

Abstract
Normal tissue toxicity reduces the therapeutic index of radiotherapy and decreases the quality of life for cancer survivors. Apoptosis is a key element of the radiation response in normal tissues like the hippocampus and small intestine, resulting in neurocognitive disorders and intestinal malabsorption. The Early Growth Response 1 (Egr1) transcription factor mediates radiation-induced apoptosis by activating the transcription of pro-apoptosis genes in response to ionizing radiation (IR). Therefore, we hypothesized that the genetic abrogation of Egr1 and the pharmacological inhibition of its transcriptional activity could attenuate radiation-induced apoptosis in normal tissues. We demonstrated that Egr1 null mice had less apoptosis in the hippocampus and intestine following irradiation as compared to their wild-type littermates. A similar result was achieved using Mithramycin A (MMA) to prevent binding of Egr1 to target promoters in the mouse intestine. Egr1 expression using shRNA dampened apoptosis and enhanced the clonogenic survival of irradiated HT22 hippocampal neuronal cells and IEC6 intestinal epithelial cells. Mechanistically, these events involved an abrogation of p53 induction by IR and an increase in the ratio of Bcl-2/Bax expression. In contrast, targeted silencing of Egr1 in two cancer cell lines (GL261 glioma cells, HCT116 colorectal cancer cells) was not radioprotective, since it reduced their growth while also sensitizing them to radiation-induced death. Further, Egr1 depletion delayed the growth of heterotopically implanted GL261 and HCT116 tumors. These results support the potential of silencing Egr1 in order to minimize the normal tissue complications associated with radiotherapy while enhancing tumor control.
Keywords: Egr1, radiation-induced apoptosis, radioprotection, radiation therapeutic index, normal tissue injury
INTRODUCTION
The ability to deliver a sufficient dose and volume of ionizing radiation (IR) during radiotherapy to control tumors is limited by normal tissue injury. Radiotherapy decreases quality of life by causing long-term side effects such as cognitive impairments and intestinal malabsorption (1). Cranial irradiation results in variable degrees of cognitive decline in both children (2-4) and adults (5,6). Hippocampus-dependent functions, such as learning, memory, and spatial information processing, are often impacted due to the loss of progenitor cells in the subgranular zone of the hippocampus—an active site of neurogenesis (7). Likewise, abdominal and pelvic radiotherapy results in chronic intestinal malabsorption. Radiation-induced apoptosis in the progenitor cells of intestinal crypts is largely responsible for intestinal tissue damage (8-10). Since intestinal homeostasis relies on the frequent renewal of the epithelium by progenitor cells, the loss of these cells due to radiotherapy prevents subsequent regeneration.
Agents that block radiation damage to normal tissues are being investigated as possible radioprotectors (1). One of the major challenges of implementing radioprotectors in clinical practice is that many radiation modulators have nonspecific targets, which enable protection of cancer cells as well as normal cells (11). Recently, we and others have demonstrated that targeting mediators of cellular radiosensitivity can selectively dampen radiation-induced apoptosis and improve normal organ function after irradiation without protecting cancer cells (11-14).
Early Growth Response 1 (Egr1), also known as NGFI-A, ZIF268, Krox24, or TIS8, is a zinc-finger transcription factor that binds and regulates transcription through a GC-rich consensus sequence of 5’-GCG(T/G)GGGCG-3’ (15). Egr1 is known as an immediate-early response gene due to the rapid kinetics of its induction by various signals, including growth factors, cytokines, and stress (16). One such stress is IR, which rapidly and transiently induces Egr1 expression (17,18). In response to these cues, Egr1 can regulate growth, differentiation, growth inhibition, and apoptosis (15,19,20). Egr1's pro-apoptotic role is carried out through the binding to its target genes that encode p53 (20), PTEN (21), Bim (22), and Bax (23). Notably, Egr1−/− mouse embryonic fibroblasts (MEFs) are resistant to apoptosis after irradiation, indicating a potential role for Egr1 in regulating radiation-induced apoptosis (24).
We have investigated the effect of Egr1 suppression on cell death in normal tissues by taking advantage of the Egr1 knockout (KO) mouse and the Mithramycin A (MMA) drug. Both genetic abrogation and pharmacological inhibition of Egr1 attenuated radiation-induced apoptosis in irradiated hippocampus and small intestine. The mechanism underlying the radioprotective effects of Egr1 suppression involves a loss of p53 induction by IR and an increase in the Bcl-2/Bax expression ratio. These cytoprotective effects did not extend to cancer cells. Knocking down Egr1 suppressed the growth and survival of glioma and colorectal cancer cells, which was compounded with radiation. The antitumor effect of Egr1 silencing was also observed in glioma and colorectal tumor models in vivo. Therefore, we demonstrate a potential molecular target that enables selective radioprotection of normal tissue and cells while enhancing cancer cell death by radiation.
METHODS AND MATERIALS
Mice and Treatment
Animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University in St. Louis. Heterozygous Egr1+/− mice maintained on a C57/BL/6J background were provided by Michelle LeBeau at University of Chicago (originally from Jeffrey Milbrandt). Heterozygous mice were crossed to obtain Egr1+/+ and Egr1−/− mice. For drug studies, ten-week old wild-type (WT) C57/BL/6J mice received two intraperitoneal (i.p.) injections of either DMSO or 150 μg/kg MMA dissolved in DMSO at 24 h and 30 min prior to irradiation. Mice receiving partial body irradiation to the cranium or abdomen were anesthetized with 2% isoflurane for immobilization and shielded with lead. Mice were irradiated using the RS-2000 (Rad Source) irradiator at a dose rate of 1 Gy/min with 160 kVp X-rays using a 0.3mm copper filter. All irradiation doses were monitored using Accudose (Radcal) to deliver the exact dose to each mouse.
Tumor growth delay
One million GL261 or HCT116 cells bearing either scrambled or Egr1-targeting shRNA were injected into the hind limb of each Nu/Nu mouse (Jackson Laboratories). Tumor volumes in mouse hind limbs were measured with digital calipers. The mean tumor volume was calculated for each treatment group (4 mice).
TUNEL assay
TUNEL assays were performed as previously described (12,13) with slight modifications. For studying apoptosis in the hippocampus, postnatal day 10 WT or Egr1−/− mouse pups were sham irradiated or cranially irradiated with 7 Gy. Twenty-four hours after irradiation, brains were harvested and fixed, embedded in paraffin, and coronally sectioned. To study apoptosis in the small intestine, ten week-old WT or Egr1−/− mice were sham irradiated or abdominally irradiated with 8 Gy. The proximal part of the jejunum was harvested at 8 h or 24 h after irradiation, fixed, cut into 1 cm long segments, which were embedded vertically and sectioned. Brain and intestinal tissue sections were stained with either the Colorimetric or Fluorometric DeadEnd™ TUNEL System (Promega) and counterstained with hematoxylin or DAPI. Tissue sections were viewed using an Olympus BX60 fluorescence microscope equipped with a Retiga 2000R digital camera. TUNEL-positive cells (TPC) in the subgranular zone of the hippocampus were counted (400x). To show the stereologic nature of the apoptosis, at least five sections at different depths of sectioning were counted per mouse. The average number of TPC per high-power field (HPF) was calculated. For the small intestine the average number of TPC per crypt was calculated from at least five HPF. At least three mice were used in each experimental group.
Cell Culture
HT22 mouse hippocampal neuronal cells were obtained in 2006 from David Schubert (whose laboratory at The Salk Institute authenticates by examining cell line-specific markers) and maintained in DMEM with 10% FBS and 1% penicillin/streptomycin (P/S). IEC6 rat small intestine epithelial crypt cells were obtained from ATCC in 2008 (which performs authentication using Short Tandem Repeat (STR) Profiling) and maintained in DMEM with 10% FBS and 1% P/S. GL261 mouse glioma cells were obtained from the National Cancer Institute, which performs authentication through Applied Biosystems AmpFISTR Identifiler testing. They were maintained in DMEM/F12 with 10% FBS and 1% P/S. HCT116 human colorectal cancer cells were obtained from ATCC (authenticated by STR Profiling) and maintained in McCoy's 5A modified media with 10% FBS and 1% P/S. All cell lines were passaged under 20 times from receipt and routinely tested for mycoplasma contamination. All cell lines were incubated at 37°C in a humidified 5% CO2/95% air incubator. For drug studies, cells were treated with either 0.1% DMSO or 250nM of MMA dissolved in DMSO.
Silencing of Egr1 with shRNA
To identify shRNA sequences that could knockdown Egr1 in HT22, IEC6, and GL261 mouse cell lines, we screened five MISSION shRNA clones (Sigma) targeted against the mouse Egr1 sequence. Two clones with the best knockdown of Egr1 were selected (mE3: CCGGCACTCCACTATCCACTATTAACTCGAGTTAATAGTGGATAGTGGAGTGTTTTT G and mE4: CCGGCATCGCTCTGAATAATGAGAACTCGAGTTCTCATTATTCAGAGCGATGTTTTT G). A similar approach was taken to screen shRNA clones that could knockdown Egr1 in the HCT116 human colorectal cancer cell line. The clones with the best knockdown were selected (hE3: CCGGCGGTTACTACCTCTTATCCATCTCGAGATGGATAAGAGGTAGTAACCGTTTTT and hE4: CCGGCATCTCTCTGAACAACGAGAACTCGAGTTCTCGTTGTTCAGAGAGATGTTTTT) . The MISSION pLKO.1-puro non-target shRNA was used as a control. MISSION shRNA clones together with packaging and envelope plasmids pUMVC and pCMV-VSV-G (Sheila Stewart, Washington University) were transfected into HEK293T packaging cells using Fugene 6 (Roche Applied Science). At 48 h post-transfection, virus-containing medium was used to transduce target cells. After selection with puromycin (2 μg/ml) for 36-48 h, cells were analyzed for Egr1 expression by immunoblotting and then used in assays for clonogenic survival, proliferation, and apoptosis.
Clonogenic survival assay
Clonogenic survival was monitored as previously described (13). Calculated numbers of cells were plated to enable normalization for plating efficiencies. Cells were allowed to attach overnight and then irradiated. After a 7-10-day incubation, plates were fixed with 70% EtOH and stained with 1% methylene blue. Colonies consisting of >50 cells were counted. Survival fractions were calculated as (number of colonies / number of cells plated) / (number of colonies for corresponding control / number of cells plated).
Apoptosis assays for cultured cells
Apoptosis was determined by Annexin V-FITC and propidium iodide (PI) staining (BD PharMingen) according to the manufacturer's instructions. Cells were analyzed by flow cytometry, using a two-color FACS analysis on the BD LSR II instrument. For each treatment, the average percent of Annexin V-positive cells was calculated. Alternatively, the apoptotic nuclei of cells were counted after nuclear staining with DAPI. The treated cells were washed with PBS, fixed in 70% ethanol for 10 minutes, and stained with DAPI. Nuclear morphology was observed using fluorescence microscopy as previously described above. Apoptosis was quantified by scoring the percentage of cells with apoptotic nuclear morphology. Condensed or fragmented nuclei were scored as apoptotic; the average percentage of apoptotic cells was calculated in 5-7 randomly selected HPF.
Cell growth and viability assays
Cell growth was determined by direct cell counting. Cells were plated at a density of 5×104 cells/well in 12-well plates. The cells were collected daily by trypsin digestion, stained with Trypan Blue, and counted using the Vi-Cell Automatic Cell Analyzer (Beckman Coulter). Growth curves were generated based on the average number of cells per day over a four-day period. The Presto Blue Assay and the MTS Assay (Promega) were performed to measure cell viability. Equal numbers of cells from various treatments were plated into a 96-well plate and irradiated the following day. Cell viability was determined 72 h post-irradiation using the SpectraMax i3 Microplate Reader (Molecular Devices).
Immunoblot analysis
Total protein was extracted from treated cells using the M-PER mammalian protein extraction reagent (Pierce). For hippocampal proteins, WT and Egr1−/− mice at postnatal day 10 were euthanized and the hippocampus was rapidly dissected. The hippocampal tissues were homogenized in 20μL of M-PER buffer per mg tissue using a tissue homogenizer (Omni International). For both cellular and tissue extracts, protein concentration was quantified using the BCA Reagent (Pierce). Protein extracts (40 μg) were subjected to immunoblot analysis using the following antibodies: anti-Egr1 (C-19) and anti-Bax (B-9) (both from Santa Cruz), anti-Egr1 (44D5), anti-p53 (1C12), anti-Bim (C34C5), and Bcl-2 (50E3) (all from Cell Signaling Technologies). Antibody against actin (Sigma) was used to monitor protein loading in each lane. Immunoblots were visualized with the Western Lightning Chemiluminescence Plus detection kit (PerkinElmer) using either film or the BioRad ChemiDoc MP Imaging System.
Statistical analyses
The mean and standard error of the mean (SEM) of each treatment group were calculated based on at least three replicates for all experiments. Experiments were repeated at least three times. All pairwise comparisons, including calculation of P values, were done using the Student's t-test. Comparisons between two Egr1 knockdown cell lines, each expressing a distinct clone of Egr1-targeting shRNA and a control cell line expressing a scrambled shRNA were performed using One Way Analysis of Variance (ANOVA). The Wilcoxon Rank Sum Test was used to compare treatment effectiveness on tumor growth delay. A P value of <0.05 was considered significant.
RESULTS
Targeting Egr1 attenuates radiation-induced apoptosis in the mouse hippocampus and small intestine
We used the Egr1 KO mouse to assess whether the absence of Egr1 could attenuate radiation-induced apoptosis in the hippocampus and the small intestine. To study the hippocampus we irradiated the cranium of Egr1 KO (Egr1−/−) and WT (Egr1+/+) mouse pups. Hippocampal sections were analyzed for apoptosis in the subgranular zone using TUNEL staining (Fig. 1A). Twenty-four hours post-irradiation, Egr1−/− mice showed significantly fewer TUNEL-positive cells as compared to WT controls (Fig. 1B). We also studied apoptosis in the intestinal crypts of Egr1−/− and WT mice irradiated in the abdomen. Twenty-four hours post-irradiation, there were significantly fewer TUNEL-positive cells per crypt in Egr1−/− mice as compared to WT mice (Fig. 1A&B). A similar effect was also observed at 8 h post-irradiation (Supplementary Fig. S1 A&B).
Figure 1. Targeting Egr1 lessens radiation-induced apoptosis in vivo.

(A) Representative micrographs of TUNEL-stained mouse hippocampus and intestinal crypts from Egr1+/+ or Egr1−/− mice. (B) Quantification of TPC per HPF in the hippocampus and TPC per intestinal crypt for each treatment group. (C) WT mice were treated with DMSO or MMA. Representative micrographs of TUNEL-stained hippocampus and intestinal crypts of mice. (D) Quantification of TPC per HPF in hippocampus and TPC per intestinal crypt. ns, not significant, **p<0.005, ****p<0.0001.
The function of Egr1 was also inhibited by treating mice with MMA, a drug that represses transcription by selectively displacing GC-rich binding transcription factors like Egr1 (22). WT mice that were administered i.p. injections of MMA prior to abdominal irradiation had significantly fewer apoptotic cells per crypt than vehicle-treated mice (Fig. 1C&D). In contrast, histological assessment of apoptosis revealed no significant differences in apoptosis in the hippocampal subgranular zones of MMA-treated mice compared with those in the vehicle cohort. Taken together, suppressing Egr1 through genetic manipulation or pharmacological inhibition can attenuate radiation-induced apoptosis in normal tissues.
Suppressing Egr1 levels protects cultured normal cells from radiation-induced cell killing
To elucidate the mechanism by which Egr1 depletion reduced cell death in the hippocampal sub-granular zone and the intestinal crypts in mice, we knocked down Egr1 in cultured HT22 hippocampal neuronal progenitor cells and IEC6 intestinal epithelial crypt cells using different Egr1-specific shRNA clones. Cells stably expressing the shRNA clones were screened for knockdown by immunoblotting (Fig. 2A). Of the Egr1 shRNA clones screened, clone mE3 had the most efficient knockdown in both cell lines. We then determined the colony forming abilities of the irradiated HT22/mE3 and IEC6/mE3 knockdown cells. Both Egr1 knockdown cell lines were protected from radiation-induced cell death when compared to their corresponding scrambled controls at all radiation doses tested (Fig. 2B). Silencing Egr1 resulted in the radioprotection of the cells with a dose modifying factor of 1.2 for HT22 and 1.4 for IEC6 cells. Thus, our results indicate that suppressing Egr1 levels protects cultured hippocampal neuronal progenitors and intestinal crypt cells from radiation-induced cell death.
Figure 2. Knockdown of Egr1 increases clonogenic survival and attenuates radiation-induced apoptosis in cultured normal cells.

(A) HT22 and IEC6 cells expressing scrambled (Scr) or five Egr1-specific shRNAs (mE1-5). Cellular proteins were immunoblotted using antibodies against Egr1 and Actin (loading control). Densitometry values represent the ratio of Egr1 to Actin normalized to the Scr control. (B) Scr and knockdown HT22 and IEC6 cells were irradiated and plated for a clonogenic survival assay. Shown are the survival fractions. (C) Scr and knockdown HT22 and IEC6 cells were irradiated. Bar graphs of the average percentage of Annexin V-positive cells. (D) HT22 and IEC6 cells were treated with DMSO or MMA 30 min prior to irradiation and washed out 2 h later. Bar graphs of the average percentage of Annexin V-positive cells for each treatment; *p<0.05, **p<0.005, ***p<0.001.
Apoptosis in irradiated normal cells is attenuated by Egr1 knockdown and inhibition
Since Egr1 is known to transcriptionally regulate proteins involved in apoptosis, we hypothesized that one cellular mechanism by which Egr1 suppression enhanced clonogenic survival after irradiation involves attenuation of apoptosis. We tested this hypothesis by comparing apoptotic fractions between irradiated HT22/mE3 or IEC6/mE3 cells and their scrambled controls using Annexin V/PI staining. We have previously demonstrated the maximum effective dose to evaluate apoptosis by Annexin V assays in HT22 (13) and IEC6 (12) cells at 24 h after irradiation is 4 or 6 Gy, respectively. HT22/mE3 and IEC6/mE3 knockdown cells had smaller fractions of apoptotic cells as compared to the scrambled controls after irradiation (Fig. 2C). Evaluation of the nuclear morphology of dying cells using DAPI staining indicated similar outcomes (Supplementary Fig. S2 A&B). Similar reductions in apoptotic fractions were observed in irradiated HT22 and IEC6 cells treated with MMA prior to irradiation (Fig. 2D). Smaller fractions of irradiated HT22 and IEC6 cells pre-treated with MMA underwent apoptosis as compared to the vehicle-treated cells. Hence, both knockdown of Egr1 and functional inhibition by MMA protect hippocampal neuronal progenitors and intestinal crypt cells from radiation-induced apoptosis.
Egr1 suppression alters the expression of molecular mediators of radiosensitivity in normal cells
We also examined how Egr1 status can affect its downstream transcriptional targets involved in apoptosis to shed light on the molecular mechanisms by which silencing Egr1 attenuates radiation-induced apoptosis. Using ex vivo protein lysates made from hippocampal tissues of Egr1−/− and WT mice, we first verified that the expression of Egr1 was inducible one hour after irradiation in the WT tissues but not in the KO tissues (Figure 3A). We also analyzed the expression of p53, as well as Bax, a positive regulator of mitochondrial apoptosis, and Bcl-2, a negative regulator of mitochondrial apoptosis. The hippocampal extracts from Egr1−/− mice showed a loss of p53 induction by IR, blunted Bax induction by IR, and enhanced basal expression and IR-induction of Bcl-2, as compared to the hippocampal extracts from the WT mice (Figure 3A). We also investigated whether similar protein expression changes occurred in cultured cells. Egr1 depletion in both HT22 and IEC6 cell lines led to an abrogation of p53 induction by IR, downregulation of Bax, and upregulation of Bcl-2 (Figure 3B). Taken all together, the expression changes of these apoptotic proteins seen in the ex vivo tissue model and the cultured cell models of Egr1 depletion are consistent with their phenotype of diminished radiation-induced apoptosis.
Figure 3. Knocking down Egr1 modifies the expression of apoptotic proteins.

(A) Egr1 WT and KO mouse pups were sham irradiated or cranially irradiated with 7 Gy. Total cellular lysate from hippocampal tissue was isolated 1 h and 6 h after irradiation. Cellular proteins were immunoblotted using antibodies against Egr1, p53, Bax, Bcl-2, and Actin (loading control). Densitometry values represent the ratio of the various proteins to Actin normalized to the WT 0Gy control. (B) Scr and knockdown HT22 and IEC6 cells were sham irradiated or irradiated with 4 or 6 Gy, respectively, and harvested after 6 h. Cellular proteins were immunoblotted using antibodies to p53, Bax, Bcl-2. Actin was used to evaluate protein loading. Densitometry values represent the ratio of the various proteins to Actin normalized to the Scr 0Gy control.
Targeting Egr1 abrogates the growth and survival of cancer cells
To ascertain if targeting Egr1 could produce the undesired effect of also protecting cancer cells from radiation-induced death, we knocked down Egr1 in the GL261 glioma cell line and the HCT116 colorectal cell line. These cells were chosen because they represent tumors that are typically treated with cranial or pelvic radiation and are associated with neurocognitive deficits or chronic malabsorption. Using the same approach as described above for normal cell lines, we established knockdown cell lines by stably expressing two Egr1-specific shRNAs in mouse GL261 (mE3, mE4) and human HCT116 (hE3, hE4) cells. For comparison, a control cell line expressing a scrambled shRNA was established for both GL261 and HCT116 cells. Knockdown by the Egr1-targeting shRNAs was validated by western blotting for both GL261 and HCT116 cell lines (Figure 4A). To assess if the knockdown of Egr1 had an effect on cell growth and proliferation, we generated growth curves for the GL261 and HCT116 knockdown cells. Silencing Egr1 reduced the cell growth of the GL261 and HCT116 cells, as evidenced by the slower rate of proliferation of the cancer cells expressing the Egr1-targeting shRNAs as compared to those expressing the scrambled shRNA (Figure 4B).
Figure 4. Silencing Egr1 reduces the growth of glioma and colorectal cancer cell lines.
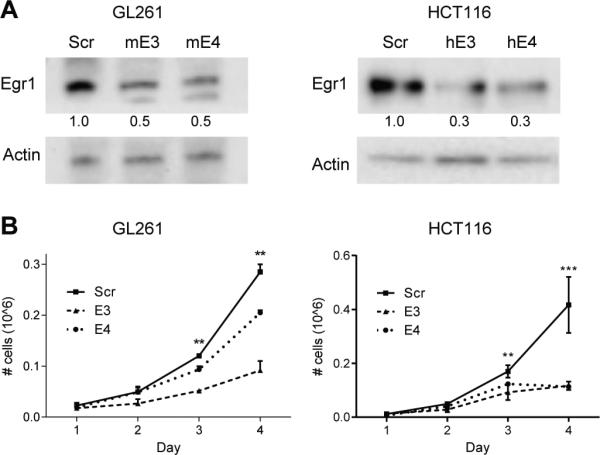
(A) GL261 glioma and HCT116 colorectal cells were infected with lentiviruses expressing scrambled or two Egr1-specific shRNAs (GL261: mE3, mE4; HCT116: hE3, hE4). Cellular proteins were immunoblotted using antibodies to Egr1 and Actin (loading control). Densitometry values represent the ratio of Egr1 to Actin normalized to the Scr control. (B) Growth curves show the average numbers of viable cells on each day. **p<0.005, ***p<0.001.
Next, we determined if suppressing Egr1 expression would protect GL261 and HCT116 cells from radiation-induced death. The results reveal a lack of radioprotection; instead, Egr1 knockdown caused a significant abrogation in cell survival in unirradiated GL261 and HCT116 cell lines (Figure 5A). Survival was further decreased in the knockdown cells following irradiation, as compared to the control cells (Figure 5A). In a cell viability assay, silencing Egr1 reduced viability in unirradiated cells, which was further dampened in the presence of radiation (Figure 5B). We also evaluated the effect of silencing Egr1 on radiation-induced apoptosis in GL261 and HCT116 cells using Annexin V/PI staining. Knocking down Egr1 increased the apoptotic fraction of unirradiated GL261 and HCT116 cells as compared to the control cells, which was further amplified when combined with irradiation (Figure 5C). Taken together, these data suggest that silencing Egr1 blunts the growth and survival of GL261 glioma and HCT116 colorectal cancer cell lines, as well as sensitizes them to radiation-induced killing.
Figure 5. Knockdown of Egr1 reduces survival and viability while enhancing apoptosis in cancer cell lines.

(A) Scr and knockdown GL261 (mE3, mE4) and HCT116 (hE3, hE4) cells were irradiated and plated for a clonogenic survival assay. Shown is the plating efficiencies normalized to Scr 0Gy. (B) Scr and knockdown GL261 and HCT116 cells were irradiated with the indicated doses. Shown is the viability relative to the control (Scr 0Gy) (C) Scr and knockdown GL261 and HCT116 cells were sham irradiated or irradiated with 4 Gy. Shown is the average percentage of Annexin V-positive cells. *p<0.05, **p<0.005, ***p<0.001, ****p<0.0001.
Egr1 knockdown suppresses the growth of glioma and colorectal tumors in mice
To determine the in vivo efficacy of Egr1 knockdown, heterotopic mouse tumors were established from mouse glioma GL261 cells or human colorectal cancer HCT116 cells that expressed either scrambled or Egr1-targeting shRNA. The tumor volumes were measured over time (Figure 6A). There was a significant difference in the average number of days required to reach a tumor volume of 0.25 cm3 between GL261 tumors expressing scrambled (GL261 Scr) versus Egr1-targeting shRNA (GL261 mE3), as shown in Figure 6B. An average of 20 days was required to reach this tumor volume of 0.25 cm3 in mice bearing GL261 Scr tumors compared to 27.3 days in mice bearing GL261 mE3 tumors. Similarly there was a delay in tumor growth between HCT116 tumors expressing scrambled shRNA (HCT116 Scr) and those expressing Egr1-targeting shRNA (HCT116 hE4). An average of 27 days was required to reach this tumor volume of 1cm3 in mice bearing HCT116 Scr tumors compared to 31.6 days in mice bearing HCT116 hE4 tumors; the difference was not statistically significant (p=0.07).
Figure 6. Silencing Egr1 suppresses tumor growth in a glioma and colorectal mouse tumor model.

(A) Mean volumes of GL261 and HCT116 tumors bearing either scrambled or Egr1-targeting shRNA at various time points following implantation (B) Average number of days it took for tumors to reach 0.25cm3 (GL261) and 1.0cm3 (HCT116); *p<0.05.
DISCUSSION
The mechanisms of injury in normal tissues after irradiation include progenitor cell depletion, microvascular injury, necrosis, and inflammation (25-27). This study focuses specifically on radiation-induced apoptosis in normal tissue progenitor cells of the hippocampus and small intestine. Irradiation results in the impairment of hippocampus-dependent functions, partly owing to the apoptosis and loss of neuronal progenitor cells that normally participate in neurogenesis (28). In the gastrointestinal system, irradiation induces apoptosis of the small intestinal crypts, which leads to denudation of the intestinal mucosa and reduces the surface for nutrient absorption (29,30). Several pathways have been implicated in radiation-induced apoptosis in these tissues, including those of the tumor suppressor p53 (31) and p53 targets, such as PUMA (9), ATM (8), Bax (32), and Bcl-2 (33-35). Here we show that Egr1 is a key regulator of radiation-induced apoptosis and could be exploited for the development of protective agents to lessen radiation-induced apoptosis. Egr1 plays a major role in radiation-induced apoptosis in normal tissues, as evidenced by the radioprotective phenotype in the hippocampus and the small intestine when Egr1 was either genetically abrogated or pharmacologically suppressed. The molecular mechanism underlying the radioprotection was then investigated in cultured neuronal progenitor cells and intestinal epithelial crypt cells.
Many studies have elucidated Egr1's role in radiation-induced apoptosis in cultured cells, including one in which the knockdown of Egr1 in MEFs led to resistance from radiation-induced apoptosis (24). The present work demonstrates Egr1's role in radiation-induced apoptosis in normal tissues. To determine the molecular mechanisms by which Egr1 knockout conferred radioprotection to the hippocampus and small intestine, we analyzed expression levels of downstream transcriptional targets of Egr1 that are important for apoptotic signaling following irradiation: p53, Bax, and Bcl-2. Egr1 depletion in the knockout mouse hippocampal tissues and knockdown cell lines resulted in an abrogation of p53 induction by IR. p53 is a major mediator of radiation-induced apoptosis that lies downstream of Egr1 (20,24). Its induction by IR is dependent on Egr1, as evidenced by the failure to induce p53 in Egr1−/− MEFs following irradiation (24). This observation correlated with the finding that these MEFs were also resistant to radiation-induced apoptosis (24). Egr1's other downstream targets, Bax and Bcl-2, are members of the Bcl-2 family of proteins that regulate mitochondrial apoptosis. Proteins of the Bcl-2 family can be either pro- or anti-apoptotic and the balance between these factors can determine if a cell commits to apoptosis (36). The main mechanism of action of the Bcl-2 family of proteins is the regulation of cytochrome C release from the mitochondria via increased mitochondrial membrane permeability. Egr1 can trans-activate the gene encoding the pro-apoptotic factor Bax (23) and trans-repress the gene encoding the anti-apoptotic factor Bcl-2 (37). In accordance with this, our results showed that Egr1 depletion led to the downregulation of Bax and upregulation of Bcl-2. This finding was based on experiments in both ex vivo hippocampal tissue lysates from Egr1−/− mice and cultured cells in which Egr1 was knocked down. The former experiment enabled us to learn the mechanistic details of normal tissues. Taken together, Egr1 is important for radiation-induced apoptosis. The suppression of Egr1 in both normal tissues and cells resulted in a radioprotective effect that was correlated with the loss of p53 induction by IR and an increase in the Bcl-2/Bax expression ratio.
Due to the lack of specific inhibitors to disrupt Egr1 function, we chose to use MMA as a complement to our Egr1 knockout mouse model and shRNA knockdown strategies. MMA was first tested as a cancer treatment in the 1960's and has been shown to be useful against some cancers like glioma and testicular cancer. MMA had antitumor activity in a mouse glioma model (38) and combining radiation with MMA achieved better tumor control than MMA alone in a heterotopic mouse glioma model (39). However, its toxic side effects have limited its clinical use. The typical human dose is 25-30 μg/kg/day and is only given up to ten days (Cancer Drug Manual). Despite the toxicities, MMA is currently being investigated in the treatment of lung cancers, esophageal cancer, and mesothelioma in a clinical trial (NCT01624090; clinicaltrials.org). A recent study showed that low doses (600μg/kg) for three days a week had an anticancer effect with low toxicities (40).
In recent years there has been an interest in using MMA to ameliorate neuronal cell death after a variety of stresses (22,41,42). MMA has been shown to prevent Egr1-dependent trans-activation of pro-apoptosis genes, which resulted in an abrogation of apoptosis in neuronal cells. In our studies, we used a dose of MMA (150μg/kg) that could be safely administered to mice and that has been used to achieve cytoprotection in a previous study (43). We found that MMA attenuated radiation-induced apoptosis in small intestinal crypts and in cultured normal cells. Due to the non-specific nature of MMA's action, however, we cannot exclude the possibility that MMA conferred radioprotection in this study by inhibiting the binding of other GC-rich binding transcription factors. For example, Sp1 is another transcription factor that can bind to GC-rich sequences in DNA and is a target of MMA. It is possible that one or more of these factors contributes to the radioprotection we observed. While MMA treatment radioprotected the small intestines of mice, we did not find radioprotection of the hippocampus. In a previous study, daily injections of 150 μg/kg MMA for 52 days improved the neurological outcomes in a Huntington's disease mouse model (43). This study suggests that MMA can penetrate the blood-brain barrier. However, since we treated mice with only two doses of 150 μg/kg MMA, it is possible that prolonged dosing may be required for the radioprotection of the hippocampus.
Ideally, a clinically useful molecular target for radioprotection should confer selective protection to normal cells without impairing the cytotoxic effects of radiation on cancer cells. In marked contrast to the protective effects observed in normal tissues, knocking down Egr1 did not radioprotect the glioma and colorectal cancer cell lines. Instead, silencing Egr1 had a negative effect on the growth and survival of GL261 glioma and HCT116 colorectal cancer cells, which was enhanced in combination with radiation treatment. The antitumor effect of Egr1 knockdown was also observed in both gliomas (GL261) and colorectal (HCT116) tumor models in vivo. We speculate that targeting Egr1 produced differential responses in normal versus cancer cells because Egr1 is known to play either a pro-death or pro-life role depending on the cellular context and stimulus. Although Egr1 plays a pro-death role in normal tissues and cells, Egr1 is required for the growth of various cancers, including prostate (16), gastric (44), and kidney cancers (45). Egr1 participates in cellular growth and survival and can be induced by growth factors and mitogens (16). The fact that Egr1 can transactivate a number of pro-survival genes, including those encoding growth factors like insulin-like growth factor II, platelet-derived growth factor A and B, and transforming growth factor β1 lends support to its pro-life function (16). Insofar as Egr1 is a regulator of the epidermal growth factor receptor, Egr1 is thought to be important for colorectal cancer growth (46). A direct role for Egr1 in cell proliferation has also been proposed for glioma cells (47). Additional studies will be required to determine the mechanism by which Egr1 knockdown reduced the growth and enhanced radiation-induced apoptosis in these cancer cells. In summary, this work has identified and characterized Egr1 as a novel molecular target for selectively radioprotecting normal cells and radiosensitizing glioma and colorectal cancer cells.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Michelle Le Beau for the generous gift of the Egr1 knockout mice, Ana Heermann, Sophie Hu, Jarrett Joubert, and Rowan Karvas for their technical assistance, Andrei Laszlo for editing the manuscript, and Todd DeWees for assistance with statistical calculations.
Financial Support - National Cancer Institute grants 5RO1CA174966-03, Elizabeth and James McDonnell III Endowment (D.E. Hallahan), Washington University Department of Radiation Oncology Startup Funds (D. Thotala), and 5T32 GM07200 National Research Science Award-Medical Scientist (D.Y. Zhao).
Footnotes
The authors have no conflicts of interests to declare.
REFERENCES
- 1.Greenberger JS. Radioprotection. In Vivo. 2009;23:323–36. [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson VA, Godber T, Smibert E, Weiskop S, Ekert H. Cognitive and academic outcome following cranial irradiation and chemotherapy in children: a longitudinal study. Br J Cancer. 2000;82:255–62. doi: 10.1054/bjoc.1999.0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redmond KJ, Mahone EM, Terezakis S, Ishaq O, Ford E, McNutt T, et al. Association between radiation dose to neuronal progenitor cell niches and temporal lobes and performance on neuropsychological testing in children: a prospective study. Neuro oncology. 2013;15:360–9. doi: 10.1093/neuonc/nos303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Padovani L, André N, Constine LS, Muracciole X. Nature Reviews Neurology. Vol. 8. Nature Publishing Group; 2012. Neurocognitive function after radiotherapy for paediatric brain tumours. pp. 578–88. [DOI] [PubMed] [Google Scholar]
- 5.Surma-aho O, Niemela M, Vilkki J, Kouri M, Brander A, Salonen O, et al. Adverse long-term effects of brain radiotherapy in adult low-grade glioma patients. Neurology. 2001;56:1285–90. doi: 10.1212/wnl.56.10.1285. [DOI] [PubMed] [Google Scholar]
- 6.Crossen JR, Garwood D, Glatstein E, Neuwelt EA. Neurobehavioral sequelae of cranial irradiation in adults: a review of radiation-induced encephalopathy. J Clin Oncol. 1994;12:627–42. doi: 10.1200/JCO.1994.12.3.627. [DOI] [PubMed] [Google Scholar]
- 7.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci. 2010;11:339–50. doi: 10.1038/nrn2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ch'ang H-J, Maj JG, Paris F, Xing HR, Zhang J, Truman J-P, et al. ATM regulates target switching to escalating doses of radiation in the intestines. Nat Med. 2005;11:484–90. doi: 10.1038/nm1237. [DOI] [PubMed] [Google Scholar]
- 9.Qiu W, Carson-Walter EB, Liu H, Epperly M, Greenberger JS, Zambetti GP, et al. PUMA Regulates Intestinal Progenitor Cell Radiosensitivity and Gastrointestinal Syndrome. Cell Stem Cell. 2008;2:576–83. doi: 10.1016/j.stem.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Stem Cell. Elsevier Inc; 2013. Stem Cells Are Indispensablefor Radiation-Induced Intestinal Regeneration. pp. 1–11. [DOI] [PubMed] [Google Scholar]
- 11.Citrin D, Cotrim AP, Hyodo F, Baum BJ, Krishna MC, Mitchell JB. Radioprotectors and Mitigators of Radiation-Induced Normal Tissue Injury. The Oncologist. 2010;15:360–71. doi: 10.1634/theoncologist.2009-S104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thotala DK, Geng L, Dickey AK, Hallahan DE, Yazlovitskaya EM. A New Class of Molecular Targeted Radioprotectors: GSK-3β Inhibitors. International Journal of Radiation Oncology*Biology*Physics. 2010;76:557–65. doi: 10.1016/j.ijrobp.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thotala DK, Hallahan DE, Yazlovitskaya EM. Inhibition of Glycogen Synthase Kinase 3 Attenuates Neurocognitive Dysfunction Resulting from Cranial Irradiation. Cancer Research. 2008;68:5859–68. doi: 10.1158/0008-5472.CAN-07-6327. [DOI] [PubMed] [Google Scholar]
- 14.Yazlovitskaya EM, Edwards E, Thotala D, Fu A, Osusky KL, Whetsell WO, et al. Lithium Treatment Prevents Neurocognitive Deficit Resulting from Cranial Irradiation. Cancer Research. 2006;66:11179–86. doi: 10.1158/0008-5472.CAN-06-2740. [DOI] [PubMed] [Google Scholar]
- 15.Cao XM, Koski RA, Gashler A, McKiernan M, Morris CF, Gaffney R, et al. Identification and characterization of the Egr-1 gene product, a DNA-binding zinc finger protein induced by differentiation and growth signals. Molecular and Cellular Biology. 1990;10:1931–9. doi: 10.1128/mcb.10.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gitenay D, Baron VT. Is EGR1 a potential target for prostate cancer therapy? Future Oncology. 2009;5:993–1003. doi: 10.2217/fon.09.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datta R, Rubin E, Sukhatme V, Qureshi S, Hallahan D, Weichselbaum RR, et al. Ionizing radiation activates transcription of the EGR1 gene via CArG elements. Proc Natl Acad Sci USA. 1992;89:10149–53. doi: 10.1073/pnas.89.21.10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hallahan DE, Sukhatme VP, Sherman ML, Virudachalam S, Kufe D, Weichselbaum RR. Protein kinase C mediates x-ray inducibility of nuclear signal transducers EGR1 and JUN. Proc Natl Acad Sci USA. 1991;88:2156–60. doi: 10.1073/pnas.88.6.2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallahan DE, Dunphy E, Virudachalam S, Sukhatme VP, Kufe DW, Weichselbaum RR. C-jun and Egr-1 participate in DNA synthesis and cell survival in response to ionizing radiation exposure. J Biol Chem. 1995;270:30303–9. doi: 10.1074/jbc.270.51.30303. [DOI] [PubMed] [Google Scholar]
- 20.Nair P, Muthukkumar S, Sells SF, Han SS, Sukhatme VP, Rangnekar VM. Early growth response-1-dependent apoptosis is mediated by p53. J Biol Chem. 1997;272:20131–8. doi: 10.1074/jbc.272.32.20131. [DOI] [PubMed] [Google Scholar]
- 21.Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, et al. The Egr-1 transcription factor directly activates PTEN during irradiation-induced signalling. Nat Cell Biol. 2001;3:1124–8. doi: 10.1038/ncb1201-1124. [DOI] [PubMed] [Google Scholar]
- 22.Xie B, Wang C, Zheng Z, Song B, Ma C, Thiel G, et al. Egr-1 Transactivates Bim Gene Expression to Promote Neuronal Apoptosis. Journal of Neuroscience. 2011;31:5032–44. doi: 10.1523/JNEUROSCI.5504-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zagurovskaya M, Shareef MM, Das A, Reeves A, Gupta S, Sudol M, et al. EGR-1 forms a complex with YAP-1 and upregulates Bax expression in irradiated prostate carcinoma cells. Oncogene. 2009;28:1121–31. doi: 10.1038/onc.2008.461. [DOI] [PubMed] [Google Scholar]
- 24.Das A. Ionizing Radiation Down-regulates p53 Protein in Primary Egr-1−/− Mouse Embryonic Fibroblast Cells Causing Enhanced Resistance to Apoptosis. Journal of Biological Chemistry. 2000;276:3279–86. doi: 10.1074/jbc.M008454200. [DOI] [PubMed] [Google Scholar]
- 25.Harfouche G, Martin MT. Response of normal stem cells to ionizing radiation: a balance between homeostasis and genomic stability. Mutat Res. 2010;704:167–74. doi: 10.1016/j.mrrev.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Kirsch DG, Santiago PM, di Tomaso E, Sullivan JM, Hou WS, Dayton T, et al. p53 Controls Radiation-Induced Gastrointestinal Syndrome in Mice Independent of Apoptosis. Science. 2010;327:593–6. doi: 10.1126/science.1166202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson E, Monje M. Effect of cancer therapy on neural stem cells: implications for cognitive function. Curr Opin Oncol. 2012;24:672–8. doi: 10.1097/CCO.0b013e3283571a8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizumatsu S, Monje ML, Morhardt DR, Rola R, Palmer TD, Fike JR. Extreme sensitivity of adult neurogenesis to low doses of X-irradiation. Cancer Research. 2003;63:4021–7. [PubMed] [Google Scholar]
- 29.Potten CS. Extreme sensitivity of some intestinal crypt cells to X and gamma irradiation. Nature. 1977;269:518–21. doi: 10.1038/269518a0. [DOI] [PubMed] [Google Scholar]
- 30.Paris F, Fuks Z, Kang A, Capodieci P, Juan G, Ehleiter D, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science. 2001;293:293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 31.Chow BM, Li YQ, Wong CS. Radiation-induced apoptosis in the adult central nervous system is p53-dependent. Cell Death Differ. 2000;7:712–20. doi: 10.1038/sj.cdd.4400704. [DOI] [PubMed] [Google Scholar]
- 32.Chong MJ, Murray MR, Gosink EC, Russell HR, Srinivasan A, Kapsetaki M, et al. Atm and Bax cooperate in ionizing radiation-induced apoptosis in the central nervous system. Proc Natl Acad Sci USA. 2000;97:889–94. doi: 10.1073/pnas.97.2.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Przemeck SMC, Duckworth CA, Pritchard DM. Radiation-induced gastric epithelial apoptosis occurs in the proliferative zone and is regulated by p53, bak, bax, and bcl-2. Am J Physiol Gastrointest Liver Physiol. 2007;292:G620–7. doi: 10.1152/ajpgi.00391.2006. [DOI] [PubMed] [Google Scholar]
- 34.Rotolo JA, Maj JG, Feldman R, Ren D, Haimovitz-Friedman A, Cordon-Cardo C, et al. Bax and Bak Do Not Exhibit Functional Redundancy in Mediating Radiation-Induced Endothelial Apoptosis in the Intestinal Mucosa. International Journal of Radiation Oncology*Biology*Physics. 2008;70:804–15. doi: 10.1016/j.ijrobp.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 35.Pritchard DM, Potten CS, Korsmeyer SJ, Roberts S, Hickman JA. Damage-induced apoptosis in intestinal epithelia from bcl-2-null and bax-null mice: investigations of the mechanistic determinants of epithelial apoptosis in vivo. Oncogene. 1999;18:7287–93. doi: 10.1038/sj.onc.1203150. [DOI] [PubMed] [Google Scholar]
- 36.Volkmann N, Marassi FM, Newmeyer DD, Hanein D. Cell Death Differ. Vol. 21. Nature Publishing Group; 2013. The rheostat in the membrane: BCL-2 family proteins and apoptosis. pp. 206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang RP, Fan Y, Peng A, Zeng ZL, Reed JC, Adamson ED, et al. Suppression of human fibrosarcoma cell growth by transcription factor, Egr-1, involves down-regulation of Bcl-2. Int J Cancer. 1998;77:880–6. doi: 10.1002/(sici)1097-0215(19980911)77:6<880::aid-ijc14>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy BJ, Yarbro JW, Kickertz V, Sandberg-Wollheim M. Effect of mithramycin on a mouse glioma. Cancer Res. 1968;28:91–7. [PubMed] [Google Scholar]
- 39.McNulty TB, Dirks VA, Yarbro JW, Kennedy BJ. Combination Therapy with Radiation and Mithramycin or Actinomycin D in a Transplanted Mouse Glioma. Cancer. 1969;23:1273–9. doi: 10.1002/1097-0142(196906)23:6<1273::aid-cncr2820230605>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 40.Ohgami T, Kato K, Kobayashi H, Sonoda K, Inoue T, Yamaguchi S-I, et al. Low-dose mithramycin exerts its anticancer effect via the p53 signaling pathway and synergizes with nutlin-3 in gynecologic cancers. Cancer Sci. 2010;101:1387–95. doi: 10.1111/j.1349-7006.2010.01543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chatterjee S, Zaman K, Ryu H, Conforto A, Ratan RR. Sequence-selective DNA binding drugs mithramycin A and chromomycin A3 are potent inhibitors of neuronal apoptosis induced by oxidative stress and DNA damage in cortical neurons. Ann Neurol. 2001;49:345–54. [PubMed] [Google Scholar]
- 42.Hagiwara H, Iyo M, Hashimoto K. Mithramycin protects against dopaminergic neurotoxicity in the mouse brain after administration of methamphetamine. Brain Res. 2009;1301:189–96. doi: 10.1016/j.brainres.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 43.Ferrante RJ. Chemotherapy for the Brain: The Antitumor Antibiotic Mithramycin Prolongs Survival in a Mouse Model of Huntington's Disease. Journal of Neuroscience. 2004;24:10335–42. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun T, Tian H, Feng Y-G, Zhu Y-Q, Zhang W-Q. Egr-1 promotes cell proliferation and invasion by increasing β-catenin expression in gastric cancer. Dig Dis Sci. 2013;58:423–30. doi: 10.1007/s10620-012-2356-4. [DOI] [PubMed] [Google Scholar]
- 45.Scharnhorst V, Menke AL, Attema J, Haneveld JK, Riteco N, van Steenbrugge GJ, et al. EGR-1 enhances tumor growth and modulates the effect of the Wilms' tumor 1 gene products on tumorigenicity. Oncogene. 2000;19:791–800. doi: 10.1038/sj.onc.1203390. [DOI] [PubMed] [Google Scholar]
- 46.Chen A, Xu J, Johnson AC. Curcumin inhibits human colon cancer cell growth by suppressing gene expression of epidermal growth factor receptor through reducing the activity of the transcription factor Egr-1. Oncogene. 2006;25:278–87. doi: 10.1038/sj.onc.1209019. [DOI] [PubMed] [Google Scholar]
- 47.Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002;193:287–92. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.