyopathy, genetics, heart failure, pediatrics, whole exome sequencing
Subject Categories: Genetics, Cardiomyopathy, Pediatrics, Heart Failure
Introduction
Dilated cardiomyopathy (DCM), characterized by left ventricular dilation and systolic dysfunction, is a degenerative myocardial disorder resulting in heart failure and premature death. Consequentially, it is the most common indication for cardiac transplantation in adults and children.1, 2 DCM is typically diagnosed in the fourth to fifth decade of life subsequent to symptomatic presentation after an unknown period of insidiously silent disease progression.3 Identification of idiopathic DCM as a familial disorder in 20% to 48% of cases has provided the rational for screening echocardiography to detect presymptomatic DCM in family members and launched efforts to discover its genetic underpinnings.3, 4, 5 DCM has proved to be genetically heterogeneous, attributable to mutations in over 50 genes.6, 7, 8 DCM is associated with variable age‐dependent penetrance, even among carriers of the same heterozygous mutation segregating within a family, attributable to poorly understood molecular mechanisms. Moreover, a subset of patients develops symptomatic DCM and advanced heart failure in childhood in the absence of myocardial disease in their adult parents. Whereas sporadic DCM in an infant or child should prompt a broad differential diagnosis and testing for reversible causes of heart failure,8 it remains an idiopathic and likely genetic disorder in 66% of children.9 Emerging data from clinical gene panel testing suggest that the genetic landscape of DCM in children and adults is different.10 Indeed, pediatric DCM provides a unique opportunity to identify mutations with major effect on cardiac structure and function in the absence of acquired age‐dependent risk factors for heart failure. Whole exome sequencing (WES) is a transformative technology that overcomes the limitations of a candidate gene approach, enabling discovery of new and unsuspected genetic underpinnings of DCM, including sporadic cases.
Methods
Study Subjects
Subjects provided written informed consent under a research protocol approved by the Mayo Clinic Institutional Review Board and were phenotypically classified by transthoracic echocardiography. Diagnostic criteria for DCM were left ventricular diastolic and/or systolic short‐axis chamber dimension Z‐score ≥2.0 and left ventricular ejection fraction <50%. Genomic DNA was isolated from peripheral blood white cells. Formalin‐fixed, paraffin‐embedded cardiac tissue was available from the patient and 3 additional pediatric subjects, procured at cardiac transplantation or autopsy.
Array Comparative Genomic Hybridization
Array comparative genomic hybridization using a custom 180K oligonucleotide microarray (Agilent, Santa Clara, CA) was performed on a DNA sample from the affected patient with a genome‐wide functional resolution of 100 kilobases (kb). Deletions ≥200 kb and duplications ≥500 kb were considered clinically relevant. Those below the size threshold were considered relevant if sufficient evidence supporting pathogenicity from Online Mendelian Inheritance in Man (OMIM) (http://ncbi.nlm.nih.gov/omim), PubMed (http://ncbi.nlm.nih.gov/pubmed), or ClinGen Dosage Sensitivity Map (http://ncbi.nlm.nih.gov/projects/dbvar/clingen/index.shtml) was present.
WES and Bioinformatics
WES and variant annotation were performed on DNA samples from the affected patient and both clinically unaffected parents, utilizing the Mayo Clinic Medical Genome Facility and Bioinformatics Core. The Agilent SureSelect Human All Exon v4+UTRs capture kit (Agilent, Santa Clara, CA) was used for exome capture. Samples from the family trio were multiplexed on a single lane and 101‐bp, paired‐end sequencing was performed on Illumina's HiSeq2000 platform (Illumina, Inc, San Diego, CA). Reads were aligned to the hg19 reference genome with Novoalign (http://novocraft.com) followed by sorting and marking of duplicate reads using Picard (http://picard.sourceforge.net). Local realignment of insertions/deletions (INDELs) and base quality score recalibration were then performed using the Genome Analysis Toolkit (GATK).11 Single‐nucleotide variants (SNVs) and INDELs were called across all 3 samples simultaneously using GATK's UnifiedGenotyper with variant quality score recalibration.12 The resultant variant call format files were analyzed with QIAGEN's Ingenuity Variant Analysis software (QIAGEN, Redwood City, CA). To determine rarity of variants, minor allele frequencies from 3 publicly available databases were utilized, collectively comprised of WES or whole genome sequences (WGS) from 7664 individuals: 1000 Genomes, WGS data from 1092 individuals13; Exome Variant Server (EVS), WES data from 6503 individuals (http://evs.gs.washington.edu/EVS/); and Complete Genomics Genome (CG), WGS data from 69 individuals.14 To exclude false positives, in‐house WGS or WES data from 115 individuals not affected with DCM were utilized. Genes were then prioritized based on fulfilling 1 or more of the following criteria.
Striated muscle enrichment
The Human Protein Atlas15 was utilized to determine RNA expression levels across a range of tissues. Specifically, RNA tissue expression was analyzed to determine whether striated‐muscle–specific expression was observed in any of the 8 candidate genes.
Murine cardiac phenotype
The Mouse Genome Informatics database16 was utilized to query all 8 candidate genes to determine whether murine ortholog knockout data were available and, if so, resulted in either an abnormal cardiac phenotype, or an unrelated phenotype.
≤1 Node upstream of DCM genes
A gene list comprised of 50 genes (45 on Partners Healthcare DCM gene panel [http://personalizedmedicine.partners.org], EYA4, TMPO, PSEN1, PSEN2, and SYNE1)6, 7, 8 associated with DCM was uploaded to Ingenuity Variant Analysis, and variants were filtered to include those within genes either directly associated with disease or 1 node removed from an associated DCM gene.
Sanger Sequencing
Sanger sequencing was performed on DNA samples from the family trio to confirm mutations identified by WES. The primer pairs used for the polymerase chain reaction were: TNNT2 c.421C>T (NM_001001430.2) forward 5′– GGCACCATTGCTTCAAGACT –3′, reverse 5′– CTCAAGTGATCTACCCGCCTT –3′; XIRP2 c.5460delT (NM_152381.5) forward 5′– TCAAATGAAACACTGACAGC –3′, reverse 5′– TGACAATCTTTACAGTGCCTT –3′; XIRP2 c.6968dupA (NM_152381.5) forward 5′– ATCCAATCAACTTTAACCCT –3′, reverse 5′– CCTGTTATGATTTTAGCCTGA –3′.
Histochemistry and Immunohistochemistry
Left ventricular tissue, procured from the patient's explanted heart at the time of transplantation (age 12), was formalin‐fixed and paraffin embedded by standard clinical laboratory protocols. Sections (5 μm thick) were obtained from the paraffin tissue blocks, and hematoxylin and eosin (H&E) and Masson trichrome‐stained slides were generated using standard clinical histochemistry protocols. Additional 5‐μm sections were obtained for immunohistochemical staining. Three additional specimens served as controls. Left ventricular tissue was obtained at autopsy from a 13‐year‐old male who died from noncardiac causes and had no evidence of DCM or other cardiac disease. Explanted left ventricular tissue was available from 2 individuals with genetically defined DCM who underwent cardiac transplantation: a 9‐year‐old female with a mutation in LMNA, encoding lamin A/C, and a 12‐year‐old female with a mutation in RBM20, encoding RNA‐binding motif protein 20. Staining was performed using the Leica Bond RX Stainer (Leica Microsystems, Buffalo Grove, IL). Tissue slides were dewaxed and antigens were retrieved online using Bond Dewax and Epitope Retrieval 2 (Leica Microsystems). For N‐cadherin immunostaining, tissue slides were subjected to antigen retrieval for 20 minutes. The primary antibody directed against N‐cadherin (Clone IAR06; Leica Microsystems) was used at 1:75 dilution and slides were incubated for 15 minutes. For β‐catenin immunostaining, tissue slides were subjected to antigen retrieval for 10 minutes. The primary antibody directed against β‐catenin (clone E5; Santa Cruz Biotechnology, Santa Cruz, CA) was used at 1:1000 dilution and slides were incubated for 15 minutes. The detection system used for both primary antibodies was the Polymer Refine Detection System (Leica Microsystems). This system includes a hydrogen peroxidase blocking step, a secondary antibody conjugated to HRP, a DAB‐based reaction, and hematoxylin counterstain. Once completed, slides were dehydrated in increasing concentrations of ethyl alcohol and rinsed for 5 minutes in tap water. Slides were then dehydrated in increasing concentrations of ethyl alcohol and xylene before permanent cover slipping in xylene based media.
Transmission Electron Microscopy
Formalin‐fixed cardiac tissue was obtained from the paraffin tissue blocks and deparaffinized by standard clinical laboratory protocols. Tissues were rinsed 3 times in 0.1 mol/L of sodium phosphate buffer, postfixed, and stained in 1% osmium tetroxide. Tissues were then rinsed 3 times in distilled water, en bloc stained in 2% aqueous uranyl acetate, and dehydrated in a graded series of ethanol followed by absolute acetone. Tissues were subsequently infiltrated and embedded in epoxy resin. Semithin (0.6 μm) sections for light microscopy were cut with an ultramicrotome and stained with toluidine blue. Thin sections, ≈100 nm in thickness, were cut with an ultramicrotome, poststained with 0.3% aqueous lead citrate, and examined in a FEI Tecnai G2 12 transmission electron microscope operated at 80 kV. Digital images were captured with a Gatan Model 785 ES1000W Erlangshen side mount 4k×2.7k CCD camera and Digital Micrograph software.
Results
Clinical Presentation of Sporadic, End‐Stage DCM in a Pediatric Patient
The patient was diagnosed with DCM at age 11, subsequent to the incidental finding of cardiomegaly on a computed tomography scan obtained for abdominal pain. Echocardiography and cardiac catheterization revealed a severe form of DCM with marked systolic and diastolic dysfunction: left ventricular diastolic/systolic short‐axis chamber dimensions=62/58 mm (Z‐scores=7.4/12.5); left ventricular ejection fraction=12% (Z‐score=−10.9); mild‐moderate mitral valve regurgitation; severe left atrial enlargement; and left ventricular end‐diastolic pressure=27 mm Hg (normal <12 mm Hg). Circulating N‐terminal pro‐brain natriuretic peptide was significantly elevated at 3874 pg/mL (normal, <140 pg/mL). Electrocardiography demonstrated normal sinus rhythm, left atrial enlargement, left axis deviation, prolonged QTc interval, and abnormal Q waves and T‐wave inversion in the lateral leads. Screening echocardiograms in her parents revealed normal left ventricular size and systolic function (father, age 53; mother, age 49). The patient developed progressive, end‐stage heart failure and ventricular tachycardia despite maximal medical therapy, culminating in treatment with a continuous milrinone infusion, implantable cardioverter‐defibrillator, and ultimately cardiac transplantation within 1 year of diagnosis. Weight of the explanted heart was 2.6 times greater than the expected mean for the patient's age and height/weight, with severe biventricular hypertrophy, marked biventricular dilation, and moderate interstitial fibrosis.
Exome Sequencing Reveals De Novo TNNT2 and Compound Heterozygous XIRP2 Mutations
Array comparative genomic hybridization ruled out chromosomal aneuploidy in the patient. To identify a pathogenic mutation(s), targeted exome capture and WES were performed on genomic DNA samples from the family trio. All 3 samples had over 98% of reads map to the hg19 reference genome, and, on average, 95% of the 71 megabase capture region had ≥×20 coverage. Similarly, post‐hoc analysis of DCM genes from GeneDx's DCM/LVNC (http://genedx.com/) and Ambry Genetics DCMNEXT panels (http://ambrygen.com/) revealed coverage of 94% of the coding and untranslated regions. Approximately 73 000 SNVs and 9500 INDELs were identified in the coding region of each sample, which was within an expected range for our standard variant calling workflow. An iterative variant filtering approach was then employed (Figure 1). Variants that mapped to the coding region and passed quality score recalibration were filtered to exclude those located in the top 0.5% of the most exonically variable genes and present in 3 or more in‐house, non‐DCM controls. Variants were then filtered for rarity, excluding variants with a minor allele frequency ≥1.0% in 1000 Genomes,13 EVS (http://evs.gs.washington.edu/EVS/), or CG.14 Next, all truncation, canonical splice‐site, and missense variants were retained. Filtered data from the patient were analyzed in conjunction with parental WES data to model all potential modes of inheritance for sporadic DCM, including homozygous recessive, compound heterozygous, uniparental disomy, and de novo, culminating in a candidate list of 12 variants in 8 genes (Table S1). Genes were then prioritized based on striated muscle‐specific RNA expression,15 abnormal cardiac phenotype upon murine ortholog knockout,16 and connection to a known DCM gene (n=50 genes).
Figure 1.
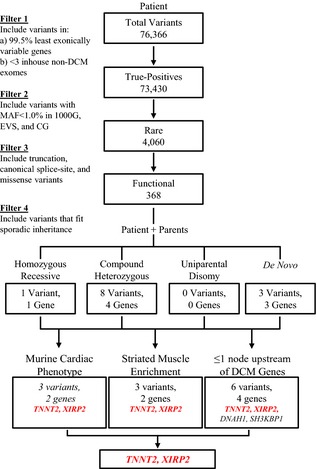
To identify a pathogenic mutation among variants identified by whole exome sequencing, an iterative filtering scheme was employed. Twelve rare coding variants in 8 genes fit plausible modes of inheritance for sporadic DCM. Of the 12 variants (Table S1), only 3 variants in 2 candidate genes, TNNT2 and XIRP2, fit >1 of the following criteria: showed striated muscle enrichment; had an associated cardiac phenotype in mice; and/or were located within 1 node of a known DCM gene. 1000G indicates 1000 genomes; CG, complete genomics; DCM, dilated cardiomyopathy; EVS, exome variant server; MAF, minor allele frequency.
Four of the 8 candidate genes, ZYG11B, PMPCB, ZBTB10, and BRCA2, did not fulfill any of the 3 criteria. Two of the 8 candidate genes, DNAH1 and SH3KBP1, fulfilled the sole criterion of being connected to a known DCM gene. DNAH1, harboring compound heterozygous missense variants, was linked to CRYAB, an established DCM gene, from a proteomic study performed on brain samples from patients with Alzheimer's disease.17 Knockout of the murine ortholog results in phenotypes unrelated to DCM.18 SH3KBP1, harboring a de novo missense mutation, was linked to 2 established DCM genes, ACTC1 and LMNA. The connection to ACTC1 was from a study that showed its involvement in cross‐linking F‐actin in podosomes of fibroblasts,19 whereas the connection to LMNA was a false‐positive text‐mining link based on a study utilizing LMNA as a negative control when determining binding of SH3BP2 to SH3KBP1.20 Murine ortholog knockout restricted to B cells elicited a definitive B‐ and T‐cell phenotype,21 which was not evident in our patient in a pretransplant quantitative B‐ and T‐cell surface marker assay. Consequently, neither DNAH1 nor SH3KBP1 were prioritized for further investigation.
The remaining 2 candidate genes, TNNT2 and XIRP2, fulfilled all 3 criteria, with robust supportive evidence for each. Troponin T type 2 (cardiac) (TNNT2, OMIM 191045) had over 30 murine cardiac identifiers, was categorized as being tissue enriched in cardiac muscle, and had multiple reports associating it with DCM, 1 of which reported the exact substitution observed in our patient.22, 23 Xin actin‐binding repeat containing 2 (XIRP2, OMIM 609778) had 18 murine cardiac identifiers, was categorized as being heart/skeletal muscle enriched, and was connected to NEBL, a known DCM gene.24 In examining the role of XIRP2 and its importance in striated muscle development and myofibril assembly, coimmunoprecipitation and cocrystallization assays showed binding of XIRP2 through a proline‐rich sequence to the SH3 domain of NEBL,24 providing robust supportive evidence linking XIRP2 to a known DCM gene.
The patient was found to harbor a pathogenic de novo missense mutation in TNNT2 (c.421C>T p. R141W; Figure 2) and compound heterozygous truncating mutations in XIRP2 (c.5460delT p.Phe1820fs*2 on the paternal allele; c.6968dupA p.Asn2323fs*15 on the maternal allele; Figure 3). The truncation mutations were recessive, given that each was inherited from an unaffected parent, and added an anomalous peptide tail of 2 and 15 residues, respectively.
Figure 2.
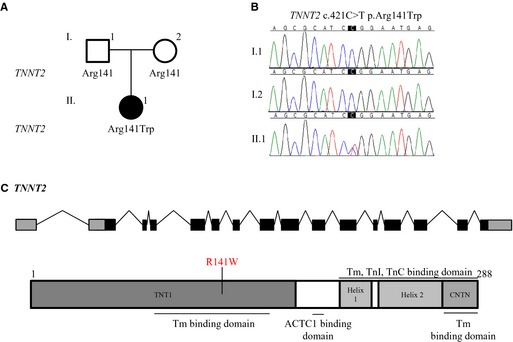
Whole exome sequencing identified a de novo mutation in TNNT2. A, Family pedigree. Square=male; circle=female; solid=affected; open=unaffected. B, Sanger sequencing of TNNT2 confirmed the R141W mutation. C, Gene and protein topology for TNNT2. The identified substitution lies within a known Tm binding domain. ACTC1 indicates actin, alpha, cardiac muscle 1; Tm, tropomyosin; TnC, troponin C type 1 (slow); TnI, troponin I type 3 (cardiac).
Figure 3.
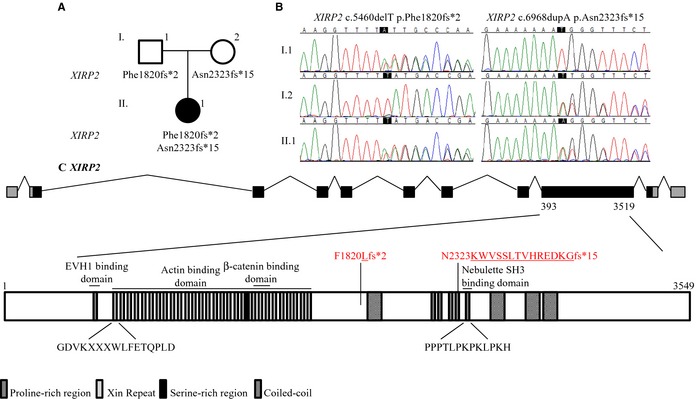
Whole exome sequencing identified compound heterozygous truncating mutations in XIRP2. A, Family pedigree. B, Sanger sequencing of XIRP2 confirmed the truncation mutations. C, Gene and protein topology for XIRP2. XIRP2 contains 28 xin repeats, 6 proline‐rich regions, a serine‐rich region, and 4 coiled‐coil domains. The Asn2323fs*15 mutation adds a 15‐residue anomalous peptide tail and truncates the protein within the 5th proline‐rich region before the last proline‐rich region, which binds to the SH3 domain of nebulette, and 3 of the coiled‐coil domains. The Phe1820fs*2 occurs even further upstream, truncating 3 additional proline‐rich regions and a coiled‐coil domain. EVH1 indicates Ena/VASP homology 1 domain.
Immunohistochemistry Demonstrates Misregistration, Mislocalization, and Shortening of ICDs
Histomorphologic analysis of H&E and trichrome‐stained sections of left ventricular myocardium revealed similar chronic changes in the patient harboring TNNT2 and XIRP2 mutations and 2 pediatric DCM controls, compared to a normal pediatric control (Figure 4A through 4H). Alterations included moderate‐to‐severe myocyte hypertrophy with marked nuclear enlargement and mild patchy interstitial fibrosis—nonspecific findings that are characteristically observed in DCM.
Figure 4.
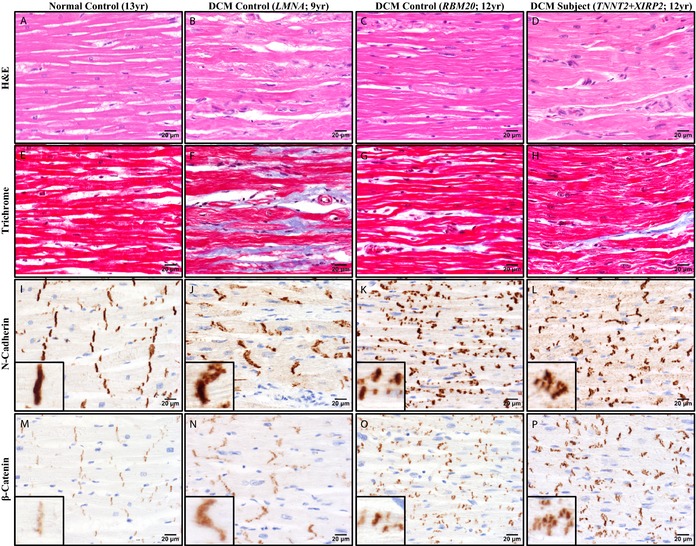
Histochemistry and immunohistochemistry revealed abnormalities in ICD structure in pediatric DCM. Representative photomicrographs of left ventricular myocardial tissue from a normal male control, age 13 (A, E, I, and M); a DCM female control harboring a LMNA mutation, age 9 (B, F, J, and N); a DCM female control harboring a RBM20 mutation, age 12 (C, G, K, and O); and the female DCM patient harboring TNNT2 and XIRP2 mutations, age 12 (D, H, L, and P). All 3 DCM patients showed nonspecific alterations (B through D, H&E=hematoxylin & eosin; F through H, Masson trichrome). N‐cadherin (I through L) and β‐catenin (M through P) immunohistochemistry revealed ICD abnormalities that differed among the DCM patients. The LMNA DCM control showed many irregular ICDs oriented obliquely, but with ICD length relatively preserved (J and N). In contrast, the RBM20 DCM control showed many short ICDs, including unusual linear arrays of multiple short parallel ICDs distributed along the sides of myocytes (K and O). The patient with TNNT2 and XIRP2 mutations showed severely disorganized ICDs, with numerous clustered groups composed of a mixture of oblique, short, and irregular forms (L and P). Immunoreactivity of ICDs for β‐catenin was increased in all DCM individuals relative to the normal control (M through P). Scale bars (A through P)=20 μm. DCM indicates dilated cardiomyopathy; ICD, intercalated disc.
Because N‐cadherin and β‐catenin are important structural and signaling components of ICDs and a β‐catenin‐binding domain has been mapped within the Xin repeats of XIRP225 (Figure 3C), immunohistochemistry was performed to evaluate alterations in their expression and/or localization. Immunostaining for N‐cadherin revealed abnormalities of localization and microanatomic structure of ICDs that differed among the individuals with DCM (Figure 4I through 4L). By light microscopy, ICDs in the normal control were linear and well organized at the ends of each myocyte, oriented perpendicular to the long axis of the myocytes. Furthermore, myocytes were well organized relative to their neighbors, with groups of adjacent ICDs tending to line up in loose registry with one another. This registry of ICDs was lost to varying degrees in the individuals with DCM, particularly in our patient and the RBM20 control. In addition to misregistration of ICDs, DCM hearts also showed mislocalization and/or shortening of ICDs that differed by genetic background. The LMNA DCM control showed the subtlest abnormalities, with many ICDs oriented obliquely, rather than perpendicular, to the longitudinal axis of the myocytes, but with ICD length relatively preserved. The RBM20 DCM control had more‐striking abnormalities, with many myocytes showing multiple short ICDs distributed along their sides. Our patient showed marked disorganization of ICDs with a mixture of oblique, short, and long linear forms oriented parallel to the longitudinal axis of the myocyte, with groups of short ICDs forming a stair‐step pattern across myocytes. Immunostaining for β‐catenin revealed increased immunoreactivity of ICDs in all DCM individuals relative to the normal control, suggesting increased expression and/or localization of β‐catenin at the ICD, as has been described previously in patients with hypertrophic cardiomyopathy26 (Figure 4M through 4P).
Electron Microscopy Confirms Abnormal ICD Ultrastructure
Transmission electron microscopy performed on myocardial tissue demonstrated several ultrastructural abnormalities in the DCM tissues. All exhibited variably increased numbers of mitochondria (mitochondriosis) and sarcotubular dilatation, nonspecific changes that are characteristically observed in DCM and other types of myocardial disease. Changes were also present that differed among the individuals, mirroring abnormalities observed by light microscopy and immunohistochemistry. The LMNA DCM control again showed subtle abnormalities, with only mildly irregular ICDs and increased amplitude of the undulations of the fascia adherens (Figure 5B and 5F). The RBM20 DCM control had more‐severe abnormalities, with marked irregularity and decreased amplitude of undulations of the fascia adherens, shortening of overall ICD length, and interruption and displacement of ICDs along the long axis of the myocyte (Figure 5C and 5G). Our patient, however, showed the most pronounced ultrastructural changes, with extensive interruption and displacement or stair‐stepping of ICDs, including long segments oriented parallel to the myofilaments, along with marked irregularity and decreased amplitude of undulations of the fascia adherens (Figure 5D and 5H).
Figure 5.

Transmission electron microscopy performed on left ventricular myocardial tissue from each patient revealed several abnormalities. All diseased individuals showed variably increased numbers of mitochondria (B through D, F through H) relative to normal control (A and E), a nonspecific finding characteristic of diseased myocardium. DCM patients also showed ICD ultrastructure changes that mirrored abnormalities seen by light microscopy (shown in Figure 4). The LMNA DCM control showed relatively subtle abnormalities, with only mildly irregular ICDs and increased amplitude of the undulations of the fascia adherens (B and F). The RBM20 DCM control had more pronounced abnormalities, with irregularity and decreased amplitude of undulations, and interruption and displacement of ICDs along the long axis of the myocyte (C and G). The patient with TNNT2 and XIRP2 mutations showed the most severe changes, with extensive interruption and displacement or stair‐stepping of ICDs including long segments oriented parallel to the myofilaments, with marked irregularity and decreased amplitude of undulations (D and H). Boxes in A through D indicate region further magnified and displayed in E through H, respectively. Scale bars A through D=2 μm; E through H=600 nm. DCM indicates dilated cardiomyopathy; ICD, intercalated disc.
Discussion
TNNT2‐R141W Causes DCM in Humans and Mice
The TNNT2 R141W mutation, unreported in publicly available population databases, has been previously identified as a cause for familial DCM.23 The mutation alters a highly conserved residue within an alpha‐tropomyosin‐binding domain,27 and extensive functional assays have demonstrated defects in thin filament Ca2+ sensitivity.28, 29, 30, 31, 32 In addition, transgenic cTnTR141W mice develop decreased calcium sensitivity and DCM.33, 34
TNNT2‐R141W Is Associated With Incomplete Penetrance
The TNNT2 R141W mutation was first reported in a 5‐generation family comprised of 14 living individuals with DCM.23 Onset of cardiac symptoms typically occurred in the 2nd decade of life, but varied widely, with the youngest relative diagnosed at 1 year and the oldest at 84. In addition, 5 family members who carried the R141W mutation did not have echocardiographic manifestations of DCM at ages 1, 15, 29, 42, and 47 years. TNNT2 mutations in general have been associated with earlier disease onset, representing one of the most common known causes for pediatric DCM.10, 27 However, unexplained variability in penetrance of many mutations has been noted.35, 36, 37 This suggests that a modifier gene(s) may contribute to the age‐dependent penetrance of TNNT2‐mediated DCM and account for the severe disease phenotype in our preadolescent patient.
XIRP2 Is Necessary for Proper Cardiac Development and Growth
XIRP2 belongs to the ancient Xin gene family, initially identified as the ortholog cXin in chickens found to have striated muscle‐restricted expression.38 Subsequent studies have confirmed that mouse and human homologs, mXinα/Xirp1 and mXinβ/Xirp2 and hXinα/XIRP1 and hXinβ/XIRP2, respectively, also show preferential expression in striated muscle and localize to ICDs.39, 40 Analogous to our patient's parents, who are clinically unaffected heterozygous mutation carriers, mXinβ/Xirp2 +/− mice are indistinguishable from wild‐type mice. Knockout of both mXinβ/Xirp2 alleles in mice, however, leads to diastolic dysfunction and postnatal death before weaning.41 In addition, mice fail to form mature ICDs, with myocardial mislocalization of N‐cadherin and β‐catenin.41, 42 Interestingly, mXinβ/Xirp2‐null pups were found to have a significant delay in switching off slow skeletal troponin I, hypothesized to allow for increased Ca2+‐activated myofilament tension.41
XIRP2 Is a Novel Candidate Modifier of TNNT2‐Mediated DCM
The human ortholog, hXinβ/XIRP2, encoding a 3549‐amino‐acid protein, was initially identified as a gene coexpressed with 13 known cardiomyopathy‐associated genes.43 Analysis of XIRP2 in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org) and with the genic intolerance program44 reveals a relatively high frequency of natural, functional variation. However, frameshift and nonsense variants resulting in protein truncation (5.7%) are far less common than missense variants (63.7%). This pattern of genetic variation is reminiscent of ALMS1 and the specificity for recessive truncating mutations as a cause for Alström syndrome (OMIM 203800), a syndromic form of pediatric DCM.45 Individuals with compound heterozygous mutations cannot be determined from the ExAC database. However, among the 60 706 individuals in ExAC, only 2 are homozygous for a truncating XIRP2 variant, a state analogous to compound heterozygosity in our patient. The data set is not known to contain samples from DCM patients, and the chance that these individuals also carry a pathogenic DCM mutation would be extremely low. We speculate, however, that individuals homozygous or compound heterozygous for XIRP2 truncating variants may be at increased risk for developing heart failure if triggered by additional genetic and/or environmental risk factors.46 Future studies will be necessary to identify additional individuals with recessive truncating mutations and confirm the role XIRP2 plays in ICD maturation and DCM‐mediated heart failure.
TNNT2‐XIRP2 Coupled Mutations in Sporadic, Early‐Onset DCM
ICDs are critical for cardiac mechanotransduction,47 and mutations in the ICD protein metavinculin have been identified in adult‐onset DCM.48 ICD disruption was observed to variable degrees in our patient and DCM controls, suggesting that it may be a general feature of end‐stage myocardial disease irrespective of etiology. Notably, the DCM control harboring a RBM20 mutation displayed extensive ICD disruptions, consistent with this genetic subtype's association with early‐onset DCM.49 The patient harboring TNNT2 and XIRP2 mutations, however, showed marked irregularity of ICDs, with a stair‐stepping pattern reminiscent of mXinβ/Xirp2‐null mice, described as numerous, miniature ICD‐like structures arranged in tandem.42 mXinβ/Xirp2 plays an indispensable role in terminal redistribution of ICD components, such as N‐cadherin, during maturation.42 The consistent mislocalization of N‐cadherin in our patient and mXinβ/Xirp2‐null mice suggests a similar defect in biphasic ICD maturation.
Immunoreactivity of β‐catenin was increased in all pediatric DCM cardiac samples, but did not differ among genetic subtypes. Whereas mislocalization of β‐catenin was observed in mXinβ/Xirp2‐null mice,41 both frameshift mutations identified in our patient occur downstream of the β‐catenin‐binding domain (Figure 3C). This suggests that truncated XIRP2 may be expressed and retain partial activity within the myocardium. An antibody targeting XIRP2 upstream of the identified frameshift mutations is not currently available, precluding the ability to determine truncated XIRP2 expression and localization in our patient. The identified frameshift mutations occur upstream of several protein domains, including a sequence within the last proline‐rich region, PPPTLPKPKLPKH, that binds to the SH3 domain of nebulette during early stages of myofibril development.24 Nebulette, a cardiac‐specific isoform of nebulin50 and a known DCM gene,51 binds to troponin and tropomyosin and plays an essential role in maintaining thin filament integrity.52 Both truncation mutations would disrupt this interaction as well as 3 downstream coiled‐coil domains.53 The full spectrum of XIRP2 domains and interacting partners is unknown,54 but premature truncation and introduction of an anomalous peptide tail in the recessive state could eliminate domains necessary for ICD maturation within the myocardium.
mXinβ/Xirp2‐null pups displayed diastolic dysfunction with preserved systolic function, prompting examination of contractile and regulatory protein isoform switches. A significant delay in switching off slow skeletal troponin I was observed.41 It was hypothesized that this would result in increased Ca2+‐activated myofilament tension within cardiomyocytes, a plausible explanation for diastolic dysfunction. Interestingly, our patient had both systolic and diastolic dysfunction, with strikingly high left ventricular end‐diastolic pressure and severe left atrial enlargement. We postulate that effects of the identified pathogenic TNNT2 mutation, previously shown to result in decreased Ca2+‐sensitivity and contractile force development, were exacerbated by ICD‐altering mutations in XIRP2, which would impair force transmission between neighboring cardiomyocytes.47 TNNT2‐XIRP2 coupled mutations would thus lead to defects in both contractile force generation and transmission and result in an early‐onset phenotype, necessitating cardiac transplantation in childhood.
Sources of Funding
This work was supported by the National Institutes of Health (R01 HL071225 [Olson]), Pre‐Doctoral Training Program in Molecular Pharmacology (T32GM072474 [Long]), and an American Heart Association Pre‐Doctoral Fellowship (14PRE18070007 [Long]).
Disclosures
The authors have no conflicts of interest relevant to this article to disclose.
Supporting information
Table S1. Twelve variants in 8 candidate genes identified through whole exome sequencing. Chr indicates chromosome; DCM, dilated cardiomyopathy; ExAC, Exome Aggregation Consortium; FPKM, number of fragments per kilobase gene model and million reads; HPA, Human Protein Atlas; MAF, minor allele frequency; MGI, Mouse Genome Informatics; OMIM, Online Mendelian Inheritance in Man. Red text indicates pathogenic and modifier mutations.
Acknowledgments
The authors thank the family and patients who participated in this study.
(J Am Heart Assoc. 2015;4:e002443 doi: 10.1161/JAHA.115.002443)
An accompanying Table S1 is available at http://jaha.ahajournals.org/content/4/12/e002443/suppl/DC1
References
- 1. Dipchand AI, Kirk R, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dobbels F, Lund LH, Rahmel AO, Yusen RD, Stehlik J; International Society for Heart and Lung Transplantation . The Registry of the International Society for Heart and Lung Transplantation: sixteenth official pediatric heart transplantation report–2013; focus theme: age. J Heart Lung Transplant. 2013;32:979–988. [DOI] [PubMed] [Google Scholar]
- 2. Lund LH, Edwards LB, Kucheryavaya AY, Dipchand AI, Benden C, Christie JD, Dobbels F, Kirk R, Rahmel AO, Yusen RD, Stehlik J; International Society for Heart and Lung Transplantation . The Registry of the International Society for Heart and Lung Transplantation: thirtieth official adult heart transplant report—2013; focus theme: age. J Heart Lung Transplant. 2013;32:951–964. [DOI] [PubMed] [Google Scholar]
- 3. Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, Burnett JC, Rodeheffer RJ, Chesebro JH, Tazelaar HD. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. [DOI] [PubMed] [Google Scholar]
- 4. Baig MK, Goldman JH, Caforio AL, Coonar AS, Keeling PJ, McKenna WJ. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol. 1998;31:195–201. [DOI] [PubMed] [Google Scholar]
- 5. Grünig E, Tasman JA, Kücherer H, Franz W, Kübler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol. 1998;31:186–194. [DOI] [PubMed] [Google Scholar]
- 6. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. [DOI] [PubMed] [Google Scholar]
- 7. McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Olson TM, Chan DP. Dilated cardiomyopathy In: Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, eds. Moss and Adams' Heart Disease in Infants, Children, and Adolescents: Including the Fetus and Young Adult. 8th ed Vol. II. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013:1235–1246. [Google Scholar]
- 9. Towbin JA, Lowe AM, Colan SD, Sleeper LA, Orav EJ, Clunie S, Messere J, Cox GF, Lurie PR, Hsu D, Canter C, Wilkinson JD, Lipshultz SE. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA. 2006;296:1867–1876. [DOI] [PubMed] [Google Scholar]
- 10. Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–608. [DOI] [PubMed] [Google Scholar]
- 11. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a mapreduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennel TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. The 1000 Genomes Consortium . An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, Dahl F, Fernandez A, Staker B, Pant KP, Baccash J, Borcherding AP, Brownley A, Cedeno R, Chen L, Chernikoff D, Cheung A, Chirita R, Curson B, Ebert JC, Hacker CR, Hartlage R, Hauser B, Huang S, Jiang Y, Karpinchyk V, Koenig M, Kong C, Landers T, Le C, Liu J, McBride CE, Morenzoni M, Morey RE, Mutch K, Perazich H, Perry K, Peters BA, Peterson J, Pethiyagoda CL, Pothuraju K, Richter C, Rosenbaum AM, Roy S, Shafto J, Sharanhovich U, Shannon KW, Sheppy CG, Sun M, Thakuria JV, Tran A, Vu D, Zaranek AW, Wu X, Drmanac S, Oliphant AR, Banyai WC, Martin B, Ballinger DG, Church GM, Reid CA. Human genome sequencing using unchained base reads on self‐assembling DNA nanoarrays. Science. 2010;327:78–81. [DOI] [PubMed] [Google Scholar]
- 15. Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F. Tissue‐based map of the human proteome. Science. 2015;347:1260419 doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 16. Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE; The Mouse Genome Database Group . The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014;42:D810–D817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cottrell BA, Galvan V, Banwait S, Gorostiza O, Lombardo CR, Williams T, Schilling B, Peel A, Gibson B, Koo EH, Link CD, Bredesen DE. A pilot proteomic study of amyloid precursor interactors in Alzheimer's disease. Ann Neurol. 2005;58:277–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Neesen J, Kirschner R, Ochs M, Schmiedl A, Habermann B, Mueller C, Holstein AF, Nuesslein T, Adham I, Engel W. Disruption of an inner arm dynein heavy chain gene results in asthenozoospermia and reduced ciliary beat frequency. Hum Mol Genet. 2001;10:1117–1128. [DOI] [PubMed] [Google Scholar]
- 19. Gaidos G, Soni S, Oswald DJ, Toselli PA, Kirsch KH. Structure and function analysis of the CMS/CIN85 protein family identifies actin‐bundling properties and heterotypic‐complex formation. J Cell Sci. 2007;120:2366–2377. [DOI] [PubMed] [Google Scholar]
- 20. Le Bras S, Moon C, Foucault I, Breittmayer JP, Deckert M. Abl‐SH3 binding protein 2, 3BP2, interacts with CIN85 and HIP‐55. FEBS Lett. 2007;581:967–974. [DOI] [PubMed] [Google Scholar]
- 21. Kometani K, Yamada T, Sasaki Y, Yokosuka T, Saito T, Rajewsky K, Ishiai M, Hikida M, Kurosaki T. CIN85 drives B cell responses by linking BCR signals to the canonical NF‐ĸB pathway. J Exp Med. 2011;208:1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. 2000;343:1688–1696. [DOI] [PubMed] [Google Scholar]
- 23. Li D, Czernuszewicz GZ, Gonzalez O, Tapscott T, Karibe A, Durand JB, Brugada R, Hill R, Gregoritch JM, Anderson JL, Quiñones M, Bachinski LL, Roberts R. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation. 2001;104:2188–2193. [DOI] [PubMed] [Google Scholar]
- 24. Eulitz S, Sauer F, Pelissier MC, Boisguerin P, Molt S, Schuld J, Orfanos Z, Kley RA, Volkmer R, Wilmanns M, Kirfel G, van der Ven PF, Fürst DO. Identification of Xin‐repeat proteins as novel ligands of the SH3 domains of nebulin and nebulette and analysis of their interaction during myofibril formation and remodeling. Mol Biol Cell. 2013;24:3215–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grosskurth SE, Bhattacharya D, Wang Q, Lin JJ. Emergence of Xin demarcates a key innovation in heart evolution. PLoS One. 2008;3:e2857 doi: 10.1371/journal.pone.0002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Masuelli L, Bei R, Sacchetti P, Scappaticci I, Francalanci P, Albonici L, Coletti A, Palumbo C, Minieri M, Fiaccavento R, Carotenuto F, Fantini C, Carosella L, Modesti A, Di Nardo P. β‐catenin accumulates in intercalated discs of hypertrophic cardiomyopathic hearts. Cardiovasc Res. 2003;60:376–387. [DOI] [PubMed] [Google Scholar]
- 27. Hershberger RE, Pinto JR, Parks SB, Kushner JD, Li D, Ludwigsen S, Cowan J, Morales A, Parvatiyar MS, Potter JD. Clinical and functional characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circ Cardiovasc Genet. 2009;2:306–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu QW, Morimoto S, Harada K, Du CK, Takahashi‐Yanaga F, Miwa Y, Sasaguri T, Ohtsuki I. Cardiac troponin T mutation R141W found in dilated cardiomyopathy stabilizes the troponin T‐tropomyosin interaction and causes a Ca2+ desensitization. J Mol Cell Cardiol. 2003;35:1421–1427. [DOI] [PubMed] [Google Scholar]
- 29. Mirza M, Marston S, Willott R, Ashley C, Mogensen J, McKenna W, Robinson P, Redwood C, Watkins H. Dilated cardiomyopathy mutations in three thin filament regulatory proteins result in a common functional phenotype. J Biol Chem. 2005;280:28498–28506. [DOI] [PubMed] [Google Scholar]
- 30. Venkatraman G, Gomes AV, Kerrick WG, Potter JD. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. J Biol Chem. 2005;280:17584–17592. [DOI] [PubMed] [Google Scholar]
- 31. Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and α‐tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–1273. [DOI] [PubMed] [Google Scholar]
- 32. Liu B, Tikunova SB, Kline KP, Siddiqui JK, Davis JP. Disease‐related cardiac troponins alter thin filament Ca2+ association and dissociation rates. PLoS One. 2012;7:e38259 doi: 10.1371/journal.pone.0038259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Juan F, Wei D, Xiongzhi Q, Ran D, Chunmei M, Lan H, Chuan Q, Lianfeng Z. The changes of the cardiac structure and function in cTnTR141W transgenic mice. Int J Cardiol. 2008;128:83–90. [DOI] [PubMed] [Google Scholar]
- 34. Lombardi R, Bell A, Senthil V, Sidhu J, Noseda M, Roberts R, Marian AJ. Differential interactions of thin filament proteins in two cardiac troponin T mouse models of hypertrophic and dilated cardiomyopathies. Cardiovasc Res. 2008;79:109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morales A, Pinto JR, Siegfried JD, Li D, Norton N, Hofmeyer M, Vallin M, Morales AR, Potter JD, Hershberger RE. Late onset sporadic dilated cardiomyopathy caused by a cardiac troponin T mutation. Clin Transl Sci. 2010;3:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campbell N, Sinagra G, Jones KL, Slavov D, Gowan K, Merlo M, Carniel E, Fain PR, Aragona P, Di Lenarda A, Mestroni L, Taylor MR. Whole exome sequencing identifies a troponin T mutation hot spot in familial dilated cardiomyopathy. PLoS One. 2013;8:e78104 doi: 10.1371/journal.pone.0078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Golbus JR, Puckelwartz MJ, Dellefave‐Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, Pytel P, McNally EM. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang DZ, Hu X, Lin JL, Kitten GT, Solursh M, Lin JJ. Differential display of mRNAs from the atrioventricular region of developing chicken hearts at stages 15 and 21. Front Biosci. 1996;1:a1–a15. [DOI] [PubMed] [Google Scholar]
- 39. Wang DZ, Reiter RS, Lin JL, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJ. Requirement of a novel gene, Xin, in cardiac morphogenesis. Development. 1999;126:1281–1294. [DOI] [PubMed] [Google Scholar]
- 40. Pacholsky D, Vakeel P, Himmel M, Löwe T, Stradal T, Rottner K, Fürst DO, van der Ven PF. Xin repeats define a novel actin‐binding motif. J Cell Sci. 2004;117:5257–5268. [DOI] [PubMed] [Google Scholar]
- 41. Wang Q, Lin JL, Reinking BE, Feng HZ, Chan FC, Lin CI, Jin JP, Gustafson‐Wagner EA, Scholz TD, Yang B, Lin JJ. Essential roles of an intercalated disc protein, mXinβ, in postnatal heart growth and survival. Circ Res. 2010;106:1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Q, Lin JL, Chan SY, Lin JJ. The Xin repeat‐containing protein, mXinβ, initiates the maturation of the intercalated discs during postnatal heart development. Dev Biol. 2013;374:264–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Walker MG. Pharmaceutical target identification by gene expression analysis. Mini Rev Med Chem. 2001;1:197–205. [DOI] [PubMed] [Google Scholar]
- 44. Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709 doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Long PA, Evans JM, Olson TM. Exome sequencing establishes diagnosis of Alström syndrome in an infant presenting with non‐syndromic dilated cardiomyopathy. Am J Med Genet A. 2015;167A:886–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Reyes S, Terzic A, Mahoney DW, Redfield MM, Rodeheffer RJ, Olson TM. KATP channel polymorphism is associated with left ventricular size in hypertensive individuals: a large‐scale community‐based study. Hum Genet. 2008;123:665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lyon RC, Zanella F, Omens JH, Sheikh F. Mechanotransduction in cardiac hypertrophy and failure. Circ Res. 2015;116:1462–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olson TM, Illenberger S, Kishimoto NY, Huttelmaier S, Keating MT, Jockusch BM. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation. 2002;105:431–437. [DOI] [PubMed] [Google Scholar]
- 49. Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. J Am Coll Cardiol. 2009;54:930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moncman CL, Wang K. Nebulette: a 107kD nebulin‐like protein in cardiac muscle. Cell Motil Cytoskeleton. 1995;32:205–225. [DOI] [PubMed] [Google Scholar]
- 51. Arimura T, Nakamura T, Hiroi S, Satoh M, Takahashi M, Ohbuchi N, Ueda K, Nouchi T, Yamaguchi N, Akai J, Matsumori A, Sasayama S, Kimura A. Characterization of the human nebulette gene: a polymorphism in an actin‐binding motif is associated with nonfamilial idiopathic dilated cardiomyopathy. Hum Genet. 2000;107:440–451. [DOI] [PubMed] [Google Scholar]
- 52. Bonzo JR, Norris AA, Esham M, Moncman CL. The nebulette repeat domain is necessary for proper maintenance of tropomyosin with the cardiac sarcomere. Exp Cell Res. 2008;314:3519–3530. [DOI] [PubMed] [Google Scholar]
- 53. UniProt Consortium . UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang Q, Lin JL, Erives AJ, Lin CI, Lin JJ. New insights into the roles of Xin repeat‐containing proteins in cardiac development, function, and disease. Int Rev Cell Mol Biol. 2014;310:89–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Twelve variants in 8 candidate genes identified through whole exome sequencing. Chr indicates chromosome; DCM, dilated cardiomyopathy; ExAC, Exome Aggregation Consortium; FPKM, number of fragments per kilobase gene model and million reads; HPA, Human Protein Atlas; MAF, minor allele frequency; MGI, Mouse Genome Informatics; OMIM, Online Mendelian Inheritance in Man. Red text indicates pathogenic and modifier mutations.